
Abstract
Increased endoplasmic reticulum (ER) stress and the activated unfolded protein response (UPR) signaling associated with it play key roles in physiological processes as well as under pathological conditions. The UPR normally protects cells and re-establishes cellular homeostasis, but prolonged UPR activation can lead to the development of various pathologies. These features make the UPR signaling pathway an attractive target for the treatment of diseases whose pathogenesis is characterized by chronic activation of this pathway. Here, we focus on the molecular signaling pathways of the UPR and suggest possible ways to target this response for therapeutic purposes.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
The endoplasmic reticulum and ER stress
The endoplasmic reticulum (ER) is an organelle that comprises a continuous membranous structure consisting of tubules, flattened sacs, and the lumen [1]. The entire ER network is interconnected, and the membranes serve to separate molecules that reside in the lumen of the ER from those in the cytoplasm [2, 3]. The ER is highly sensitive to the nutritional and energy states of the cell, and it dramatically adjusts its machinery depending on the demand created in the cell [4, 5]. The ER folds and modifies newly synthesized peptides into their tertiary, lowest energy conformation [6], following which proteins are translocated to the Golgi apparatus for packaging and secretion, or are transported to membranes [5, 7, 8] (Fig. 1). Proteins that enter the ER are subjected to post-translational modifications such as N-linked glycosylation, disulfide bond formation, and proline hydroxylation [9, 10].

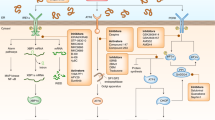
Normal state. Proteins that enter the ER are folded and transported to the Golgi apparatus or other destination. GRP78 is bound to the luminal domains of PERK, IRE1, and ATF6
The ER lumen has characteristics of the extracellular environment [10], and it is possible that the ER is evolved from an invagination of the plasma membrane [10]. This is indeed a viable possibility and is supported by the observation that secretory proteins are produced in the ER in an environment similar to that of the extracellular conditions, enabling proteins to protect their conformation when they are outside the cell. Similarly, the calcium concentration within the ER is the same as that of the extracellular environment [11]. As all secretory and membrane proteins pass through the ER before exiting the cell to face the extracellular environment, it is likely the ER provides a final control point to ensure the fidelity of proteins before exporting them out of the cell. In addition to proteins, sterols, phospholipids, and assembled lipoproteins are also synthesized in the lumen of ER [12, 13]. Finally, the ER serves as a major calcium reservoir: maintenance of a high calcium concentration within the ER is essential for ER function, and for optimizing the activity of enzymes within the ER [14–16].
The so-called misfolded proteins, which are not properly folded into their functionally active structures, are retained in the lumen of ER until they attain their proper conformations [5, 17, 18]. If this final tertiary structure cannot be achieved, misfolded proteins are then transported back to the cytosol and subjected to ubiquitination and proteasome-dependent degradation, a process referred to as ER-associated degradation (ERAD) [17–19] (Fig. 1). Under conditions wherein misfolded proteins accumulate within the lumen of the ER, or when ER capacity cannot meet the demand for protein synthesis, the organelle enters into a state called “ER stress” [5, 17, 18]. This condition is triggered by processes such as increased protein synthesis, genetic mutations that cause defects in folding, alteration in calcium homeostasis, and nutrient starvation such as glucose deprivation [5, 15, 17]. As a cellular recovery and survival mechanism, the ER responds to ER stress by activating a series of complex coordinated signaling pathways, collectively called the unfolded protein response (UPR) [4, 5, 17, 18, 20–22] (Fig. 2). The UPR is a physiological response when it occurs in acute settings, but it can lead to pathologies, or even cell death (reviewed below) under conditions of chronic, unresolved ER stress.

ER stress state. During ER stress, GRP78 dissociates from PERK, IRE1, and ATF6. PERK and IRE1 oligomerize, forming a dimeric structure with a deep groove where peptide can bind. Upon oligomerization, PERK and IRE1 are auto-phosphorylated. PERK phosphorylates eIF2α, leading to attenuation in global protein synthesis. Phosphorylated eIF2α leads to translation and nuclear translocation of ATF4 and Nrf2. Activated IRE1 mediates unconventional mRNA splicing of XBP1 to generate XBP1s. IRE1 also recruits TRAF2 and ASK1 and leads to activation of JNK. ATF6 translocates to the Golgi apparatus and the cytoplasmic tail of ATF6 acts as a transcription factor to regulate UPR target genes
The unfolded protein response
The UPR is initiated by activation of three major transducers: protein kinase RNA (PKR)-like ER kinase (PERK), inositol requiring enzyme-1 (IRE1), and activating transcription factor-6 (ATF6) [5, 23] (Fig. 2). The primary role of the UPR is to maintain and re-establish ER homeostasis [4, 23]. Under physiological conditions, the UPR is activated as a cellular survival program that protects the cell from ER stress and helps it recover from damage or increased work overloads. In essence, the UPR functions to reset the ER, from the “stress” state back to its normal condition [4, 17]. However, sustained ER stress and prolonged UPR activation can also trigger the apoptotic machinery, and ultimately lead to cell death [4, 18]. Despite the beneficial role of the UPR in the maintenance of cellular homeostasis, prolonged ER stress often leads to pathological conditions [21, 24, 25]. The functional significance of the UPR is not yet fully understood, but it is known to contribute to the pathogenesis of many diseases including diabetes, cancer, atherosclerosis, neurologic diseases, and inflammatory bowel disease [4, 22, 26–28].
Under ER stress conditions, a chaperone called the glucose regulating protein 78 (GRP78) initially cues the three transducers of the UPR for activation of downstream signaling cascades [5, 29]. GRP78, also known as BiP, is a member of the Hsp70 family of chaperones, and it negatively regulates the UPR signaling pathway by physically interacting with the three UPR transducers [5]. In the basal state (no external stimulus or stress condition), GRP78 is bound to the luminal domains of PERK, IRE1, and ATF6, and the three UPR transducers remain in a state of low activity [5]. Under stress conditions (accumulation of unfolded and misfolded proteins in the ER), GRP78 is released from all three transducers and binds to unfolded and misfolded proteins in the lumen of ER [30]. Exactly how the transducers sense ER stress is still under investigation [23]. Several models have been proposed for activation of the UPR and suggest that each branch of the UPR is separately regulated [17, 18]. Below, we briefly outline how the UPR signaling elements are regulated in order to understand potential therapeutic approaches.
Protein kinase RNA (PKR)-like ER kinase
PERK is a type I transmembrane kinase that resides in the ER. Activation of PERK during ER stress reduces protein synthesis globally [29, 31], thereby decreasing the speed of entry of new polypeptides into the ER. This mechanism facilitates the process of homeostasis, by enabling the existing unfolded proteins in the lumen to attain their folded conformation. While PERK plays a central role in the regulation of the UPR, we do not fully understand the mechanism by which PERK senses ER stress, although several models have been suggested [18]. For example, the binding of GRP78 to PERK may maintain PERK in an inactive state, as supported by reports that GRP78 and PERK form a complex under normal conditions [30], and that PERK is activated by reduction in the expression of GRP78 and is inhibited when GRP78 is overexpressed [30]. Thus, one model posits that as a result of an increase in the amount of unfolded proteins in the lumen of ER, GRP78 is titrated away from PERK by the misfolded proteins, and this dissociation leads to oligomerization and activation of PERK [17, 18]. Another model proposes that accumulation of unfolded proteins leads to a dissociation of GRP78 from the unfolded proteins that it is bound to in the lumen. This leads to direct binding of the unfolded proteins to the luminal domain of PERK, which in turn results in oligomerization and autophosphorylation of PERK [18]. The deep groove formed by the dimeric structure of PERK can support the direct binding of a peptide [17, 18]. Additional detailed studies are needed to unravel the mechanisms of PERK activation.
The kinase activity of PERK leads to phosphorylation of Serine51 of its main downstream effector, the eukaryotic initiation factor 2 (eIF2α), which then leads to global attenuation of protein synthesis [29, 31]. However, proteins such as activating transcription factor 4 (ATF4) escape from this global inhibition of protein synthesis [32]. ATF4 contains inhibitory upstream open reading frames (uORFs) in its mRNA that normally suppress the initiation of translation in the absence of ER stress [33]. However, when ER stress is elevated, phosphorylated eIF2α leads to ribosomal skipping of the uORFs and active translation of ATF4 mRNA [33]. ATF4 acts as a transcription factor for genes that contribute to ER function and apoptosis. An example of an immediate ATF4 target gene is the transcription factor C/EBP homologous protein (CHOP) [34, 35], which is believed to participate in the initiation of apoptotic pathways by upregulating the transcription of genes when accumulation of unfolded/misfolded proteins goes beyond the capacity of the ER [36]. Another gene induced by ATF4 is growth arrest and DNA damage-inducible 34 (GADD34) [37, 38]. GADD34 interacts with the catalytic subunit of protein phosphatase (PP1c) [37] and serves as a negative feedback loop to deactivate PERK action by dephosphorylating eIF2α, by which it resets the UPR to the basal state once ER stress is resolved [37, 38].
eIF2α phosphorylation also increases translation of CCAAT/enhancer-binding protein (C/EBP) in in vitro models [39]. Likewise, reduced phosphorylation of eIF2α, achieved by overexpression of GADD34 in the liver, leads to decreased expression of C/EBPα and C/EBPβ, and of their downstream effector PPARγ [39]. Accordingly, GADD34 transgenic mice display lower blood glucose levels, improved glucose tolerance, lower levels of liver glycogen, and diminished hepatosteatosis [39].
Another effector protein that is phosphorylated by PERK is nuclear factor erythroid2-related factor 2 (Nrf2) [40], which forms a complex with Kelch-like erythroid-cell-drived protein with cap’n’collar homology-associating protein 1 (Keap1). The Nrf2/Keap1 complex is maintained in the cytoplasm under normal conditions (without ER stress) and is subjected to degradation by ubiquitin–proteasome pathway [40]. Upon ER stress, PERK phosphorylates Nrf2 and results in dissociation of the Nrf2/Keap1 complex. The released free Nrf2 stably translocates to the nucleus, and acts as a transcription factor for genes that encode antioxidant proteins and detoxifying enzymes [40].
Inositol requiring enzyme 1
The second arm of the UPR is IRE1. Like PERK, IRE1 is also a type I transmembrane kinase, and the two transmembrane proteins share similar structures in their luminal domains [18, 41]. IRE1 is highly conserved from yeast to humans [42, 43]. Two homologues of IRE1 have been identified: IRE1α and IRE1β [41]. IRE1α is expressed in all cells throughout the body, while expression of IRE1β is restricted to the intestinal epithelium and lung [44, 45]. IRE1 has endoribonuclease as well as kinase activity [46–48]. During ER stress, IRE1-bound GRP78 is released from IRE1 [49]. Studies in yeast document that, once this release occurs, the structure of IRE1 allows it to bind to unfolded proteins, providing insight into the mechanism of the IRE1 activation that follows ER stress [17, 30, 49]. IRE1α has a conserved cLD, which contains interface 1 and interface 2 [50]. Interface 1 forms a deep groove where peptides can bind, while interface 2 induces further oligomerization [50]. As IRE1 senses ER stress and oligomerizes, its kinase and endoribonuclease activities are activated [51], and it is auto-phosphorylated, which increases its kinase activity, initiating a signaling cascade that can activate c-Jun amino terminal kinase (JNK) [52]. The endoribonuclease domain of IRE1 cleaves the mRNA of a transcription factor called X-box binding protein-1 (XBP1) [53–55], and leads to the translation of a higher molecular weight protein, which is the spliced form of XBP1 (XBP1s) [43, 56–58].
XBP1 is a member of the CREB/ATF basic region-leucine zipper family of transcription factors, and is ubiquitously expressed in adult tissues [59]. The full-length XBP1 mRNA is referred to as unspliced XBP1 (XBP1u) [53, 54]. IRE1 cleaves the mRNA of XBP1u and initiates the excision of 26 nucleotides from the mRNA [53], which in turn results in a frame shift and ultimately in the generation of the spliced form XBP1s [53–55]. While XBP1u is extremely unstable, and is subjected to proteasome-dependent and -independent degradation soon after translation [60], XBP1s is a highly active transcription factor and a master regulator of ER capacity [4, 5, 18, 57, 58, 61]. XBP1s upregulates expression of ER chaperones [57, 62] and the components of ER-associated degradation [57], and also plays a key role in ER expansion [58, 63]. While XBP1s target genes are fairly well known [57], the exact role of XBP1u is debated. XBP1u was initially believed to negatively regulate the transcriptional activity of XBP1s by directly interacting with the spliced form, forming a complex, and directing XBP1s to proteasome-mediated degradation [64, 65]. However, a subsequent report suggested that XBP1u is capable of inducing the UPR and increasing expression of XBP1s target genes as well as other non-target genes, but only if XBP1u is stabilized and its degradation is blocked as implicated by a XBP1u mutant with improved stability [60]. It has been documented that the half-life of XBP1u is extremely short and the protein is usually subjected to rapid proteasomal degradation soon after synthesis, at a rate that is almost the same as its level of synthesis [60]. The same study also concluded that the short half-life of XBP1u abrogates a physiologically significant role for the unspliced protein and proposed that the rapid degradation of XBP1u is required to prevent uncontrolled activation of the UPR [60]. We also agree that XBP1u does not have any physiological importance as we have been unable to detect the unspliced protein in any tissue or cells that we have examined to date (unpublished observations).
Activating transcription factor 6
The third arm of the UPR is ATF6, which is categorized as a type II transmembrane protein with two homologs, ATF6α and ATF6β [66]. Structurally, ATF6 contains a DNA-binding domain with the bZIP and a transcriptional activation domain in the cytoplasmic portion [66]. In its N-terminus, ATF6 carries two Golgi localization signals (GLS), referred to as GLS1 and GLS2 [67, 68]. Under normal conditions without ER stress, GRP78 binds to GLS1, and this interaction retains ATF6 in an inactive state in the ER membrane [67, 68]. Once activated by ER stress, GRP78 is released from ATF6, and triggers the translocation of ATF6 to the Golgi apparatus, a process that requires the GLS2 domain [67]. The transported ATF6 is subjected to intramembrane proteolysis, whereby it is cleaved by SP1 and SP2 [69–71], resulting in the generation of a 50-kDa DNA-binding domain of ATF6 derived from its cytoplasmic tail, referred to as ATF6F or ATF6N [68]. The cleaved fragment translocates to the nucleus and acts as a transcription factor that activates UPR target genes. ATF6 regulates the expression of a variety of genes that contain that CRE and ERSE [62, 72]. While little is known about the negative regulation or deactivation of ATF6, XBP1u also reportedly binds ATF6α and leads to proteasomal degradation of ATF6α [65].
The UPR elements in pathophysiology
The exact functions of the UPR under physiological and pathological conditions, and its role in disease pathogenesis, are not fully understood yet. However, what we do know is that the UPR is involved in various processes, including development, differentiation, maintenance of homeostasis, and apoptosis [17, 25]. The physiological role of the UPR is well defined from studies with animal models. In this section, we review the knockout models that have been developed for elucidating the role of the major UPR signaling molecules in pathophysiology, focusing mainly on metabolic diseases.
PERK in pathophysiology
An important characteristic of the Perk −/− mouse is that it develops diabetes, due to the destruction of pancreatic β cells [73]. Under normal conditions, insulin is synthesized in response to increased glucose levels in the blood, and is secreted into the circulation to keep glucose levels within a very tight range [74]. Insulin biosynthesis takes place in the ER of pancreatic β cells, and is dynamically controlled by ER capacity and stress conditions. When the demand for insulin synthesis exceeds the capacity of the ER, it triggers ER stress, and activates PERK [4] and its downstream effector eIF2α, to reduce the workload. In Perk −/− mice, however, protein synthesis cannot be controlled by PERK even under conditions of ER stress [73]. In fact, when Perk −/− mice are challenged with glucose, uncontrolled synthesis of insulin further triggers ER stress [31, 73]. The prolonged unresolved ER stress initiates the apoptotic pathway, culminating in the destruction of β cells and the development of diabetes [73].
While the study of whole body knockout mouse models of PERK points out an important role this kinase activity in pancreatic β cell biology, β cell-specific PERK-deficient mice surprisingly do not develop diabetes [75]; rather they have a normal number of β cells and display normal glucose tolerance with low of blood glucose level [75]. Further investigation is needed to clarify the relationship between PERK, insulin biosynthesis, and its regulation in β cells.
eIF2α in pathophysiology
Mice in which Serine51 of eIF2α is mutated to Alanine (eIF2αS51A mice) have a complete defect in eIF2α phosphorylation at Serine51. They display defects in pancreatic β cells during late embryonic development, and develop severe hypoglycemia and a failure to survive for more than 18 h after birth [76]. Multiple factors are believed to be responsible for the hypoglycemia in eIF2αS51A mice, including lower levels of gluconeogenic enzymes and diminished glycogen storage in the liver [76]. Heterozygous eIF2αS51A mice, on the contrary, have a functionally active pancreas, plus normal basal glucose and insulin levels [77]. They display normal glucose tolerance levels and insulin sensitivity under normal diet feeding conditions. However, when heterozygous eIF2αS51A mice are challenged with a high-fat diet (HFD), they develop a higher level of obesity compared to control animals [77]. A combination of an eIF2α mutation and HFD leads to abnormal ER function, glucose intolerance, and reduced insulin sensitivity [77]. Note that transgenic mice, in which the function of eIF2α is impaired, exhibit features that are similar to those observed in humans with type 2 diabetes. The absence of eIF2α in mice results in increased ER stress and malfunction of the pancreas, resulting from β cell destruction [77].
CHOP in pathophysiology
Depletion of CHOP, a factor that promotes programmed cell death, improves β cell function and cell survival in mice [78]. The type 2 diabetes that is induced by a high fat diet (HFD) and treatment with a moderate dose of streptozotocin (STZ) is reversed by deletion of the CHOP gene [78]. Deficiency in CHOP maintains insulin secretion and prevents hyperglycemia in the HFD/STZ mouse model [78]. These features are also observed in the leptin receptor-deficient obese db/db mouse model [78]. CHOP deletion alone leads to an increase in body weight with augmented adiposity, but without any disturbance in glucose metabolism [78–80]. The contributions of CHOP function to other organs have also been reported [81, 82]. In particular, the induction of apoptosis in lung tissue following intraperitoneal treatment with LPS is suppressed in CHOP knockout mice [82], as is ischemia-associated apoptosis of neurons in the brain [81].
Another example of the pathological effects of a chronic UPR is seen in the AkitaIns2 mouse model [83]. This mouse has a missense mutation in the proinsulin 2 (Ins2) gene (Cysteine96 residue to Tyrosine) [84, 85], which disrupts disulfide bonds between chains of insulin and leads to improper folding of insulin. The resulting unfolded insulin is retained in the ER of pancreatic β cells, induces ER stress, and activates UPR signaling [83]. Phenotypic features of the AkitaIns2 mouse include hypoinsulinemia and hyperglycemia [83]. These features of type I diabetes are believed to be primarily due to chronic activation of the UPR in β cells, triggered by the accumulation of unfolded insulin protein [83, 86]. This notion is substantiated in studies of AkitaIns2 mice that are crossed with Chop −/− mice. Prolonged ER stress in the pancreatic β cells is known to induce expression of CHOP, an ER stress-associated apoptosis factor [87]; thus, increased expression of CHOP, as in the AkitaIns2 mouse, causes programmed cell death of pancreatic β cells and the development of progressive hypoinsulinemia and diabetes [83]. However, when the AkitaIns2 mouse is bred with Chop −/− mice, the offspring AkitaIns2 Chop −/− mice exhibit decreased apoptosis of pancreatic β cells and a resulting delay in the progression of diabetes [83].
In parallel, deletion of CHOP prevents UPR-induced apoptosis and improves glucose homeostasis in HFD-fed eIF2αS/A mutant mice that are obese and diabetic [78]. CHOP deletion improves β cell function and preserves pancreatic β cell mass [78]. These phenotypic features are also observed in STZ-treated mice and leptin receptor-deficient db/db mouse model wherein CHOP deletion improves glucose tolerance and prevents hyperglycemia in the fasting state [78].
ATF6α in pathophysiology
ATF6α knockout mice do not display significant differences in the expression of ER chaperones relative to controls [88]. mRNA expression profiling of Atf6α −/− cells demonstrates no changes except in expression of the Atf6α gene. Moreover, mice in which ATF6α is deleted do not display any developmental defects, leading to the conclusion that ATF6α does not have an important role in embryonic and postnatal development [88]. However, in vitro as well as in vivo studies show that ATF6α is needed for folding, secretion, and degradation during ER stress condition [88], suggesting that ATF6α is involved in mediating adaptation to chronic ER stress. Atf6α −/− mice also display persistent ER stress in the liver and kidneys upon induction of ER stress with chemicals [89], and unresolved ER stress in the liver of these mice results in a loss of lipid homeostasis and microvesicular steatosis [89].
XBP1 in pathophysiology
A physiological role for XBP1 was first identified for the differentiation of plasma cells [90]. XBP1 expression was found to be upregulated by IL-4 when plasma cells were induced to differentiate [91]. Accordingly, XBP1 overexpression in B cells induces terminal differentiation of these cells into antibody-producing plasma cells [90]. Likewise, XBP1 depletion results in fewer numbers of plasma cells, while the B cells themselves display a defect in immunoglobulin production in in vitro [90]. The importance of XBP1 in physiological conditions is substantiated by reports that germline deletion of XBP1 is lethal [92, 93]. These initial studies on XBP1 were performed prior to the recognition that XBP1 is the mammalian homolog of Hac1p, and that it plays a key role in UPR signaling. Germline Xbp1 −/− mice die from severe liver hypoplasia [92] or necrosis of cardiac myocytes [93]. On the other hand, heterozygous Xbp1 +/− mice are viable and appear to possess no phenotype until they are challenged with a HFD (discussed further below under “Obesity and type 2 diabetes” section) [26].
The UPR in disease
As outlined above, elements of the UPR participate in various physiological conditions. Although precise genetic and mechanistic association of the UPR with human disease has not been fully investigated yet, considerable ongoing basic research is focused on uncovering how the UPR contributes to human disease, and results of such studies are rapidly being translated to clinical settings. Malfunction of the ER and failure in localization of proteins to their correct cellular destinations are widely believed to cause a number of diseases in humans [4, 5, 25, 94, 95]. Some of these examples, which are related to metabolic, neurodegenerative, and oncologic disorders, are discussed briefly below and of potential therapeutic relevance to UPR-directed strategies are outlined below.
Obesity and type 2 diabetes
Obesity is a complex metabolic disorder that contributes to the development of many other life-threatening diseases, including heart disease, type 2 diabetes, and cancer [96–98]. Type 2 diabetes is a highly debilitating condition that arises in obesity [96, 99]. Over the last decade, increased ER stress signaling has been implicated in the development of insulin resistance and type 2 diabetes [26, 27, 100, 101]: initial observations indicated that ER stress parameters, such as phosphorylation of PERK and IRE1, are increased in the liver and adipose tissues of obese and type 2 diabetic mice [26, 100, 102–105]. Increased ER stress signaling, through activation of IRE1, leads to inhibition of insulin receptor signaling, which in turn results in insulin resistance and type 2 diabetes [26]. Furthermore, when Xbp1 +/− mice, which are on a background that is completely resistant to the development of obesity, insulin resistance, and type 2 diabetes, are fed a HFD, these mice in fact develop obesity, severe insulin resistance, and type 2 diabetes [26]. These observations provide the first evidence that ER stress has a key role in the pathology of type 2 diabetes. Since then, other groups have also reported that ER stress is increased in obesity, and have established a causal relationship between the protein folding capacity of the ER and insulin sensitivity in obesity [103, 105].
Intense efforts are currently aimed at understanding why ER stress develops in obesity. It was reported that free fatty acids (FFA), which are implicated in the development of insulin resistance in obesity, cause ER stress and activate the UPR [106–108]. For example, the saturated FFA palmitate creates ER stress in pancreatic β cells, hepatocytes, and cardiomyoblasts. Excess palmitate causes perturbations in the ER system, and activates the UPR by altering the integrity of the ER membrane [109–112]. Another possible link between obesity and the development of ER stress is the mammalian target of rapamycin (mTOR) signaling pathway [113]. mTOR is involved in regulating a wide range of cellular events, such as growth, proliferation, metabolism, autophagy, and apoptosis [114, 115]. mTOR functions in two different complexes called mTOR complex 1 and 2 (mTORC1 and mTORC2) [116, 117]. mTORC1 comprises mTOR, raptor, mLST8, Deptor, and PRAS40; and mTORC2 is composed of mTOR, rictor, Deptor, Protor, mSIN1, and mLST8 [117]. Increased activation of the mTORC1 pathway blocks insulin and IGF1 signaling pathways. In tuberous sclerosis complex (TSC)-deficient cells, the insulin-induced tyrosine phosphorylation of insulin receptor substrate 1 and 2 (IRS1 and IRS2) is blocked, and consequently, activation of phosphotidyl inositol 3-kinase (PI3K) and its downstream Akt is inhibited [118, 119]. A deficiency of the TSC1 or TSC2 genes leads to constitutive activation of the mTORC1 complex [120, 121]. ER stress levels in these TSC-deficient cells are elevated in an mTORC1-dependent manner, and activation of the UPR contributes to mTORC1-mediated inhibition of insulin signaling via degradation of IRS1 [113]. Considering the fact that obesity is characterized by increased mTORC1 activity, it is possible that this pathway contributes to the development of ER stress in obesity.
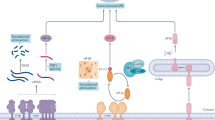
As discussed above, even heterozygous deficiency of XBP1s is sufficient to create severe ER stress, insulin resistance, and type 2 diabetes in mice fed a HFD [26]. Our recent observations led us to identify an interesting pathology that plays a central role in the development of ER stress in obesity. We have shown that p85α and p85β, the regulatory subunits of PI3K, interact with XBP1s, and that this interaction plays an important role in nuclear translocation of XBP1s [122] (Fig. 3). This interaction is also extremely important for driving the nuclear translocation of XBP1s during postprandial states to inhibit gluconeogenesis, and also to reduce ER stress as a result of nutrient fluxes. However, the interaction between p85s and XBP1s is disrupted in obesity, and there is a major reduction in nuclear translocation of XBP1s and in the upregulation of chaperones [122]. These results indicate that obesity is characterized by loss of XBP1s activity or by an XBP1s-deficient state. Given these observations, it is possible to postulate that loss of XBP1s activity contributes significantly to the development of ER stress in obesity, and consequently to insulin resistance and type 2 diabetes. Indeed, reestablishing the activity of XBP1s in the liver of obese and diabetic mice greatly enhances glucose tolerance, increases insulin sensitivity, and reduces blood glucose levels to euglycemia [123].
Calcium levels in the ER also contribute to the development of obesity-related ER stress. Normally, the ER stores free calcium, and the high calcium levels in its lumen are essential for ER functions and ER homeostasis [14, 16]. Perturbation of luminal calcium concentrations in the ER creates severe ER stress as it interferes with the activities of enzymes and chaperones [14, 124]. The Ca2+ pump sarco(endo)plasmic reticulum Ca2+-ATPase (SERCA) resides in the ER membrane and functions to reuptake cytosolic calcium into the lumen of the ER [16]. Inhibition of SERCA activity by thapsigargin blocks calcium reuptake from the cytoplasm into the ER lumen, bringing about severe ER stress and triggering activation of the UPR [16, 124]. Notably, SERCA2b, the main isoform of SERCA2 in the liver [124], is significantly reduced in the liver of obese and diabetic mice, and overexpression of this isoform in the liver of obese mice markedly reduces ER stress and blood glucose levels, resulting in improved glucose tolerance and insulin sensitivity [15]. This role of SERCA2b is likely mediated via increased chaperone activity as a result of restoration of calcium levels in the ER by SERCA2b [15].
A recent surprising finding was that the inflammatory kinase p38 mitogen-activated protein kinase (p38 MAPK) interacts with XBP1s [125] (Fig. 3). In general, inflammatory pathways are believed to be detrimental for metabolic homeostasis [126–128]. Indeed, recent reports indicate that ER stress in obesity could be induced by inflammation and inflammatory signaling cascades [129–134]. However, activation of inflammatory signaling cascades by TNF-α during ER stress conditions has a completely different effect, and in fact may lead to a reduction in ER stress [125]. This outcome is mediated through activation of p38 MAPK. Despite being one of the main inflammatory nodules in the cell, p38 MAPK greatly enhances nuclear translocation and activation of XBP1s by phosphorylating it on Threonine48 and Serine61 [125]. Furthermore, p38 MAPK is normally activated in the liver after refeeding, but this signaling mechanism is blunted in obesity. Thus, phosphorylation of XBP1s is also diminished in obesity and its activity is reduced [125]. Gain-of-function experiments in the liver of obese and diabetic mice support the finding that activation of p38 MAPK enhances XBP1s activity, relieves ER stress, greatly reduces glucose intolerance and insulin resistance, and ultimately normalizes the blood glucose levels [125]. Collectively, these observations raise the important issue of the various roles played by inflammatory signaling in the development of metabolic diseases, and indicate that inflammation itself might even be beneficial for ER stress states and for metabolic homeostasis under certain circumstances.
Increased ER stress in hypothalamic neurons of obese mice significantly contributes to the development of leptin resistance and obesity [27]. In this context, brain-specific XBP1 depletion in mice leads to the development of obesity and severe leptin resistance when the animals are challenged with a HFD [27].
Taken together, current evidence that links ER stress to obesity and obesity-associated diseases suggests that reducing ER stress in obesity would be an attractive strategy for the treatment of obesity and type 2 diabetes. How can this be achieved by targeting elements of the UPR signaling pathways? Inhibition of IRE1 kinase activity without effecting its endoribonuclease activity could be a possible therapeutic modality aimed at reducing ER stress-mediated insulin resistance. Increasing the activity of XBP1 also holds great promise for increasing ER capacity and reducing ER stress in obesity. Indeed, as discussed above, XBP1s activity is also reduced in obesity, and re-establishment of this activity has robust anti-diabetic effects [123]. A major challenge in this area of research, however, is that inhibition of a single molecule or arm of the UPR signaling network might be compensated by other mechanisms. However, the strategy of targeting the ER as a whole, and increasing its capacity and efficiency, may yield new therapeutic approaches for treatment of type 2 diabetes and obesity.

ER stress in obesity and type 2 diabetes. a The regulatory subunits of PI3K, p85α and p85β, form a heterodimer, which dissociate from each other during insulin receptor signaling. Binding of p85α or β to XBP1s leads to nuclear translocation of XBP1s. In obesity conditions, interaction of p85α or β with XBP1s is disrupted, and it results in defective XBP1s nuclear translocation. b XBP1s interacts with FoxO1 and leads to proteasome-mediated degradation of FoxO1. Defective XBP1s nuclear translocation in obesity conditions results in accumulation of FoxO1 in the nucleus. c p38 MAPK phosphorylates XBP1s on Thr48 and Ser61 residues, and these phosphorylations are required for XBP1s nuclear translocation, which is defective in obesity conditions
Wolcott-Rallison syndrome
The possible contribution of UPR elements in the development of human disease has direct relevance for the pathogenesis of Wolcott-Rallison (WR) syndrome, a disorder characterized by early-onset diabetes and multiple epiphyseal dysplasias [135, 136]. WR syndrome is an autosomal recessive disease caused by mutations in the EIF2AK3 gene, which encodes PERK in humans [135, 136]. The mutations in WR syndrome impair the ability of PERK to phosphorylate eIF2α [136]. Patients with this syndrome exhibit features that are similar to those reported for PERK-deficient mice, which is now an accepted model for studying the pathologies associated with this disorder [73, 135]. Patients with WR syndrome develop hypoinsulinemia and hyperglycemia, due to a progressive destruction of pancreatic β cells [135, 137]. Strikingly, however, WR patients often display acute severe hypoglycemia, which is not reported for Perk −/− mice [138]. WR syndrome has not been investigated in depth at a molecular level, but the fact that PERK is a major contributor to the development of type 1 diabetes mellitus in this syndrome underscores the significance of the UPR in the pathophysiology of WR-related diabetes.
α1-Antitrypsin deficiency
Deficiency of α1-antitrypsin (α1-AT) is an example of a human disease caused by protein misfolding in the ER. α1-AT is a protease inhibitor that is synthesized in the liver, and functions primarily to protect cells from neutrophil elastase activity, which enzymatically destroys tissues, especially in lung alveoli. α1-AT deficiency is a genetic disorder caused by a mutation in the α1-AT gene [139]. The mutant α1-ATZ molecule is one of the most common deficiency variants [139, 140], arising from a point mutation at the Glutamate342 which is substituted with a Lysine residue [140]. The substitution disrupts the structure of α1-AT such that mutant α1-ATZ molecules favor the formation of a dimer [140]. α1-ATZ is functionally active even after it has been secreted [141–143]. However, the ER machinery tends to retain the misfolded α1-ATZ proteins in the lumen until such time as it may be properly folded [142]. α1-ATZ is degraded by the proteosome-dependent pathway [144], but also by autophagy [145]. The α1-ATZ that is trapped in the ER of liver cells triggers elevated ER stress and inflammation [146].
Levels of ER stress and activation of the UPR have not yet been extensively studied within the context of α1-AT deficiency in humans. However, increased levels of XBP1s and ATF4 are reported in monocytes from humans with α1-AT deficiency [147]. Overexpression of active ATF6 in cells promotes the disposal of α1-ATZ by ER-associated degradation pathways [148], directing to a strategy that could limit hepatic damage caused by α1-ATZ that is trapped in the ER. However, the lack of functional α1-AT in patients by this approach would present an obstacle. Methods that induce proper folding of α1-ATZ, or improve release of α1-ATZ from the ER, might be more viable in terms of therapeutic applications because the mutated form would be active in this case.
Cystic fibrosis
Cystic fibrosis (CF) is caused by a mutation in the gene that encodes the cystic fibrosis transmembrane conductance regulator (CFTR) protein [149], an ion channel that regulates the transport of chloride and sodium ions across epithelial membranes. A mutation in CFTR results in the abnormal movement of ions, and severely affects organs such as the lung, liver, and pancreas [149]. As for other transmembrane proteins, CFTR is synthesized in the ER [150]. The most common CF mutation is the deletion of the phenylalanine508 residue (ΔF508-CFTR) [151, 152]. While most of the misfolded ΔF508-CFTR protein is subjected to proteasome-mediated degradation [150, 153], some of it is retained in the ER and ER-Golgi intermediate compartment [153, 154]. Newly synthesized CFTR is glycosylated at Asparagine897 and 900 residues and transported to the Golgi apparatus for further modification before the transport to the plasma membrane [154]. The 508 aa resides in one of the two NBD, which is found in the cytoplasmic region of the CFTR protein. The ΔF508 mutation results in an overall conformational defect due to alterations in domain–domain interactions within the protein. While the detailed mechanism by which mutant CFTR is retained in the ER is not fully understood, it is suggested that there are diarginine (RXR) ER retention/retrieval signal motifs in the cytoplasmic domain of CFTR. These motifs reside inside of the properly folded CFTR protein. However, ΔF508 mutation leads to exposure of these motifs to the ER lumen, and blocks complete folding of the protein, which ultimately leads to retention of CFTR protein in the ER and subsequent degradation [155]. In support, replacing the arginine residues with a lysine restores the trafficking and function of CFTR [156].
CF is often accompanied by chronic airway infection and inflammation. Bronchial epithelia in humans with CF display increased levels of XBP1s and ATF4 [157, 158], and in vitro studies show that ATF6 levels are increased in ΔF508-CFTR-expressing cells [159]. Several studies suggest that activation of the UPR in CF protects airway epithelia by increasing the concentration of stored calcium [157, 158]. Activation of the UPR protects from the amino acid loss and oxidative stress caused by inflammation in CF [157, 158]. Meanwhile, CF exhibits mechanisms to overcome increased ER stress and inhibit further activation of the UPR. Mutant CFTR proteins that are trapped in the ER are directly subjected to ubiquitin-dependent proteasomal degradation [160]. In addition, CFTR is transcriptionally repressed during ER stress and activation of the UPR [161, 162]. Calreticulin, an ER stress-responsive molecular chaperone found in the ER, decreases the expression and membrane localization of CFTR [163, 164]. Accordingly, downregulation of calreticulin increases CFTR expression in the membrane, both in in vitro and in vivo settings [163, 164]. Thus, calreticulin likely traps mutant CFTR in the ER lumen. Use of siRNA to downregulate ATF6 also leads to increased membrane CFTR and better ion flux through the CFTR [159]. The contribution of elements of the UPR to the regulation of CF suggests that the modulation of UPR might yield opportunities for developing novel CF therapeutics. As with α1-AT deficiency, because the mutant CFTR molecule still retains some functional capacity [165], a rewarding approach might be to increase the membrane trafficking of CFTR with use of chemical chaperones that facilitate its release from the ER.
Neurodegenerative diseases
ER stress and the UPR signaling are closely linked with many neurodegenerative diseases, including Parkinson’s (PD), Huntington’s (HD), and Alzheimer’s (AD) diseases.
Parkinson’s disease
PD is characterized by impairment of movement due to the loss of dopaminergic neurons in the brain. Genetic studies reveal that a familial form of PD, known as autosomal recessive juvenile parkinsonism (AR-JP), results from defects in the Parkin gene that encodes the ubiquitin protein ligase E3 [166–168], which tags proteins for degradation. It has been suggested that the development of AR-JP, caused by a mutation of the Parkin gene, is related to regulation of the UPR. In this case, defective E3 activity in AR-JP leads to a failure in the tagging of Parkin substrates for degradation, their accumulation in the ER, and the triggering of ER stress in neurons [169]. Prolonged ER stress ultimately leads to neuronal cell death and the development of PD [169–171].
In vitro studies document that Parkin protein actively responds to the UPR. Parkin is upregulated during ER stress to induce protein degradation [166, 167], and overexpression of Parkin in dopaminergic neuroblastoma cells reduces ER stress and suppresses neuronal cell apoptosis induced by the UPR [166, 167]. Several studies indicate that CHOP mediates cell death of dopaminergic neurons in PD [81, 172, 173]. For example, the levels of CHOP are increased following administration of 6-hydroxydopamine (6-OHDA), a neurotoxin that induces apoptosis of dopaminergic neurons [174]. In parallel, Chop knockout mice are resistant to 6-OHDA treatment-induced apoptosis of dopaminergic neurons [174]. Thus, within the context of apoptosis and the development of neurodegenerative diseases, CHOP may emerge as an attractive target for a therapeutic modality. However, because current data were obtained from whole body Chop knockout mice, further research is essential to confirm the role of CHOP in specific populations of neurons.
Huntington’s disease
HD is also a genetic neurodegenerative disorder caused by an increase in the number of CAG trinucleotide repeats in the huntingtin (HTT) gene [175, 176]. Mutations result in abnormally long huntingtin protein [177]. Fragments of long huntingtin protein bind with each other and accumulate in regions of the cytoplasm and in the perinuclear space [178, 179]. In most cases, these accumulations form nuclear inclusions in neurons and disrupt the function of the brain region that mediates movement, thinking, and emotions [180, 181]. Aggregated huntingtin proteins impair the proteasome degradation system, which leads to further accumulation of other misfolded proteins [182, 183] and contributes to the development of ER stress [177].
An in vitro study with the use of siRNA initially suggested that the ER is involved in the development of HD. Specifically, deletion of the HTT gene by siRNA disrupts the structure and networks of the ER [184]. The finding that HTT proteins associate with microtubules [185] led to the suggestion that HTT interferes with the ER network by disrupting the configuration of the cytoskeleton [184].
More direct evidence for the correlation of HD and ER function is derived from postmortem brain samples of HD patients, which display elevated expression levels of UPR target genes such as CHOP, GRP78, and Herp [186]. Expression of these ER stress-related genes is augmented in an HD mouse model [186]. In vitro studies by other investigators also confirm that ER stress and expression of UPR target genes are increased in HD [187, 188]. Specifically, expression of CHOP, GRP78, and PDI is higher (relative to control) in a striatal cell line that was established from an HTT knock-in mouse model [187]. Another group also reports on elevated JNK activity in cells that overexpress expanded poly(Q) peptides, which form aggregates resembling those of HTT protein in HD [188]. JNK activation is followed by caspase-12 activation and apoptosis [188].
Mutant HTT also disturbs ER calcium homeostasis [189]. Perturbation of high intraluminal calcium concentrations in the ER interferes with the activity of chaperones, and creates severe ER stress [14, 124]. A recent study showed that XBP1 deletion decreases accumulation of HTT protein, and protects against the development of HD symptoms in the YAC128 mouse model, which carries the human HTT gene with 128 CAG repeats [190]. The mechanism that links reduced HTT protein with XBP1 deficiency is enhanced autophagy, which is regulated by increased expression of Forkhead box protein O1 (FoxO1) [190, 191]. This observation is in parallel with the notion that XBP1s mediates proteasome-mediated degradation of FoxO1 [123] and by the observation that XBP1 deficiency results in increased levels of FoxO1 [123]. Taken together, the above observations that ER homeostasis is disrupted in HD could be incorporated into future studies aimed at developing novel treatments for HD.
Alzheimer’s disease
Elevated ER stress is a feature of AD, a disorder characterized by formation of insoluble fibrous protein aggregates in the brain. A major component of such aggregates is the amyloid β (Aβ) peptide [192, 193]. Aβ is generated via cleavage of the transmembrane glycoprotein amyloid precursor protein (APP) by presenilin (PS), which is a component of the γ-secretase complex, a membrane-resident protease [192].
The length of Aβ varies from 36 to 43 amino acid residues, depending on the site where APP is cleaved. The most common peptides are Aβ40 and Aβ42, which arise from cleavage after residues 40 and 42, respectively [194]. In neurons, generation of Aβ40 occurs in the Golgi apparatus, while Aβ42 is generated in the ER [194]. Generation of Aβ42 in the ER may be an initial event in the development of AD. Thus, inhibition of Aβ42 production may arrest the development or progression of disease [194]. Accumulation of unfolded proteins and activation of the UPR is also seen in patients with AD [195], and the levels of GRP78 and phosphorylated PERK are elevated in the temporal cortex and hippocampus of these patients at different stages of AD [195]. Moreover, the UPR is activated in pre-tangle hippocampal neurons of AD patients, as shown by the report of increased levels of phosphorylated PERK, eIF2α, and IRE1 [196].
Genetic studies reveal more than 100 mutations in the PS gene that are associated with an autosomal dominant familial AD (FAD) [197, 198]. Recent evidence indicates that PS forms an ion channel for calcium trafficking [199] and mutations of PS that are associated with FAD is important for ER calcium homeostasis [199]. Therefore, mounting evidence points to dysregulation of ER function and ER stress signaling as having a key role in AD pathology.
Cancer
The link between the UPR and cancers has been amply established as the UPR is highly activated in a number of cancers. The expression of GRP78 and other glucose-regulated proteins that are induced during tumor growth [200] was documented in the 1990s. Since then, increased GRP78 levels have been reported in several cancers, including malignant human breast cancer [201], lung cancer [202], colon cancer [203], and ovarian cancer [204, 205]. In agreement with this notion, suppression of GRP78 inhibits cancer cell growth [206, 207]. Increased levels of chaperones could be protective for tumor cells and enable them to grow faster and have a more solid ER homeostasis.
A role for IRE1 in cancer biology has also been highlighted by investigations of malignant gliomas [208], which are deadly, highly proliferative brain tumors. Angiogenesis is a particular hallmark of gliomas. Inhibition of IRE1 signaling results in decreased angiogenesis, slower tumor growth rates, and reduced invasiveness of the glioma cells [208]. Similarly, depletion of PERK or ATF4 in human tumor cells also reduces tumor growth rate and angiogenesis [209]. Furthermore, PERK induces the translation of pro-angiogenic genes, and PERK deletion in MEFs impairs vasculogenesis and decreases cancer cell proliferation [210]. In addition, ER stress leads to degradation of p53, a tumor suppressor gene [211]. Taken together, many tumors depend on an intact UPR for survival.
Another example of the association between the UPR and cancer is seen in multiple myeloma (MM), which is characterized by excess production of monoclonal proteins in bone marrow plasma cells. MM cells display increased ER stress and elevated levels of XBP1 [212]. Earlier reports identified a requirement of XBP1 in plasma cell differentiation [90], which makes it plausible that XBP1 is involved in the development of MM. In support of this notion, transgenic mice with overexpression of XBP1s in B cells and plasma cells develop pathology, including subendothelial immunoglobulin deposition, similar to that reported for human MM [213]. Furthermore, these mice exhibit aberrant expression of genes that are also dysregulated in human MM [213]. In an effort to treat MM by manipulating the UPR, treatment of MM cell lines with a small molecule IRE1α endoribonuclease inhibitor called MKC-3946 [214] resulted in reduced ER stress and a reduced rate of tumor cell growth [214].
Bortezomib, a 26S proteasome inhibitor that is used in the treatment of MM [215, 216], has an anti-cancer activity that prevents degradation of pro-apoptotic factors such as IκB, and suppresses production of anti-apoptotic proteins such as Bcl-2 [217, 218]. Degradation of IκB by the proteasome promotes the translocation of nuclear factor κB (NFκB) to the nucleus, and increases the expression of genes involved in cell growth and cell survival. NFκB activity is reportedly elevated in MM patients [219]. Another mechanism responsible for bortezomib-induced apoptosis of MM cancer cells involves ER stress [220, 221]. Prolonged ER stress created by bortezomib-induced proteasome inhibition disturbs calcium homeostasis, and results in the release of calcium from the ER. Uptake of this calcium by mitochondria is followed by the release of cytochrome c, which activates caspases and induces apoptosis [220]. Bortezomib is known to trigger apoptosis in MM cells by activation of caspase-2 [221]. Bortezomib promotes ER stress-induced apoptosis in pancreatic cancer cells through activation of JNK [222]. Many other reagents, including MKC-3946, enhance the cytotoxic effects of bortezomib in MM [214]. MKK-3946 blocks the splicing of XBP1, induced by bortezomib by inhibiting endoribonuclease activity of IRE1 [214], thereby creating further ER stress. It is not clear how exactly MKK-3946 blocks XBP1 splicing without affecting phosphorylation of IRE1. However, through this mechanism, IRE1 still has apoptotic effect via activation of JNK in the presence of MKK-3946. Interestingly, low levels of total XBP1 mRNA are correlated with resistance to bortezomib treatment, whereas high XBP1 mRNA levels increase sensitivity to bortezomib [223]. This suggests that the total XBP1 mRNA levels prior to bortezomib treatment are important for the response to therapy [223].
Chemical chaperones
What are chemical chaperones?
Chemical or pharmaceutical chaperones comprise a group of low molecular weight compounds that are known to stabilize protein conformation against thermally and chemically induced denaturation [224, 225]. Agents that have chemical chaperone activity include polyols, amines, glycerol, trimethylamine N-oxide, and dimethyl sulfoxide [225–228], plus compounds such as 4-phenylbutyric acid (PBA) and tauroursodeoxycholic acid (TUDCA) [104, 229].

The action of chemical chaperones. a Current model. Chemical chaperones nonspecifically coat the surface of newly synthesized proteins and enhance their process of secretion. b Other possible model. Chemical chaperones enhance the transcription of genes that are involved in ER capacity, ER folding activity, and ERAD, by leading to activation of transcription factors or binding to transcription machinery. Chemical chaperones may act on calcium homeostasis by manipulating calcium influx into the ER lumen
The mechanism underlying chemical chaperone functioning
The mechanisms that mediate the functioning of chemical chaperones in protein folding are not fully understood. We discuss here two categories of action (Fig. 4): (1) compounds such as DMSO or glycerol have the ability to coat proteins in the ER, and mask hydrophobic patches on unfolded proteins, thereby increasing the secretion of the proteins [230]. These compounds do not increase the folding capacity of the ER; rather they create a detergent effect, which increases the release of unfolded proteins from the ER by allowing them to escape quality control mechanisms of the ER. As discussed below, one example of such a mechanism of action is the release of mutated CFTR protein from the ER before it is completely folded (see below in section on “Cystic fibrosis”) [224, 231]; (2) compounds indirectly affect ER folding capacity. For example, molecules that activate transcriptional programs leading to increased expression of chaperones in the ER can also act as chemical chaperones. We believe it is unlikely that chemical chaperones increase the folding of proteins in the ER directly, thereby increasing their folding. To the best of our knowledge, no examples of such effects of direct binding exist in the literature. The suggestion of direct binding having an influence on protein folding probably is derived from the observations that some chemicals can increase the secretion of the unfolded proteins, which led to the belief that these compounds (chemical chaperones), like the known molecular chaperones, assist protein folding.
In addition to these two possibilities, agents that regulate ER calcium homeostasis could also serve as chemical chaperones. Recent observations indicate that increased SERCA2b function could be beneficial for ER homeostasis [15]. This approach, without changing the expressions of chaperones, may increase the activity of molecular chaperones or folding enzymes in the ER, thereby increasing ER folding capacity.
Possible therapeutic implications of chemical chaperones for disease
Type 2 diabetes and insulin resistance
The potential use of chemical chaperones for the treatment of type 2 diabetes was first demonstrated in the study where 4-PBA and TUDCA were shown to reduce ER stress and improve insulin sensitivity [104]. Administration of 4-PBA and TUDCA, which have distinct structures but share similar chemical chaperone activity, decreased PERK and IRE1 phosphorylation in the liver of obese and diabetic mice, and greatly enhanced glucose tolerance and the diabetic phenotype [104]. While this study was a proof of principle, 4-PBA and TUDCA are both weak chemical chaperones and must be administered at high doses to reduce ER stress. Our current working hypothesis with regard to the action of these chemical chaperones is that they upregulate a complex transcriptional program that increases ER capacity, and ultimately reduces ER stress. Indeed, 4-PBA activates PPARα [232], and a more recent report documents that PPARγ regulates ER function [233]. Furthermore, TUDCA also affects other transcription factors [234, 235]. Further and more detailed work is required to understand the mechanism of action of these compounds in regulating ER homeostasis.
The effects of TUDCA on ER stress and insulin sensitivity have been studied in humans [236]. TUDCA was administered orally to obese and insulin-resistant human subjects for 4 weeks, to determine whether it could effectively treat insulin resistance in obese individuals by improving ER capacity. However, our previous observations have shown that TUDCA does not have a good oral availability in mice, and high doses of the compound are required to reduce ER stress in obese mice even when delivered via intraperitoneal injection. Because it would be a challenge to achieve a working dose of TUDCA through oral administration, the experimental design was not optimally suited for exploring possible beneficial effects of TUDCA in terms of reducing ER stress in obese humans. Indeed, administration of 1,750 mg/day of TUDCA was not enough to reduce ER stress in obese humans [236]. Nonetheless, significant increases in insulin sensitivity were recorded following TUDCA administration in this report [236]. The fact that TUDCA caused increased insulin sensitivity in the absence of effects on ER stress may indicate that the reduction in ER stress was too low to detect by the techniques used or that TUDCA exerted ER stress-dependent as well as -independent effects on the insulin signaling pathway.
Leptin resistance
Increasing leptin sensitivity in obesity could provide a unique strategy for treating this debilitating disease. Nevertheless, despite extensive research efforts, no effective leptin sensitizers have been described. Recent reports indicate that reducing ER stress in the hypothalamus of obese mice via the use of chemical chaperones can provide a novel approach for increasing leptin sensitivity and influencing the treatment of obesity [27]. However, the same issues discussed above with regard to a requirement for very high dose administration of 4-PBA and TUDCA also apply in the case of leptin sensitization. A critical next step will be to create more potent chemical chaperones with better pharmacological availability to enable translation of these studies from mice to humans.
α1-Antitrypsin deficiency
The first attempts at using chemical chaperones to stabilize α1-ATZ and increase its secretion were with the use of glycerol [143]. The simple addition of glycerol enhanced the fidelity of protein folding, and resulted in better secretion of α1-ATZ in in vitro models [143]. PBA treatment also significantly increases the secretion of ER-trapped mutant α1-ATZ. Moreover, this effect of PBA was demonstrated in transgenic mice that carry the human α1-ATZ gene. Oral administration of PBA significantly increased the release of human α1-ATZ protein into the circulation [143, 237]. However, only the end result, namely whether or not the chemical chaperone enhances secretion of α1-ATZ, was monitored in these experiments. To understand the mechanistic underpinnings of how chemical chaperones act, and to translate this approach to the clinical setting, it is also important to verify whether the levels of ER stress and activation of the UPR are affected.
Cystic fibrosis
As discussed above, the mutant ΔF508-CFTR protein in CF is retained in the ER [150, 160]. Earlier work showed that glycerol facilitates the folding of mutant CFTR protein, and increases the localization of CFTR to the membrane [238]; this work also examined the effect of PBA on CF in humans [151]. Patients with the ΔF508-CFTR mutation, who received 1 week of PBA therapy, exhibited partial improvements of CFTR activity in nasal epithelia [151]. Nasal potential difference responses, used to examine basal chloride transport as an indication of the channel’s activity, were also improved following PBA treatment. However, the test scores for sweat chloride concentration, which serves as an index of CFTR channel function, were unaffected by the treatment [151]. With regard to this seeming discrepancy in the results, it has been suggested that sweat chloride concentration does not necessarily predict the severity of lung disease in CF patients [239]. The use of another compound called benzo(c)quinolizinium has also been proposed for use in increasing ΔF508-CFTR expression or for increasing apical membrane trafficking of ΔF508-CFTR in the membrane [240, 241]. The detailed mechanism of action of the compound is not fully understood.
Neurodegenerative diseases
There is intense interest in how ER stress contributes to the development of neurodegenerative changes, and in identifying potential therapeutics that can target this process. A number of studies have explored the effect of chemical chaperones on neurodegenerative diseases. For example, administration of PBA in in vitro and in in vivo models of AD leads to a reduction in amyloid plaques in the brain and improves phenotypic behaviors [242]. In vitro assays demonstrated that PBA prevents apoptosis in neuronal cells [243]. Similarly, amyloid plaques in the brain are also decreased with use of TUDCA [244], and apoptosis is reduced in cell lines [245, 246]. Administration of TUDCA in a rat model of HD improves the phenotypic features of HD [247]. TUDCA also has neuroprotective effects against MPTP, a neurotoxin used to generate the rodent model of PD [248].
Cancer
Deficiency of tuberous sclerosis complex (TSC) genes constitutively activates mTOR signaling [120, 121] and contributes to the development of tumors [219, 249]. It was shown that loss of TSC activity elevates ER stress levels and promotes susceptibility to ER stress-induced apoptosis [113]. In this study, more apoptosis was observed in kidney adenomas from thapsigargin-treated Tsc2 +/− mice when compared to normal kidney tissues; in other words, TSC-deficient tumor cells respond better to ER stress-induced apoptosis. This observation provides an alternate strategy for targeting tumors with TSC deficiency and dysregulated mTOR signaling, and highlight that acute ER stress-induced apoptosis may be stimulated with ER stress-inducing agents in certain circumstances [113].
On the other hand, PBA treatment can induce apoptosis in cancer cells, including those from colon cancer, prostate cancer, and gastric cancer [250–252]. In fact, PBA has been already tested in clinical trials for the treatment of malignant glioma [253] and hematological malignancies [254]. However, the requirement for very high doses of PBA for successful treatment remains as a challenge for this approach. Nonetheless, these studies suggest that in certain tumors, the UPR provides anti-tumor or tumor suppressor functions.
Conclusions
The UPR is a sophisticated and highly sensitive signaling pathway that influences a broad range of activities within cells. Its primary roles are to protect cells from undergoing ER stress and to maintain ER homeostasis. However, prolonged activation of the UPR can lead to apoptosis and tissue damage. While significant progress has been made in understanding how the UPR is regulated at the molecular level, more detailed mechanistic knowledge is necessary for effective manipulation of UPR elements in order to develop strategies for the treatment of diseases. Reducing the effects of ER stress in disease will probably require a variety of approaches, depending on the disease. Increasing the folding capacity of the ER could be beneficial for some diseases (e.g., obesity), as well as those that benefit the escape or secretion of mutated proteins from the ER (e.g., CF). In other circumstances, activating the UPR could be more appropriate (e.g., TSC disease). It must be emphasized that there is no universal solution that is relevant for developing strategies to manipulate ER stress in different diseases. Effective solutions will have to be individualized for each condition.
Abbreviations
- ER:
-
Endoplasmic reticulum
- UPR:
-
Unfolded protein response
- ERAD:
-
ER-associated degradation
- PERK:
-
Protein kinase RNA (PKR)-like ER kinase
- IRE1:
-
Inositol requiring enzyme-1
- ATF6:
-
Activating transcription factor-6
- GRP78:
-
Glucose regulating protein 78
- Hsp70:
-
Heat shock protein 70
- eIF2α:
-
Eukaryotic initiation factor 2
- ATF4:
-
Activating transcription factor 4
- uORFs:
-
Upstream open reading frames
- CHOP:
-
C/EBP homologous protein
- GADD34:
-
Growth arrest and DNA damage-inducible 34
- PP1c:
-
The catalytic subunit of protein phosphatase
- C/EBP:
-
CCAAT/enhancer-binding protein
- PPARγ:
-
Peroxisome proliferator-activated receptor γ
- Nrf2:
-
Nuclear factor erythroid2-related factor 2
- Keap1:
-
Kelch-like erythroid-cell-drived protein with cap’n’collar homology-associating protein 1
- cLD:
-
Core of yeast IRE1 ER-luminal domain
- XBP1:
-
X-box binding protein-1
- XBP1s:
-
Spliced form of XBP1
- CREB/ATF:
-
cAMP response element binding/activating transcription factor
- XBP1u:
-
Unspliced form of XBP1
- bZIP:
-
The basic-leucine zipper motif
- GLS:
-
Golgi localization signals
- SP1:
-
Serine protease site 1 protease
- SP2:
-
Metalloprotease site 2 protease
- CRE:
-
ATF/cAMP response element
- ERSE:
-
ER stress response elements
- HFD:
-
High-fat diet
- STZ:
-
Streptozotocin
- LPS:
-
Lipopolysaccharide
- IL-4:
-
Interleukin-4
- FFA:
-
Free fatty acids
- mTOR:
-
Mammalian target of rapamycin
- mTORC1 and mTORC2:
-
mTOR complex 1 and 2
- IGF1:
-
Insulin-like growth factor
- TSC:
-
Tuberous sclerosis complex
- IRS1 and IRS2:
-
Insulin receptor substrate 1 and 2
- PI3K:
-
Phosphotidyl inositol 3-kinase
- SERCA:
-
Sarco(endo)plasmic reticulum Ca2+-ATPase
- p38 MAPK:
-
p38 Mitogen-activated protein kinase
- TNF-α:
-
Tumor necrosis factor α
- WR:
-
Wolcott-Rallison
- α1-AT:
-
α1-antitrypsin
- CF:
-
Cystic fibrosis
- CFTR:
-
Cystic fibrosis transmembrane conductance regulator
- NBD:
-
Nucleotide-binding domain
- PD:
-
Parkinson’s disease
- HD:
-
Huntington’s disease
- AD:
-
Alzheimer’s disease
- AR-JP:
-
Autosomal recessive juvenile parkinsonism
- 6-OHDA:
-
6-Hydroxydopamine
- Herp:
-
Homocystein-induced ER protein
- JNK:
-
c-Jun N-terminal kinase
- FoxO1:
-
Forkhead box protein O1
- Aβ:
-
Amyloid β peptide
- APP:
-
Amyloid precursor protein
- PS:
-
Presenilin
- FAD:
-
Autosomal dominant familial AD
- MEFs:
-
Mouse embryonic fibroblasts
- MM:
-
Multiple myeloma
- NFκB:
-
Nuclear factor κB
- PBA:
-
4-Phenylbutyric acid
- TUDCA:
-
Tauroursodeoxycholic acid
- MPTP:
-
1-Methyl-4-phenyl-1,2,3,6-tetrahydrophyridine
References
Palade GE (1956) The endoplasmic reticulum. J Biophys Biochem Cytol 2(4 Suppl):85–98
Friedman JR, Voeltz GK (2011) The ER in 3D: a multifunctional dynamic membrane network. Trends Cell Biol 21(12):709–717. doi:10.1016/j.tcb.2011.07.004
Shibata Y, Voeltz GK, Rapoport TA (2006) Rough sheets and smooth tubules. Cell 126(3):435–439. doi:10.1016/j.cell.2006.07.019
Marciniak SJ, Ron D (2006) Endoplasmic reticulum stress signaling in disease. Physiol Rev 86(4):1133–1149. doi:10.1152/physrev.00015.2006
Schroder M, Kaufman RJ (2005) The mammalian unfolded protein response. Annu Rev Biochem 74:739–789. doi:10.1146/annurev.biochem.73.011303.074134
Honeycutt JD, Thirumalai D (1990) Metastability of the folded states of globular proteins. Proc Natl Acad Sci U S A 87(9):3526–3529
Hicke L, Schekman R (1990) Molecular machinery required for protein transport from the endoplasmic reticulum to the Golgi complex. Bioessays 12(6):253–258. doi:10.1002/bies.950120602
Hong W (1998) Protein transport from the endoplasmic reticulum to the Golgi apparatus. J Cell Sci 111(Pt 19):2831–2839
Frand AR, Cuozzo JW, Kaiser CA (2000) Pathways for protein disulphide bond formation. Trends Cell Biol 10(5):203–210
Braakman I, Bulleid NJ (2011) Protein folding and modification in the mammalian endoplasmic reticulum. Annu Rev Biochem 80:71–99. doi:10.1146/annurev-biochem-062209-093836
Koch GL (1990) The endoplasmic reticulum and calcium storage. Bioessays 12(11):527–531. doi:10.1002/bies.950121105
van Meer G, Voelker DR, Feigenson GW (2008) Membrane lipids: where they are and how they behave. Nat Rev Mol Cell Biol 9(2):112–124. doi:10.1038/nrm2330
Blom T, Somerharju P, Ikonen E (2011) Synthesis and biosynthetic trafficking of membrane lipids. Cold Spring Harb Perspect Biol 3(8):a004713. doi:10.1101/cshperspect.a004713
Vangheluwe P, Raeymaekers L, Dode L, Wuytack F (2005) Modulating sarco(endo)plasmic reticulum Ca2+ ATPase 2 (SERCA2) activity: cell biological implications. Cell Calcium 38(3–4):291–302. doi:10.1016/j.ceca.2005.06.033
Park SW, Zhou Y, Lee J, Ozcan U (2010) Sarco(endo)plasmic reticulum Ca2+-ATPase 2b is a major regulator of endoplasmic reticulum stress and glucose homeostasis in obesity. Proc Natl Acad Sci U S A 107(45):19320–19325. doi:10.1073/pnas.1012044107
Ashby MC, Tepikin AV (2001) ER calcium and the functions of intracellular organelles. Semin Cell Dev Biol 12(1):11–17. doi:10.1006/scdb.2000.0212
Walter P, Ron D (2011) The unfolded protein response: from stress pathway to homeostatic regulation. Science 334(6059):1081–1086. doi:10.1126/science.1209038
Ron D, Walter P (2007) Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol 8(7):519–529. doi:10.1038/nrm2199
Smith MH, Ploegh HL, Weissman JS (2011) Road to ruin: targeting proteins for degradation in the endoplasmic reticulum. Science 334(6059):1086–1090. doi:10.1126/science.1209235
Bernales S, Papa FR, Walter P (2006) Intracellular signaling by the unfolded protein response. Annu Rev Cell Dev Biol 22:487–508. doi:10.1146/annurev.cellbio.21.122303.120200
Zhang K, Kaufman RJ (2008) From endoplasmic-reticulum stress to the inflammatory response. Nature 454(7203):455–462
Wang S, Kaufman RJ (2012) The impact of the unfolded protein response on human disease. J Cell Biol 197(7):857–867. doi:10.1083/jcb.201110131
Gardner BM, Pincus D, Gotthardt K, Gallagher CM, Walter P (2013) Endoplasmic reticulum stress sensing in the unfolded protein response. Cold Spring Harb Perspect Biol. doi:10.1101/cshperspect.a013169
Lin JH, Walter P, Yen TS (2008) Endoplasmic reticulum stress in disease pathogenesis. Annu Rev Pathol 3:399–425. doi:10.1146/annurev.pathmechdis.3.121806.151434
Yoshida H (2007) ER stress and diseases. FEBS J 274(3):630–658. doi:10.1111/j.1742-4658.2007.05639.x
Ozcan U, Cao Q, Yilmaz E, Lee AH, Iwakoshi NN, Ozdelen E, Tuncman G, Gorgun C, Glimcher LH, Hotamisligil GS (2004) Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 306(5695):457–461
Ozcan L, Ergin AS, Lu A, Chung J, Sarkar S, Nie D, Myers MG Jr, Ozcan U (2009) Endoplasmic reticulum stress plays a central role in development of leptin resistance. Cell Metab 9(1):35–51. doi:10.1016/j.cmet.2008.12.004
Kaser A, Lee AH, Franke A, Glickman JN, Zeissig S, Tilg H, Nieuwenhuis EE, Higgins DE, Schreiber S, Glimcher LH, Blumberg RS (2008) XBP1 links ER stress to intestinal inflammation and confers genetic risk for human inflammatory bowel disease. Cell 134(5):743–756. doi:10.1016/j.cell.2008.07.021
Harding HP, Zhang Y, Ron D (1999) Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 397(6716):271–274
Bertolotti A, Zhang Y, Hendershot LM, Harding HP, Ron D (2000) Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat Cell Biol 2(6):326–332. doi:10.1038/35014014
Harding HP, Zhang Y, Bertolotti A, Zeng H, Ron D (2000) Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol Cell 5(5):897–904
Wek RC, Jiang HY, Anthony TG (2006) Coping with stress: eIF2 kinases and translational control. Biochem Soc Trans 34(Pt 1):7–11. doi:10.1042/BST20060007
Vattem KM, Wek RC (2004) Reinitiation involving upstream ORFs regulates ATF4 mRNA translation in mammalian cells. Proc Natl Acad Sci U S A 101(31):11269–11274. doi:10.1073/pnas.0400541101
Ma Y, Brewer JW, Diehl JA, Hendershot LM (2002) Two distinct stress signaling pathways converge upon the CHOP promoter during the mammalian unfolded protein response. J Mol Biol 318(5):1351–1365
Fawcett TW, Martindale JL, Guyton KZ, Hai T, Holbrook NJ (1999) Complexes containing activating transcription factor (ATF)/cAMP-responsive-element-binding protein (CREB) interact with the CCAAT/enhancer-binding protein (C/EBP)-ATF composite site to regulate Gadd153 expression during the stress response. Biochem J 339(Pt 1):135–141
Oyadomari S, Mori M (2004) Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ 11(4):381–389. doi:10.1038/sj.cdd.4401373
Lee YY, Cevallos RC, Jan E (2009) An upstream open reading frame regulates translation of GADD34 during cellular stresses that induce eIF2alpha phosphorylation. J Biol Chem 284(11):6661–6673. doi:10.1074/jbc.M806735200
Novoa I, Zeng H, Harding HP, Ron D (2001) Feedback inhibition of the unfolded protein response by GADD34-mediated dephosphorylation of eIF2alpha. J Cell Biol 153(5):1011–1022
Oyadomari S, Harding HP, Zhang Y, Oyadomari M, Ron D (2008) Dephosphorylation of translation initiation factor 2alpha enhances glucose tolerance and attenuates hepatosteatosis in mice. Cell Metab 7(6):520–532. doi:10.1016/j.cmet.2008.04.011
Cullinan SB, Zhang D, Hannink M, Arvisais E, Kaufman RJ, Diehl JA (2003) Nrf2 is a direct PERK substrate and effector of PERK-dependent cell survival. Mol Cell Biol 23(20):7198–7209
Koizumi N, Martinez IM, Kimata Y, Kohno K, Sano H, Chrispeels MJ (2001) Molecular characterization of two Arabidopsis Ire1 homologs, endoplasmic reticulum-located transmembrane protein kinases. Plant Physiol 127(3):949–962
Patil C, Walter P (2001) Intracellular signaling from the endoplasmic reticulum to the nucleus: the unfolded protein response in yeast and mammals. Curr Opin Cell Biol 13(3):349–355
Tirasophon W, Welihinda AA, Kaufman RJ (1998) A stress response pathway from the endoplasmic reticulum to the nucleus requires a novel bifunctional protein kinase/endoribonuclease (Ire1p) in mammalian cells. Genes Dev 12(12):1812–1824
Martino MB, Jones L, Brighton B, Ehre C, Abdulah L, Davis CW, Ron D, O’Neal WK, Ribeiro CM (2012) The ER stress transducer IRE1beta is required for airway epithelial mucin production. Mucosal Immunol. doi:10.1038/mi.2012.105
Bertolotti A, Wang X, Novoa I, Jungreis R, Schlessinger K, Cho JH, West AB, Ron D (2001) Increased sensitivity to dextran sodium sulfate colitis in IRE1beta-deficient mice. J Clin Invest 107(5):585–593. doi:10.1172/JCI11476
Cox JS, Shamu CE, Walter P (1993) Transcriptional induction of genes encoding endoplasmic reticulum resident proteins requires a transmembrane protein kinase. Cell 73(6):1197–1206
Mori K, Ma W, Gething MJ, Sambrook J (1993) A transmembrane protein with a cdc2+/CDC28-related kinase activity is required for signaling from the ER to the nucleus. Cell 74(4):743–756
Korennykh AV, Egea PF, Korostelev AA, Finer-Moore J, Stroud RM, Zhang C, Shokat KM, Walter P (2011) Cofactor-mediated conformational control in the bifunctional kinase/RNase Ire1. BMC Biol 9:48. doi:10.1186/1741-7007-9-48
Okamura K, Kimata Y, Higashio H, Tsuru A, Kohno K (2000) Dissociation of Kar2p/BiP from an ER sensory molecule, Ire1p, triggers the unfolded protein response in yeast. Biochem Biophys Res Commun 279(2):445–450. doi:10.1006/bbrc.2000.3987
Gardner BM, Walter P (2011) Unfolded proteins are Ire1-activating ligands that directly induce the unfolded protein response. Science 333(6051):1891–1894. doi:10.1126/science.1209126
Welihinda AA, Kaufman RJ (1996) The unfolded protein response pathway in Saccharomyces cerevisiae. Oligomerization and trans-phosphorylation of Ire1p (Ern1p) are required for kinase activation. J Biol Chem 271(30):18181–18187
Urano F, Wang X, Bertolotti A, Zhang Y, Chung P, Harding HP, Ron D (2000) Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 287(5453):664–666
Lee K, Tirasophon W, Shen X, Michalak M, Prywes R, Okada T, Yoshida H, Mori K, Kaufman RJ (2002) IRE1-mediated unconventional mRNA splicing and S2P-mediated ATF6 cleavage merge to regulate XBP1 in signaling the unfolded protein response. Genes Dev 16(4):452–466. doi:10.1101/gad.964702
Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K (2001) XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 107(7):881–891
Calfon M, Zeng H, Urano F, Till JH, Hubbard SR, Harding HP, Clark SG, Ron D (2002) IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature 415(6867):92–96
Ruegsegger U, Leber JH, Walter P (2001) Block of HAC1 mRNA translation by long-range base pairing is released by cytoplasmic splicing upon induction of the unfolded protein response. Cell 107(1):103–114
Lee AH, Iwakoshi NN, Glimcher LH (2003) XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol Cell Biol 23(21):7448–7459
Sriburi R, Jackowski S, Mori K, Brewer JW (2004) XBP1: a link between the unfolded protein response, lipid biosynthesis, and biogenesis of the endoplasmic reticulum. J Cell Biol 167(1):35–41
Liou HC, Eddy R, Shows T, Lisowska-Grospierre B, Griscelli C, Doyle C, Mannhalter J, Eibl M, Glimcher LH (1991) An HLA-DR alpha promoter DNA-binding protein is expressed ubiquitously and maps to human chromosomes 22 and 5. Immunogenetics 34(5):286–292
Tirosh B, Iwakoshi NN, Glimcher LH, Ploegh HL (2006) Rapid turnover of unspliced Xbp-1 as a factor that modulates the unfolded protein response. J Biol Chem 281(9):5852–5860. doi:10.1074/jbc.M509061200
Sriburi R, Bommiasamy H, Buldak GL, Robbins GR, Frank M, Jackowski S, Brewer JW (2007) Coordinate regulation of phospholipid biosynthesis and secretory pathway gene expression in XBP-1(S)-induced endoplasmic reticulum biogenesis. J Biol Chem 282(10):7024–7034
Kokame K, Kato H, Miyata T (2001) Identification of ERSE-II, a new cis-acting element responsible for the ATF6-dependent mammalian unfolded protein response. J Biol Chem 276(12):9199–9205. doi:10.1074/jbc.M010486200
Fagone P, Sriburi R, Ward-Chapman C, Frank M, Wang J, Gunter C, Brewer JW, Jackowski S (2007) Phospholipid biosynthesis program underlying membrane expansion during B-lymphocyte differentiation. J Biol Chem 282(10):7591–7605
Yoshida H, Oku M, Suzuki M, Mori K (2006) pXBP1(U) encoded in XBP1 pre-mRNA negatively regulates unfolded protein response activator pXBP1(S) in mammalian ER stress response. J Cell Biol 172(4):565–575. doi:10.1083/jcb.200508145
Yoshida H, Uemura A, Mori K (2009) pXBP1(U), a negative regulator of the unfolded protein response activator pXBP1(S), targets ATF6 but not ATF4 in proteasome-mediated degradation. Cell Struct Funct 34(1):1–10
Haze K, Okada T, Yoshida H, Yanagi H, Yura T, Negishi M, Mori K (2001) Identification of the G13 (cAMP-response-element-binding protein-related protein) gene product related to activating transcription factor 6 as a transcriptional activator of the mammalian unfolded protein response. Biochem J 355(Pt 1):19–28
Shen J, Chen X, Hendershot L, Prywes R (2002) ER stress regulation of ATF6 localization by dissociation of BiP/GRP78 binding and unmasking of Golgi localization signals. Dev Cell 3(1):99–111
Haze K, Yoshida H, Yanagi H, Yura T, Mori K (1999) Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol Biol Cell 10(11):3787–3799
Shen J, Prywes R (2005) ER stress signaling by regulated proteolysis of ATF6. Methods 35(4):382–389. doi:10.1016/j.ymeth.2004.10.011
Ye J, Rawson RB, Komuro R, Chen X, Dave UP, Prywes R, Brown MS, Goldstein JL (2000) ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol Cell 6(6):1355–1364
Chen X, Shen J, Prywes R (2002) The luminal domain of ATF6 senses endoplasmic reticulum (ER) stress and causes translocation of ATF6 from the ER to the Golgi. J Biol Chem 277(15):13045–13052. doi:10.1074/jbc.M110636200
Wang Y, Shen J, Arenzana N, Tirasophon W, Kaufman RJ, Prywes R (2000) Activation of ATF6 and an ATF6 DNA binding site by the endoplasmic reticulum stress response. J Biol Chem 275(35):27013–27020. doi:10.1074/jbc.M003322200
Harding HP, Zeng H, Zhang Y, Jungries R, Chung P, Plesken H, Sabatini DD, Ron D (2001) Diabetes mellitus and exocrine pancreatic dysfunction in perk-/- mice reveals a role for translational control in secretory cell survival. Mol Cell 7(6):1153–1163
Taniguchi CM, Emanuelli B, Kahn CR (2006) Critical nodes in signalling pathways: insights into insulin action. Nat Rev Mol Cell Biol 7(2):85–96. doi:10.1038/nrm1837
Zhang W, Feng D, Li Y, Iida K, McGrath B, Cavener DR (2006) PERK EIF2AK3 control of pancreatic beta cell differentiation and proliferation is required for postnatal glucose homeostasis. Cell Metab 4(6):491–497. doi:10.1016/j.cmet.2006.11.002
Scheuner D, Song B, McEwen E, Liu C, Laybutt R, Gillespie P, Saunders T, Bonner-Weir S, Kaufman RJ (2001) Translational control is required for the unfolded protein response and in vivo glucose homeostasis. Mol Cell 7(6):1165–1176
Scheuner D, Vander Mierde D, Song B, Flamez D, Creemers JW, Tsukamoto K, Ribick M, Schuit FC, Kaufman RJ (2005) Control of mRNA translation preserves endoplasmic reticulum function in beta cells and maintains glucose homeostasis. Nat Med 11(7):757–764
Song B, Scheuner D, Ron D, Pennathur S, Kaufman RJ (2008) Chop deletion reduces oxidative stress, improves beta cell function, and promotes cell survival in multiple mouse models of diabetes. J Clin Invest 118(10):3378–3389. doi:10.1172/JCI34587
Ariyama Y, Shimizu H, Satoh T, Tsuchiya T, Okada S, Oyadomari S, Mori M (2007) Chop-deficient mice showed increased adiposity but no glucose intolerance. Obesity (Silver Spring) 15(7):1647–1656. doi:10.1038/oby.2007.197
Maris M, Overbergh L, Gysemans C, Waget A, Cardozo AK, Verdrengh E, Cunha JP, Gotoh T, Cnop M, Eizirik DL, Burcelin R, Mathieu C (2012) Deletion of C/EBP homologous protein (Chop) in C57Bl/6 mice dissociates obesity from insulin resistance. Diabetologia 55(4):1167–1178. doi:10.1007/s00125-011-2427-7
Tajiri S, Oyadomari S, Yano S, Morioka M, Gotoh T, Hamada JI, Ushio Y, Mori M (2004) Ischemia-induced neuronal cell death is mediated by the endoplasmic reticulum stress pathway involving CHOP. Cell Death Differ 11(4):403–415. doi:10.1038/sj.cdd.4401365
Endo M, Oyadomari S, Suga M, Mori M, Gotoh T (2005) The ER stress pathway involving CHOP is activated in the lungs of LPS-treated mice. J Biochem 138(4):501–507. doi:10.1093/jb/mvi143
Oyadomari S, Koizumi A, Takeda K, Gotoh T, Akira S, Araki E, Mori M (2002) Targeted disruption of the Chop gene delays endoplasmic reticulum stress-mediated diabetes. J Clin Invest 109(4):525–532. doi:10.1172/JCI14550
Yoshioka M, Kayo T, Ikeda T, Koizumi A (1997) A novel locus, Mody4, distal to D7Mit189 on chromosome 7 determines early-onset NIDDM in nonobese C57BL/6 (Akita) mutant mice. Diabetes 46(5):887–894
Wang J, Takeuchi T, Tanaka S, Kubo SK, Kayo T, Lu D, Takata K, Koizumi A, Izumi T (1999) A mutation in the insulin 2 gene induces diabetes with severe pancreatic beta-cell dysfunction in the Mody mouse. J Clin Invest 103(1):27–37. doi:10.1172/JCI4431
Oyadomari S, Takeda K, Takiguchi M, Gotoh T, Matsumoto M, Wada I, Akira S, Araki E, Mori M (2001) Nitric oxide-induced apoptosis in pancreatic beta cells is mediated by the endoplasmic reticulum stress pathway. Proc Natl Acad Sci U S A 98(19):10845–10850. doi:10.1073/pnas.191207498
Wu J, Kaufman RJ (2006) From acute ER stress to physiological roles of the unfolded protein response. Cell Death Differ 13(3):374–384. doi:10.1038/sj.cdd.4401840
Wu J, Rutkowski DT, Dubois M, Swathirajan J, Saunders T, Wang J, Song B, Yau GD, Kaufman RJ (2007) ATF6alpha optimizes long-term endoplasmic reticulum function to protect cells from chronic stress. Dev Cell 13(3):351–364. doi:10.1016/j.devcel.2007.07.005
Rutkowski DT, Wu J, Back SH, Callaghan MU, Ferris SP, Iqbal J, Clark R, Miao H, Hassler JR, Fornek J, Katze MG, Hussain MM, Song B, Swathirajan J, Wang J, Yau GD, Kaufman RJ (2008) UPR pathways combine to prevent hepatic steatosis caused by ER stress-mediated suppression of transcriptional master regulators. Dev Cell 15(6):829–840. doi:10.1016/j.devcel.2008.10.015
Reimold AM, Iwakoshi NN, Manis J, Vallabhajosyula P, Szomolanyi-Tsuda E, Gravallese EM, Friend D, Grusby MJ, Alt F, Glimcher LH (2001) Plasma cell differentiation requires the transcription factor XBP-1. Nature 412(6844):300–307. doi:10.1038/35085509
Iwakoshi NN, Lee AH, Vallabhajosyula P, Otipoby KL, Rajewsky K, Glimcher LH (2003) Plasma cell differentiation and the unfolded protein response intersect at the transcription factor XBP-1. Nat Immunol 4(4):321–329. doi:10.1038/ni907
Reimold AM, Etkin A, Clauss I, Perkins A, Friend DS, Zhang J, Horton HF, Scott A, Orkin SH, Byrne MC, Grusby MJ, Glimcher LH (2000) An essential role in liver development for transcription factor XBP-1. Genes Dev 14(2):152–157
Masaki T, Yoshida M, Noguchi S (1999) Targeted disruption of CRE-binding factor TREB5 gene leads to cellular necrosis in cardiac myocytes at the embryonic stage. Biochem Biophys Res Commun 261(2):350–356. doi:10.1006/bbrc.1999.0972
Ron D (2002) Proteotoxicity in the endoplasmic reticulum: lessons from the Akita diabetic mouse. J Clin Invest 109(4):443–445. doi:10.1172/JCI15020
Minamino T, Kitakaze M (2010) ER stress in cardiovascular disease. J Mol Cell Cardiol 48(6):1105–1110. doi:10.1016/j.yjmcc.2009.10.026
Biddinger SB, Kahn CR (2006) From mice to men: insights into the insulin resistance syndromes. Annu Rev Physiol 68:123–158. doi:10.1146/annurev.physiol.68.040104.124723
Bray GA (2004) Medical consequences of obesity. J Clin Endocrinol Metab 89(6):2583–2589. doi:10.1210/jc.2004-0535
Lawrence VJ, Kopelman PG (2004) Medical consequences of obesity. Clin Dermatol 22(4):296–302. doi:10.1016/j.clindermatol.2004.01.012
Saltiel AR (2001) New perspectives into the molecular pathogenesis and treatment of type 2 diabetes. Cell 104(4):517–529
Nakatani Y, Kaneto H, Kawamori D, Yoshiuchi K, Hatazaki M, Matsuoka TA, Ozawa K, Ogawa S, Hori M, Yamasaki Y, Matsuhisa M (2005) Involvement of endoplasmic reticulum stress in insulin resistance and diabetes. J Biol Chem 280(1):847–851. doi:10.1074/jbc.M411860200
Ozawa K, Miyazaki M, Matsuhisa M, Takano K, Nakatani Y, Hatazaki M, Tamatani T, Yamagata K, Miyagawa J, Kitao Y, Hori O, Yamasaki Y, Ogawa S (2005) The endoplasmic reticulum chaperone improves insulin resistance in type 2 diabetes. Diabetes 54(3):657–663
Boden G, Duan X, Homko C, Molina EJ, Song W, Perez O, Cheung P, Merali S (2008) Increase in endoplasmic reticulum stress-related proteins and genes in adipose tissue of obese, insulin-resistant individuals. Diabetes 57(9):2438–2444. doi:10.2337/db08-0604
Boden G (2009) Endoplasmic reticulum stress: another link between obesity and insulin resistance/inflammation? Diabetes 58(3):518–519. doi:10.2337/db08-1746
Ozcan U, Yilmaz E, Ozcan L, Furuhashi M, Vaillancourt E, Smith RO, Gorgun CZ, Hotamisligil GS (2006) Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science 313(5790):1137–1140
Back SH, Kaufman RJ (2012) Endoplasmic reticulum stress and type 2 diabetes. Annu Rev Biochem 81:767–793. doi:10.1146/annurev-biochem-072909-095555
Boden G (1998) Free fatty acids (FFA), a link between obesity and insulin resistance. Front Biosci 3:d169–d175
Mayer CM, Belsham DD (2010) Palmitate attenuates insulin signaling and induces endoplasmic reticulum stress and apoptosis in hypothalamic neurons: rescue of resistance and apoptosis through adenosine 5' monophosphate-activated protein kinase activation. Endocrinology 151(2):576–585. doi:10.1210/en.2009-1122
Borradaile NM, Han X, Harp JD, Gale SE, Ory DS, Schaffer JE (2006) Disruption of endoplasmic reticulum structure and integrity in lipotoxic cell death. J Lipid Res 47(12):2726–2737. doi:10.1194/jlr.M600299-JLR200
Borradaile NM, Buhman KK, Listenberger LL, Magee CJ, Morimoto ET, Ory DS, Schaffer JE (2006) A critical role for eukaryotic elongation factor 1A-1 in lipotoxic cell death. Mol Biol Cell 17(2):770–778. doi:10.1091/mbc.E05-08-0742
Wei Y, Wang D, Topczewski F, Pagliassotti MJ (2006) Saturated fatty acids induce endoplasmic reticulum stress and apoptosis independently of ceramide in liver cells. Am J Physiol Endocrinol Metab 291(2):E275–E281. doi:10.1152/ajpendo.00644.2005
Kharroubi I, Ladriere L, Cardozo AK, Dogusan Z, Cnop M, Eizirik DL (2004) Free fatty acids and cytokines induce pancreatic beta-cell apoptosis by different mechanisms: role of nuclear factor-kappaB and endoplasmic reticulum stress. Endocrinology 145(11):5087–5096. doi:10.1210/en.2004-0478
Pfaffenbach KT, Gentile CL, Nivala AM, Wang D, Wei Y, Pagliassotti MJ (2010) Linking endoplasmic reticulum stress to cell death in hepatocytes: roles of C/EBP homologous protein and chemical chaperones in palmitate-mediated cell death. Am J Physiol Endocrinol Metab 298(5):E1027–E1035. doi:10.1152/ajpendo.00642.2009
Ozcan U, Ozcan L, Yilmaz E, Duvel K, Sahin M, Manning BD, Hotamisligil GS (2008) Loss of the tuberous sclerosis complex tumor suppressors triggers the unfolded protein response to regulate insulin signaling and apoptosis. Mol Cell 29(5):541–551. doi:10.1016/j.molcel.2007.12.023
Laplante M, Sabatini DM (2009) mTOR signaling at a glance. J Cell Sci 122(Pt 20):3589–3594. doi:10.1242/jcs.051011
Sarbassov DD, Ali SM, Sabatini DM (2005) Growing roles for the mTOR pathway. Curr Opin Cell Biol 17(6):596–603. doi:10.1016/j.ceb.2005.09.009
Loewith R, Jacinto E, Wullschleger S, Lorberg A, Crespo JL, Bonenfant D, Oppliger W, Jenoe P, Hall MN (2002) Two TOR complexes, only one of which is rapamycin sensitive, have distinct roles in cell growth control. Mol Cell 10(3):457–468
Sengupta S, Peterson TR, Sabatini DM (2010) Regulation of the mTOR complex 1 pathway by nutrients, growth factors, and stress. Mol Cell 40(2):310–322. doi:10.1016/j.molcel.2010.09.026
Harrington LS, Findlay GM, Gray A, Tolkacheva T, Wigfield S, Rebholz H, Barnett J, Leslie NR, Cheng S, Shepherd PR, Gout I, Downes CP, Lamb RF (2004) The TSC1-2 tumor suppressor controls insulin-PI3K signaling via regulation of IRS proteins. J Cell Biol 166(2):213–223. doi:10.1083/jcb.200403069
Shah OJ, Wang Z, Hunter T (2004) Inappropriate activation of the TSC/Rheb/mTOR/S6K cassette induces IRS1/2 depletion, insulin resistance, and cell survival deficiencies. Curr Biol 14(18):1650–1656. doi:10.1016/j.cub.2004.08.026
Garami A, Zwartkruis FJ, Nobukuni T, Joaquin M, Roccio M, Stocker H, Kozma SC, Hafen E, Bos JL, Thomas G (2003) Insulin activation of Rheb, a mediator of mTOR/S6K/4E-BP signaling, is inhibited by TSC1 and 2. Mol Cell 11(6):1457–1466
Zhang Y, Gao X, Saucedo LJ, Ru B, Edgar BA, Pan D (2003) Rheb is a direct target of the tuberous sclerosis tumour suppressor proteins. Nat Cell Biol 5(6):578–581. doi:10.1038/ncb999
Park SW, Zhou Y, Lee J, Lu A, Sun C, Chung J, Ueki K, Ozcan U (2010) The regulatory subunits of PI3K, p85alpha and p85beta, interact with XBP-1 and increase its nuclear translocation. Nat Med 16(4):429–437
Zhou Y, Lee J, Reno C, Sun C, Park S, Chung J, Lee J, Fisher S, White M, Biddinger S, Ozcan U (2011) Regulation of glucose homeostasis through a XBP-1–FoxO1 interaction. Nature Medicine. doi:10.1038/nm.2293
Lytton J, Westlin M, Hanley MR (1991) Thapsigargin inhibits the sarcoplasmic or endoplasmic reticulum Ca-ATPase family of calcium pumps. J Biol Chem 266(26):17067–17071
Lee J, Sun C, Zhou Y, Gokalp D, Herrema H, Park SW, Davis RJ, Ozcan U (2011) p38 MAPK-mediated regulation of Xbp1s is crucial for glucose homeostasis. Nat Med 17(10):1251–1260. doi:10.1038/nm.2449
Hotamisligil GS, Erbay E (2008) Nutrient sensing and inflammation in metabolic diseases. Nat Rev Immunol 8(12):923–934. doi:10.1038/nri2449
Olefsky JM, Glass CK (2010) Macrophages, inflammation, and insulin resistance. Annu Rev Physiol 72:219–246. doi:10.1146/annurev-physiol-021909-135846
Shoelson SE, Lee J, Goldfine AB (2006) Inflammation and insulin resistance. J Clin Invest 116(7):1793–1801. doi:10.1172/JCI29069
Gregor MF, Hotamisligil GS (2011) Inflammatory mechanisms in obesity. Annu Rev Immunol 29:415–445. doi:10.1146/annurev-immunol-031210-101322
Kawasaki N, Asada R, Saito A, Kanemoto S, Imaizumi K (2012) Obesity-induced endoplasmic reticulum stress causes chronic inflammation in adipose tissue. Sci Rep 2:799. doi:10.1038/srep00799
Hirosumi J, Tuncman G, Chang L, Gorgun CZ, Uysal KT, Maeda K, Karin M, Hotamisligil GS (2002) A central role for JNK in obesity and insulin resistance. Nature 420(6913):333–336. doi:10.1038/nature01137
Solinas G, Karin M (2010) JNK1 and IKKbeta: molecular links between obesity and metabolic dysfunction. FASEB J 24(8):2596–2611. doi:10.1096/fj.09-151340
Arkan MC, Hevener AL, Greten FR, Maeda S, Li ZW, Long JM, Wynshaw-Boris A, Poli G, Olefsky J, Karin M (2005) IKK-beta links inflammation to obesity-induced insulin resistance. Nat Med 11(2):191–198. doi:10.1038/nm1185
Solinas G, Vilcu C, Neels JG, Bandyopadhyay GK, Luo JL, Naugler W, Grivennikov S, Wynshaw-Boris A, Scadeng M, Olefsky JM, Karin M (2007) JNK1 in hematopoietically derived cells contributes to diet-induced inflammation and insulin resistance without affecting obesity. Cell Metab 6(5):386–397. doi:10.1016/j.cmet.2007.09.011
Julier C, Nicolino M (2010) Wolcott-Rallison syndrome. Orphanet J Rare Dis 5:29. doi:10.1186/1750-1172-5-29
Biason-Lauber A, Lang-Muritano M, Vaccaro T, Schoenle EJ (2002) Loss of kinase activity in a patient with Wolcott-Rallison syndrome caused by a novel mutation in the EIF2AK3 gene. Diabetes 51(7):2301–2305
Harding HP, Ron D (2002) Endoplasmic reticulum stress and the development of diabetes: a review. Diabetes 51(Suppl 3):S455–S461
Brickwood S, Bonthron DT, Al-Gazali LI, Piper K, Hearn T, Wilson DI, Hanley NA (2003) Wolcott-Rallison syndrome: pathogenic insights into neonatal diabetes from new mutation and expression studies of EIF2AK3. J Med Genet 40(9):685–689
Stoller JK, Aboussouan LS (2012) A review of alpha1-antitrypsin deficiency. Am J Respir Crit Care Med 185(3):246–259. doi:10.1164/rccm.201108-1428CI
Lomas DA, Mahadeva R (2002) Alpha1-antitrypsin polymerization and the serpinopathies: pathobiology and prospects for therapy. J Clin Invest 110(11):1585–1590. doi:10.1172/JCI16782
Bathurst IC, Travis J, George PM, Carrell RW (1984) Structural and functional characterization of the abnormal Z alpha 1-antitrypsin isolated from human liver. FEBS Lett 177(2):179–183
Greene CM, McElvaney NG (2010) Z alpha-1 antitrypsin deficiency and the endoplasmic reticulum stress response. World J Gastrointest Pharmacol Ther 1(5):94–101. doi:10.4292/wjgpt.v1.i5.94
Burrows JA, Willis LK, Perlmutter DH (2000) Chemical chaperones mediate increased secretion of mutant alpha 1-antitrypsin (alpha 1-AT) Z: a potential pharmacological strategy for prevention of liver injury and emphysema in alpha 1-AT deficiency. Proc Natl Acad Sci U S A 97(4):1796–1801
Qu D, Teckman JH, Omura S, Perlmutter DH (1996) Degradation of a mutant secretory protein, alpha1-antitrypsin Z, in the endoplasmic reticulum requires proteasome activity. J Biol Chem 271(37):22791–22795
Teckman JH, Perlmutter DH (2000) Retention of mutant alpha(1)-antitrypsin Z in endoplasmic reticulum is associated with an autophagic response. Am J Physiol Gastrointest Liver Physiol 279(5):G961–G974
Lawless MW, Greene CM, Mulgrew A, Taggart CC, O’Neill SJ, McElvaney NG (2004) Activation of endoplasmic reticulum-specific stress responses associated with the conformational disease Z alpha 1-antitrypsin deficiency. J Immunol 172(9):5722–5726
Carroll TP, Greene CM, O’Connor CA, Nolan AM, O’Neill SJ, McElvaney NG (2010) Evidence for unfolded protein response activation in monocytes from individuals with alpha-1 antitrypsin deficiency. J Immunol 184(8):4538–4546. doi:10.4049/jimmunol.0802864
Smith SE, Granell S, Salcedo-Sicilia L, Baldini G, Egea G, Teckman JH (2011) Activating transcription factor 6 limits intracellular accumulation of mutant alpha(1)-antitrypsin Z and mitochondrial damage in hepatoma cells. J Biol Chem 286(48):41563–41577. doi:10.1074/jbc.M111.280073
Pilewski JM, Frizzell RA (1999) Role of CFTR in airway disease. Physiol Rev 79(1 Suppl):S215–S255
Kopito RR (1999) Biosynthesis and degradation of CFTR. Physiol Rev 79(1 Suppl):S167–S173
Rubenstein RC, Zeitlin PL (1998) A pilot clinical trial of oral sodium 4-phenylbutyrate (Buphenyl) in deltaF508-homozygous cystic fibrosis patients: partial restoration of nasal epithelial CFTR function. Am J Respir Crit Care Med 157(2):484–490
Kalin N, Claass A, Sommer M, Puchelle E, Tummler B (1999) DeltaF508 CFTR protein expression in tissues from patients with cystic fibrosis. J Clin Invest 103(10):1379–1389. doi:10.1172/JCI5731
Gilbert A, Jadot M, Leontieva E, Wattiaux-De Coninck S, Wattiaux R (1998) Delta F508 CFTR localizes in the endoplasmic reticulum-Golgi intermediate compartment in cystic fibrosis cells. Exp Cell Res 242(1):144–152. doi:10.1006/excr.1998.4101
Cheng SH, Gregory RJ, Marshall J, Paul S, Souza DW, White GA, O’Riordan CR, Smith AE (1990) Defective intracellular transport and processing of CFTR is the molecular basis of most cystic fibrosis. Cell 63(4):827–834
Kim Chiaw P, Huan LJ, Gagnon S, Ly D, Sweezey N, Rotin D, Deber CM, Bear CE (2009) Functional rescue of DeltaF508-CFTR by peptides designed to mimic sorting motifs. Chem Biol 16(5):520–530. doi:10.1016/j.chembiol.2009.04.005
Chang XB, Cui L, Hou YX, Jensen TJ, Aleksandrov AA, Mengos A, Riordan JR (1999) Removal of multiple arginine-framed trafficking signals overcomes misprocessing of delta F508 CFTR present in most patients with cystic fibrosis. Mol Cell 4(1):137–142
Ribeiro CM, Boucher RC (2010) Role of endoplasmic reticulum stress in cystic fibrosis-related airway inflammatory responses. Proc Am Thorac Soc 7(6):387–394. doi:10.1513/pats.201001-017AW
Cattaneo M, Lotti LV, Martino S, Cardano M, Orlandi R, Mariani-Costantini R, Biunno I (2009) Functional characterization of two secreted SEL1L isoforms capable of exporting unassembled substrate. J Biol Chem 284(17):11405–11415. doi:10.1074/jbc.M805408200
Kerbiriou M, Le Drevo MA, Ferec C, Trouve P (2007) Coupling cystic fibrosis to endoplasmic reticulum stress: differential role of Grp78 and ATF6. Biochim Biophys Acta 1772(11–12):1236–1249. doi:10.1016/j.bbadis.2007.10.004
Ward CL, Omura S, Kopito RR (1995) Degradation of CFTR by the ubiquitin-proteasome pathway. Cell 83(1):121–127
Bartoszewski R, Rab A, Twitty G, Stevenson L, Fortenberry J, Piotrowski A, Dumanski JP, Bebok Z (2008) The mechanism of cystic fibrosis transmembrane conductance regulator transcriptional repression during the unfolded protein response. J Biol Chem 283(18):12154–12165. doi:10.1074/jbc.M707610200
Rab A, Bartoszewski R, Jurkuvenaite A, Wakefield J, Collawn JF, Bebok Z (2007) Endoplasmic reticulum stress and the unfolded protein response regulate genomic cystic fibrosis transmembrane conductance regulator expression. Am J Physiol Cell Physiol 292(2):C756–C766. doi:10.1152/ajpcell.00391.2006
Harada K, Okiyoneda T, Hashimoto Y, Oyokawa K, Nakamura K, Suico MA, Shuto T, Kai H (2007) Curcumin enhances cystic fibrosis transmembrane regulator expression by down-regulating calreticulin. Biochem Biophys Res Commun 353(2):351–356. doi:10.1016/j.bbrc.2006.12.036
Harada K, Okiyoneda T, Hashimoto Y, Ueno K, Nakamura K, Yamahira K, Sugahara T, Shuto T, Wada I, Suico MA, Kai H (2006) Calreticulin negatively regulates the cell surface expression of cystic fibrosis transmembrane conductance regulator. J Biol Chem 281(18):12841–12848. doi:10.1074/jbc.M512975200
Dalemans W, Barbry P, Champigny G, Jallat S, Dott K, Dreyer D, Crystal RG, Pavirani A, Lecocq JP, Lazdunski M (1991) Altered chloride ion channel kinetics associated with the delta F508 cystic fibrosis mutation. Nature 354(6354):526–528. doi:10.1038/354526a0
Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y, Shimizu N (1998) Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392(6676):605–608. doi:10.1038/33416
Shimura H, Hattori N, Kubo S, Mizuno Y, Asakawa S, Minoshima S, Shimizu N, Iwai K, Chiba T, Tanaka K, Suzuki T (2000) Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat Genet 25(3):302–305. doi:10.1038/77060
Imai Y, Soda M, Inoue H, Hattori N, Mizuno Y, Takahashi R (2001) An unfolded putative transmembrane polypeptide, which can lead to endoplasmic reticulum stress, is a substrate of Parkin. Cell 105(7):891–902
Takahashi R, Imai Y (2003) Pael receptor, endoplasmic reticulum stress, and Parkinson’s disease. J Neurol 250(Suppl 3):III25–III29. doi:10.1007/s00415-003-1305-8
Kitao Y, Imai Y, Ozawa K, Kataoka A, Ikeda T, Soda M, Nakimawa K, Kiyama H, Stern DM, Hori O, Wakamatsu K, Ito S, Itohara S, Takahashi R, Ogawa S (2007) Pael receptor induces death of dopaminergic neurons in the substantia nigra via endoplasmic reticulum stress and dopamine toxicity, which is enhanced under condition of parkin inactivation. Hum Mol Genet 16(1):50–60. doi:10.1093/hmg/ddl439
Takahashi R, Imai Y, Hattori N, Mizuno Y (2003) Parkin and endoplasmic reticulum stress. Ann N Y Acad Sci 991:101–106
Ryu EJ, Harding HP, Angelastro JM, Vitolo OV, Ron D, Greene LA (2002) Endoplasmic reticulum stress and the unfolded protein response in cellular models of Parkinson’s disease. J Neurosci 22(24):10690–10698
Holtz WA, O’Malley KL (2003) Parkinsonian mimetics induce aspects of unfolded protein response in death of dopaminergic neurons. J Biol Chem 278(21):19367–19377. doi:10.1074/jbc.M211821200
Silva RM, Ries V, Oo TF, Yarygina O, Jackson-Lewis V, Ryu EJ, Lu PD, Marciniak SJ, Ron D, Przedborski S, Kholodilov N, Greene LA, Burke RE (2005) CHOP/GADD153 is a mediator of apoptotic death in substantia nigra dopamine neurons in an in vivo neurotoxin model of parkinsonism. J Neurochem 95(4):974–986. doi:10.1111/j.1471-4159.2005.03428.x
Norremolle A, Riess O, Epplen JT, Fenger K, Hasholt L, Sorensen SA (1993) Trinucleotide repeat elongation in the Huntingtin gene in Huntington disease patients from 71 Danish families. Hum Mol Genet 2(9):1475–1476
Landles C, Bates GP (2004) Huntingtin and the molecular pathogenesis of Huntington’s disease. Fourth in molecular medicine review series. EMBO Rep 5(10):958–963. doi:10.1038/sj.embor.7400250
Vidal R, Caballero B, Couve A, Hetz C (2011) Converging pathways in the occurrence of endoplasmic reticulum (ER) stress in Huntington’s disease. Curr Mol Med 11(1):1–12
Chapple JP, Bros-Facer V, Butler R, Gallo JM (2008) Focal distortion of the nuclear envelope by huntingtin aggregates revealed by lamin immunostaining. Neurosci Lett 447(2–3):172–174. doi:10.1016/j.neulet.2008.09.075
Li SH, Li XJ (1998) Aggregation of N-terminal huntingtin is dependent on the length of its glutamine repeats. Hum Mol Genet 7(5):777–782
Cattaneo E, Zuccato C, Tartari M (2005) Normal huntingtin function: an alternative approach to Huntington’s disease. Nat Rev Neurosci 6(12):919–930. doi:10.1038/nrn1806
Rockabrand E, Slepko N, Pantalone A, Nukala VN, Kazantsev A, Marsh JL, Sullivan PG, Steffan JS, Sensi SL, Thompson LM (2007) The first 17 amino acids of Huntingtin modulate its sub-cellular localization, aggregation and effects on calcium homeostasis. Hum Mol Genet 16(1):61–77. doi:10.1093/hmg/ddl440
Hipp MS, Patel CN, Bersuker K, Riley BE, Kaiser SE, Shaler TA, Brandeis M, Kopito RR (2012) Indirect inhibition of 26S proteasome activity in a cellular model of Huntington’s disease. J Cell Biol 196(5):573–587. doi:10.1083/jcb.201110093
Jana NR, Zemskov EA, Wang G, Nukina N (2001) Altered proteasomal function due to the expression of polyglutamine-expanded truncated N-terminal huntingtin induces apoptosis by caspase activation through mitochondrial cytochrome c release. Hum Mol Genet 10(10):1049–1059
Omi K, Hachiya NS, Tokunaga K, Kaneko K (2005) siRNA-mediated inhibition of endogenous Huntington disease gene expression induces an aberrant configuration of the ER network in vitro. Biochem Biophys Res Commun 338(2):1229–1235. doi:10.1016/j.bbrc.2005.10.061
Gutekunst CA, Levey AI, Heilman CJ, Whaley WL, Yi H, Nash NR, Rees HD, Madden JJ, Hersch SM (1995) Identification and localization of huntingtin in brain and human lymphoblastoid cell lines with anti-fusion protein antibodies. Proc Natl Acad Sci U S A 92(19):8710–8714
Carnemolla A, Fossale E, Agostoni E, Michelazzi S, Calligaris R, De Maso L, Del Sal G, MacDonald ME, Persichetti F (2009) Rrs1 is involved in endoplasmic reticulum stress response in Huntington disease. J Biol Chem 284(27):18167–18173. doi:10.1074/jbc.M109.018325
Duennwald ML, Lindquist S (2008) Impaired ERAD and ER stress are early and specific events in polyglutamine toxicity. Genes Dev 22(23):3308–3319. doi:10.1101/gad.1673408
Kouroku Y, Fujita E, Jimbo A, Kikuchi T, Yamagata T, Momoi MY, Kominami E, Kuida K, Sakamaki K, Yonehara S, Momoi T (2002) Polyglutamine aggregates stimulate ER stress signals and caspase-12 activation. Hum Mol Genet 11(13):1505–1515
Varshney A, Ehrlich BE (2003) Intracellular Ca2+ signaling and human disease: the hunt begins with Huntington’s. Neuron 39(2):195–197
Vidal RL, Figueroa A, Court FA, Thielen P, Molina C, Wirth C, Caballero B, Kiffin R, Segura-Aguilar J, Cuervo AM, Glimcher LH, Hetz C (2012) Targeting the UPR transcription factor XBP1 protects against Huntington’s disease through the regulation of FoxO1 and autophagy. Hum Mol Genet 21(10):2245–2262. doi:10.1093/hmg/dds040
Zhao Y, Yang J, Liao W, Liu X, Zhang H, Wang S, Wang D, Feng J, Yu L, Zhu WG (2010) Cytosolic FoxO1 is essential for the induction of autophagy and tumour suppressor activity. Nat Cell Biol 12(7):665–675. doi:10.1038/ncb2069
Zhang YW, Thompson R, Zhang H, Xu H (2011) APP processing in Alzheimer’s disease. Mol Brain 4:3. doi:10.1186/1756-6606-4-3
Selkoe DJ (2001) Alzheimer’s disease: genes, proteins, and therapy. Physiol Rev 81(2):741–766
Hartmann T, Bieger SC, Bruhl B, Tienari PJ, Ida N, Allsop D, Roberts GW, Masters CL, Dotti CG, Unsicker K, Beyreuther K (1997) Distinct sites of intracellular production for Alzheimer’s disease A beta40/42 amyloid peptides. Nat Med 3(9):1016–1020
Hoozemans JJ, Veerhuis R, Van Haastert ES, Rozemuller JM, Baas F, Eikelenboom P, Scheper W (2005) The unfolded protein response is activated in Alzheimer’s disease. Acta Neuropathol 110(2):165–172. doi:10.1007/s00401-005-1038-0
Hoozemans JJ, van Haastert ES, Nijholt DA, Rozemuller AJ, Eikelenboom P, Scheper W (2009) The unfolded protein response is activated in pretangle neurons in Alzheimer’s disease hippocampus. Am J Pathol 174(4):1241–1251. doi:10.2353/ajpath.2009.080814
Tandon A, Fraser P (2002) The presenilins. Genome Biol 3 (11):reviews3014
Rogaeva E (2002) The solved and unsolved mysteries of the genetics of early-onset Alzheimer’s disease. Neuromolecular Med 2(1):1–10. doi:10.1385/NMM:2:1:01
Tu H, Nelson O, Bezprozvanny A, Wang Z, Lee SF, Hao YH, Serneels L, De Strooper B, Yu G, Bezprozvanny I (2006) Presenilins form ER Ca2+ leak channels, a function disrupted by familial Alzheimer’s disease-linked mutations. Cell 126(5):981–993. doi:10.1016/j.cell.2006.06.059
Cai JW, Henderson BW, Shen JW, Subjeck JR (1993) Induction of glucose regulated proteins during growth of a murine tumor. J Cell Physiol 154(2):229–237. doi:10.1002/jcp.1041540204
Fernandez PM, Tabbara SO, Jacobs LK, Manning FC, Tsangaris TN, Schwartz AM, Kennedy KA, Patierno SR (2000) Overexpression of the glucose-regulated stress gene GRP78 in malignant but not benign human breast lesions. Breast Cancer Res Treat 59(1):15–26
Uramoto H, Sugio K, Oyama T, Nakata S, Ono K, Yoshimastu T, Morita M, Yasumoto K (2005) Expression of endoplasmic reticulum molecular chaperone Grp78 in human lung cancer and its clinical significance. Lung Cancer 49(1):55–62. doi:10.1016/j.lungcan.2004.12.011
Xing X, Lai M, Wang Y, Xu E, Huang Q (2006) Overexpression of glucose-regulated protein 78 in colon cancer. Clin Chim Acta 364(1–2):308–315. doi:10.1016/j.cca.2005.07.016
Delie F, Petignat P, Cohen M (2012) GRP78 protein expression in ovarian cancer patients and perspectives for a drug-targeting approach. J Oncol 2012:468615. doi:10.1155/2012/468615
Huang LW, Lin CY, Lee CC, Liu TZ, Jeng CJ (2012) Overexpression of GRP78 is associated with malignant transformation in epithelial ovarian tumors. Appl Immunohistochem Mol Morphol 20(4):381–385. doi:10.1097/PAI.0b013e3182434113
Jamora C, Dennert G, Lee AS (1996) Inhibition of tumor progression by suppression of stress protein GRP78/BiP induction in fibrosarcoma B/C10ME. Proc Natl Acad Sci U S A 93(15):7690–7694
Koong AC, Chen EY, Lee AS, Brown JM, Giaccia AJ (1994) Increased cytotoxicity of chronic hypoxic cells by molecular inhibition of GRP78 induction. Int J Radiat Oncol Biol Phys 28(3):661–666
Auf G, Jabouille A, Guerit S, Pineau R, Delugin M, Bouchecareilh M, Magnin N, Favereaux A, Maitre M, Gaiser T, von Deimling A, Czabanka M, Vajkoczy P, Chevet E, Bikfalvi A, Moenner M (2010) Inositol-requiring enzyme 1alpha is a key regulator of angiogenesis and invasion in malignant glioma. Proc Natl Acad Sci U S A 107(35):15553–15558. doi:10.1073/pnas.0914072107
Wang Y, Alam GN, Ning Y, Visioli F, Dong Z, Nor JE, Polverini PJ (2012) The unfolded protein response induces the angiogenic switch in human tumor cells through the PERK/ATF4 pathway. Cancer Res. doi:10.1158/0008-5472.CAN-12-0474
Blais JD, Addison CL, Edge R, Falls T, Zhao H, Wary K, Koumenis C, Harding HP, Ron D, Holcik M, Bell JC (2006) Perk-dependent translational regulation promotes tumor cell adaptation and angiogenesis in response to hypoxic stress. Mol Cell Biol 26(24):9517–9532. doi:10.1128/MCB.01145-06
Pluquet O, Qu LK, Baltzis D, Koromilas AE (2005) Endoplasmic reticulum stress accelerates p53 degradation by the cooperative actions of Hdm2 and glycogen synthase kinase 3beta. Mol Cell Biol 25(21):9392–9405. doi:10.1128/MCB.25.21.9392-9405.2005
Munshi NC, Hideshima T, Carrasco D, Shammas M, Auclair D, Davies F, Mitsiades N, Mitsiades C, Kim RS, Li C, Rajkumar SV, Fonseca R, Bergsagel L, Chauhan D, Anderson KC (2004) Identification of genes modulated in multiple myeloma using genetically identical twin samples. Blood 103(5):1799–1806. doi:10.1182/blood-2003-02-0402
Carrasco DR, Sukhdeo K, Protopopova M, Sinha R, Enos M, Carrasco DE, Zheng M, Mani M, Henderson J, Pinkus GS, Munshi N, Horner J, Ivanova EV, Protopopov A, Anderson KC, Tonon G, DePinho RA (2007) The differentiation and stress response factor XBP-1 drives multiple myeloma pathogenesis. Cancer Cell 11(4):349–360. doi:10.1016/j.ccr.2007.02.015
Mimura N, Fulciniti M, Gorgun G, Tai YT, Cirstea D, Santo L, Hu Y, Fabre C, Minami J, Ohguchi H, Kiziltepe T, Ikeda H, Kawano Y, French M, Blumenthal M, Tam V, Kertesz NL, Malyankar UM, Hokenson M, Pham T, Zeng Q, Patterson JB, Richardson PG, Munshi NC, Anderson KC (2012) Blockade of XBP1 splicing by inhibition of IRE1alpha is a promising therapeutic option in multiple myeloma. Blood 119(24):5772–5781. doi:10.1182/blood-2011-07-366633
Kane RC, Dagher R, Farrell A, Ko CW, Sridhara R, Justice R, Pazdur R (2007) Bortezomib for the treatment of mantle cell lymphoma. Clin Cancer Res 13(18 Pt 1):5291–5294. doi:10.1158/1078-0432.CCR-07-0871
Kane RC, Bross PF, Farrell AT, Pazdur R (2003) Velcade: U.S. FDA approval for the treatment of multiple myeloma progressing on prior therapy. Oncologist 8(6):508–513
Mitsiades N, Mitsiades CS, Richardson PG, Poulaki V, Tai YT, Chauhan D, Fanourakis G, Gu X, Bailey C, Joseph M, Libermann TA, Schlossman R, Munshi NC, Hideshima T, Anderson KC (2003) The proteasome inhibitor PS-341 potentiates sensitivity of multiple myeloma cells to conventional chemotherapeutic agents: therapeutic applications. Blood 101(6):2377–2380. doi:10.1182/blood-2002-06-1768
Adams J, Palombella VJ, Sausville EA, Johnson J, Destree A, Lazarus DD, Maas J, Pien CS, Prakash S, Elliott PJ (1999) Proteasome inhibitors: a novel class of potent and effective antitumor agents. Cancer Res 59(11):2615–2622
Feinman R, Koury J, Thames M, Barlogie B, Epstein J, Siegel DS (1999) Role of NF-kappaB in the rescue of multiple myeloma cells from glucocorticoid-induced apoptosis by bcl-2. Blood 93(9):3044–3052
Landowski TH, Megli CJ, Nullmeyer KD, Lynch RM, Dorr RT (2005) Mitochondrial-mediated disregulation of Ca2+ is a critical determinant of Velcade (PS-341/bortezomib) cytotoxicity in myeloma cell lines. Cancer Res 65(9):3828–3836. doi:10.1158/0008-5472.CAN-04-3684
Gu H, Chen X, Gao G, Dong H (2008) Caspase-2 functions upstream of mitochondria in endoplasmic reticulum stress-induced apoptosis by bortezomib in human myeloma cells. Mol Cancer Ther 7(8):2298–2307. doi:10.1158/1535-7163.MCT-08-0186
Nawrocki ST, Carew JS, Pino MS, Highshaw RA, Dunner K Jr, Huang P, Abbruzzese JL, McConkey DJ (2005) Bortezomib sensitizes pancreatic cancer cells to endoplasmic reticulum stress-mediated apoptosis. Cancer Res 65(24):11658–11666. doi:10.1158/0008-5472.CAN-05-2370
Ling SC, Lau EK, Al-Shabeeb A, Nikolic A, Catalano A, Iland H, Horvath N, Ho PJ, Harrison S, Fleming S, Joshua DE, Allen JD (2012) Response of myeloma to the proteasome inhibitor bortezomib is correlated with the unfolded protein response regulator XBP-1. Haematologica 97(1):64–72. doi:10.3324/haematol.2011.043331
Gekko K, Timasheff SN (1981) Thermodynamic and kinetic examination of protein stabilization by glycerol. Biochemistry 20(16):4677–4686
Back JF, Oakenfull D, Smith MB (1979) Increased thermal stability of proteins in the presence of sugars and polyols. Biochemistry 18(23):5191–5196
Baskakov IV, Kumar R, Srinivasan G, Ji YS, Bolen DW, Thompson EB (1999) Trimethylamine N-oxide-induced cooperative folding of an intrinsically unfolded transcription-activating fragment of human glucocorticoid receptor. J Biol Chem 274(16):10693–10696
Gekko K, Koga S (1983) Increased thermal stability of collagen in the presence of sugars and polyols. J Biochem 94(1):199–205
Gerlsma SY, Stuur ER (1976) The effects from combining urea and an alcohol on the heat-induced reversible denaturation of ribonuclease. Int J Pept Protein Res 8(1):3–12
Perlmutter DH (2002) Chemical chaperones: a pharmacological strategy for disorders of protein folding and trafficking. Pediatr Res 52(6):832–836. doi:10.1203/00006450-200212000-00004
Vagenende V, Yap MG, Trout BL (2009) Mechanisms of protein stabilization and prevention of protein aggregation by glycerol. Biochemistry 48(46):11084–11096. doi:10.1021/bi900649t
Sawano H, Koumoto Y, Ohta K, Sasaki Y, Segawa S, Tachibana H (1992) Efficient in vitro folding of the three-disulfide derivatives of hen lysozyme in the presence of glycerol. FEBS Lett 303(1):11–14
Liu N, Qiang W, Kuang X, Thuillier P, Lynn WS, Wong PK (2002) The peroxisome proliferator phenylbutyric acid (PBA) protects astrocytes from ts1 MoMuLV-induced oxidative cell death. J Neurovirol 8(4):318–325. doi:10.1080/13550280290100699
Diano S, Liu ZW, Jeong JK, Dietrich MO, Ruan HB, Kim E, Suyama S, Kelly K, Gyengesi E, Arbiser JL, Belsham DD, Sarruf DA, Schwartz MW, Bennett AM, Shanabrough M, Mobbs CV, Yang X, Gao XB, Horvath TL (2011) Peroxisome proliferation-associated control of reactive oxygen species sets melanocortin tone and feeding in diet-induced obesity. Nat Med 17(9):1121–1127. doi:10.1038/nm.2421
Yang JS, Kim JT, Jeon J, Park HS, Kang GH, Park KS, Lee HK, Kim S, Cho YM (2010) Changes in hepatic gene expression upon oral administration of taurine-conjugated ursodeoxycholic acid in ob/ob mice. PLoS One 5(11):e13858. doi:10.1371/journal.pone.0013858
Castro RE, Sola S, Ma X, Ramalho RM, Kren BT, Steer CJ, Rodrigues CM (2005) A distinct microarray gene expression profile in primary rat hepatocytes incubated with ursodeoxycholic acid. J Hepatol 42(6):897–906. doi:10.1016/j.jhep.2005.01.026
Kars M, Yang L, Gregor MF, Mohammed BS, Pietka TA, Finck BN, Patterson BW, Horton JD, Mittendorfer B, Hotamisligil GS, Klein S (2010) Tauroursodeoxycholic acid may improve liver and muscle but not adipose tissue insulin sensitivity in obese men and women. Diabetes 59(8):1899–1905. doi:10.2337/db10-0308
Soto C, Sigurdsson EM, Morelli L, Kumar RA, Castano EM, Frangione B (1998) Beta-sheet breaker peptides inhibit fibrillogenesis in a rat brain model of amyloidosis: implications for Alzheimer’s therapy. Nat Med 4(7):822–826
Sato S, Ward CL, Krouse ME, Wine JJ, Kopito RR (1996) Glycerol reverses the misfolding phenotype of the most common cystic fibrosis mutation. J Biol Chem 271(2):635–638
Davis PB, Schluchter MD, Konstan MW (2004) Relation of sweat chloride concentration to severity of lung disease in cystic fibrosis. Pediatr Pulmonol 38(3):204–209. doi:10.1002/ppul.20054
McPherson MA, Pereira MM, Russell D, McNeilly CM, Morris RM, Stratford FL, Dormer RL (2001) The CFTR-mediated protein secretion defect: pharmacological correction. Pflugers Arch 443(Suppl 1):S121–S126. doi:10.1007/s004240100658
Dormer RL, Derand R, McNeilly CM, Mettey Y, Bulteau-Pignoux L, Metaye T, Vierfond JM, Gray MA, Galietta LJ, Morris MR, Pereira MM, Doull IJ, Becq F, McPherson MA (2001) Correction of delF508-CFTR activity with benzo(c)quinolizinium compounds through facilitation of its processing in cystic fibrosis airway cells. J Cell Sci 114(Pt 22):4073–4081
Wiley JC, Pettan-Brewer C, Ladiges WC (2011) Phenylbutyric acid reduces amyloid plaques and rescues cognitive behavior in AD transgenic mice. Aging Cell 10(3):418–428. doi:10.1111/j.1474-9726.2011.00680.x
Wiley JC, Meabon JS, Frankowski H, Smith EA, Schecterson LC, Bothwell M, Ladiges WC (2010) Phenylbutyric acid rescues endoplasmic reticulum stress-induced suppression of APP proteolysis and prevents apoptosis in neuronal cells. PLoS One 5(2):e9135. doi:10.1371/journal.pone.0009135
Nunes AF, Amaral JD, Lo AC, Fonseca MB, Viana RJ, Callaerts-Vegh Z, D’Hooge R, Rodrigues CM (2012) TUDCA, a bile acid, attenuates amyloid precursor protein processing and amyloid-beta deposition in APP/PS1 mice. Mol Neurobiol 45(3):440–454. doi:10.1007/s12035-012-8256-y
Ramalho RM, Borralho PM, Castro RE, Sola S, Steer CJ, Rodrigues CM (2006) Tauroursodeoxycholic acid modulates p53-mediated apoptosis in Alzheimer’s disease mutant neuroblastoma cells. J Neurochem 98(5):1610–1618. doi:10.1111/j.1471-4159.2006.04007.x
Sola S, Castro RE, Laires PA, Steer CJ, Rodrigues CM (2003) Tauroursodeoxycholic acid prevents amyloid-beta peptide-induced neuronal death via a phosphatidylinositol 3-kinase-dependent signaling pathway. Mol Med 9(9–12):226–234
Keene CD, Rodrigues CM, Eich T, Chhabra MS, Steer CJ, Low WC (2002) Tauroursodeoxycholic acid, a bile acid, is neuroprotective in a transgenic animal model of Huntington’s disease. Proc Natl Acad Sci U S A 99(16):10671–10676. doi:10.1073/pnas.162362299
Holtveg HM, Reimer E, Hultberg IB (1989) Hemangioendothelial sarcoma of the thyroid. Ugeskr Laeger 151(33):2088–2089
Gomez MR (1991) Phenotypes of the tuberous sclerosis complex with a revision of diagnostic criteria. Ann N Y Acad Sci 615:1–7
Carducci MA, Nelson JB, Chan-Tack KM, Ayyagari SR, Sweatt WH, Campbell PA, Nelson WG, Simons JW (1996) Phenylbutyrate induces apoptosis in human prostate cancer and is more potent than phenylacetate. Clin Cancer Res 2(2):379–387
Li LZ, Deng HX, Lou WZ, Sun XY, Song MW, Tao J, Xiao BX, Guo JM (2012) Growth inhibitory effect of 4-phenyl butyric acid on human gastric cancer cells is associated with cell cycle arrest. World J Gastroenterol 18(1):79–83. doi:10.3748/wjg.v18.i1.79
Feinman R, Clarke KO, Harrison LE (2002) Phenylbutyrate-induced apoptosis is associated with inactivation of NF-kappaB IN HT-29 colon cancer cells. Cancer Chemother Pharmacol 49(1):27–34
Phuphanich S, Baker SD, Grossman SA, Carson KA, Gilbert MR, Fisher JD, Carducci MA (2005) Oral sodium phenylbutyrate in patients with recurrent malignant gliomas: a dose escalation and pharmacologic study. Neuro Oncol 7(2):177–182. doi:10.1215/S1152851704000183
Bhalla K, List A (2004) Histone deacetylase inhibitors in myelodysplastic syndrome. Best Pract Res Clin Haematol 17(4):595–611. doi:10.1016/j.beha.2004.08.011
Acknowledgments
This work was supported by an RO1 grant (R01DK081009) provided to U.O. by the US National Institutes of Health and the Timothy Murphy funds provided to the Division of Endocrinology, Boston Children’s Hospital; and a K99 grant (K99DK093788) provided to S.W.P. by the National Institutes of Health.
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is a contribution to the special issue on “The unfolded protein response in immune diseases” - Guest Editors: Richard Blumberg and Arthur Kaser
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Park, S.W., Ozcan, U. Potential for therapeutic manipulation of the UPR in disease. Semin Immunopathol 35, 351–373 (2013). https://doi.org/10.1007/s00281-013-0370-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00281-013-0370-z