
Abstract
Significant advances in the diagnosis and therapy for uveitis have been made to improve the quality of care for patients with ocular inflammatory diseases. While traditional ophthalmic examination techniques, fluorescein angiography, and optical coherence tomography continue to play a major role in the evaluation of patients with uveitis, the advent of spectral domain optical coherence tomography and fundus autofluorescence into clinical practice provides additional information about disease processes. Polymerase chain reaction and cytokine diagnostics have also continued to play a greater role in the evaluation of patients with inflammatory diseases. The biologic agents, a group of medications that targets cytokines and other soluble mediators of inflammation, have demonstrated promise in targeted immunotherapy for specific uveitic entities. Their ophthalmic indications have continued to expand, improving the therapeutic armentarium of uveitis specialists.
Similar content being viewed by others


Introduction
Uveitis, which is derived from uva (Latin for grape) is defined as an inflammation of the uveal tract or middle coat of the eye (iris, ciliary body, and choroid). In clinical practice, the uveitis specialist is confronted with diagnostic and treatment dilemmas involving not only inflammation of the uvea but also inflammatory syndromes involving other ocular, orbital, and periorbital structures. Some of the more common nonuveal tract inflammatory processes include inflammations of the orbit, (i.e., orbital pseudotumor), sclera (scleritis), and retinal vascular inflammation (retinal vasculitis).
Determining the etiology of a uveitis syndrome may be a difficult task, but it is especially important in ophthalmic conditions associated with a systemic disease. For example, scleritis may be associated with a number of systemic conditions including rheumatoid arthritis, systemic lupus erythematosus, gout, rheumatoid arthritis, or sarcoidosis. Scleritis may also be associated with infectious diseases such as tuberculosis and syphilis. Panuveitis may be associated with sarcoidosis, Vogt–Koyanagi–Harada’s disease (VKH), Behçet’s disease, or may also represent a masquerade syndrome such as endophthalmitis from systemic bacteremia [1].
After determining the diagnosis of a particular inflammatory condition and excluding an infectious process, consideration of an immunosuppressive regimen is the next important decision faced by the ocular inflammatory specialist. While corticosteroids remain a mainstay of therapy for acute, noninfectious uveitis, the multiple side effects associated with long-term corticosteroid use warrant the consideration of other therapeutic options. The use of immunosuppressive medications such as the antimetabolites (methotrexate, mycophenolate mofetil, azathioprine), T cell calcineurin inhibitors (cyclosporine, tacrolimus), and alkylating agents (cyclophosphamide, chlorambucil) have demonstrated efficacy in the treatment of uveitis with long-term drug-free remissions in some cases [2, 3]. However, each medication has its unique side effect profile requiring laboratory testing, and stringent long-term follow-up is required to ensure that the adverse systemic effects associated with the medication do not outweigh the visual benefits. A newer class of medications, the biologic agents, comprises a number of medications directed against specific cytokines, cytokine receptors, cellular adhesion molecules, and other soluble mediators of inflammation. The biologic agents have shown promise in immunotherapy for ocular inflammatory disease, and ophthalmic indications for their use will likely continue to expand [4, 5].
Animal models of uveitis including experimental autoimmune uveitis (EAU) in rodents have been particularly instructive in unraveling the pathogenic mechanisms of uveitis and have been useful for testing novel immunomodulatory agents. While a complete discussion of the many contributions derived from EAU and other animal models of uveitis is beyond the scope of this review, some of the therapies discussed in this review will reference the EAU literature [6, 7].
Inflammatory mechanisms have also been implicated in other ophthalmic conditions including diabetic retinopathy and age-related macular degeneration (ARMD) [8, 9]. Whether an inflammatory insult is the inciting agent or a result of another pathogenic mechanism for these conditions is not clear. In ARMD, inflammatory mechanisms implicated include elements of the complement cascade, particularly complement factor H [10, 11] and cellular components including macrophages and multinucleated giant cells, which have been identified in histopathologic specimens of patients with ARMD [12, 13]. In diabetic retinopathy, the role of a number of inflammatory cytokines, chemokines, and adhesion molecules involved in leukocyte trafficking has been studied and may contribute to low-grade subclinical inflammation [9]. Research efforts continue to be performed in these areas, and immunotherapy may play a role in these conditions in the future. The pathways underlying uveitis and related ocular inflammatory syndromes have been characterized extensively, and advances in the clinical diagnosis of and immunotherapy of these conditions will be discussed in this review.
Specifically, this review focuses on recent advances in the clinical diagnosis and treatment of anterior, intermediate, posterior, and panuveitis. The diagnostic modalities available to clinicians for the evaluation of uveitis have continued to improve over the last decade with increasingly sophisticated laboratory testing (e.g., polymerase chain reaction [PCR], cytokine evaluation, flow cytometry) and ophthalmic imaging techniques (e.g., fundus autofluorescence [FAF], three-dimensional spectral domain optical coherence tomography [OCT]). Significant advances in immunotherapy have also been made, particularly with the increasing use of biologic agents. A number of local therapies including local sustained-release corticosteroid delivery systems (e.g., fluocinolone acetonide implant, intravitreal dexamethasone delivery system) have also been developed and will be discussed in this review.
Classification of uveitis
The anatomic classification of uveitis serves as a useful starting point to guide the differential diagnosis and therapy of an ocular inflammatory condition. The Standardization of Uveitis Nomenclature Working Group convened to establish a classification and grading scheme for uveitis and determined that the anatomic classification of uveitis be used. The scheme was designed not only to provide uniformity in the classification of disease processes but also to permit meta-analyses and to standardize the evaluation of outcomes of patients treated with novel immunotherapies [14].
Anterior uveitis refers to inflammation involving the iris and ciliary body, and terms such as iritis, iridocyclitis, and cyclitis are no longer used. While anterior uveitis is often idiopathic, systemic conditions that may be associated with anterior uveitis include sarcoidosis, tuberculosis, syphilis, and human leukocyte antigen (HLA)-B27-related diseases (i.e., ankylosing spondylitis, Reiter syndrome, Crohn’s disease). Anterior uveitis may also be toxic, medication related, as seen with metipranolol therapy [15].
Intermediate uveitis refers to the subset of uveitis in which inflammation is primarily observed in the vitreous cavity. Pars planitis, which had previously been used by some uveitis specialists as being synonymous with intermediate uveitis, is a designated subset of intermediate uveitis (i.e., intermediate uveitis of the pars plana subtype). While intermediate uveitis may be idiopathic, it may also reflect a systemic infection or inflammatory process such as Lyme disease, syphilis, or tuberculosis. Localized ocular infectious causes including Toxoplasmosis gondii and Toxocara canis are often included in the differential diagnosis of intermediate uveitis. However, these infectious etiologies of intermediate uveitis are more commonly implicated in cases of retinochoroiditis with an associated vitritis. Multiple sclerosis may also be associated with intermediate uveitis, and a magnetic resonance imaging scan and neurologic evaluation may be warranted in patients with a suggestive clinical history.
Posterior uveitis refers to inflammation of the choroid and retina and may result from localized ocular inflammation or from a systemic condition. For example, birdshot retinochoroidopathy and serpiginous choroidopathy only affect ophthalmic structures. Posterior uveitic syndromes with systemic associations include VKH, sarcoidosis, Behçet’s disease, tuberculosis choroidopathy, and syphilis. Sympathetic ophthalmia may present with ophthalmic manifestations alone or with both ophthalmic and systemic findings.
Panuveitis involves inflammation of the anterior, intermediate, and posterior uveal structures, and the differential diagnosis is similar to that of posterior uveitis. Herpetic viral retinitides may also present as panuveitis or posterior uveitis in both immunosuppressed and immunocompetent individuals, and diagnostic strategies are continuing to be developed for the diagnosis of these infectious viral conditions.
Another group of diseases, termed masquerade syndromes, may present with characteristics of ocular inflammatory disease such as vitritis or posterior segment inflammation but are caused by neoplastic or infectious processes. For example, primary intraocular lymphoma (PIOL) may masquerade as an intermediate or posterior uveitis. Recognition of this central nervous system neoplasm in the differential diagnosis of intermediate uveitis is critical to avoid potentially missing a life-threatening diagnosis. Endophthalmitis may present with indolent inflammation (e.g., Propiobacterium acnes) or severe intraocular inflammation (e.g., B. cereus endophthalmitis), and therapy may include antibiotics and surgical therapy in some situations.
Diagnostic evaluation for the uveitis patient, which may include ancillary ophthalmic testing (e.g., fluorescein angiography [FA], OCT), analysis of serum, aqueous or vitreous specimens, or radiographic imaging, is currently recommended for patients who present with intermediate, posterior, or panuveitis. Patients with anterior uveitis who demonstrate recurrent disease, uveitis recalcitrant to topical corticosteroids, or bilateral anterior uveitis require additional diagnostic workup for an underlying etiology. Each patient’s workup will vary depending on the history and exam features of their disease, anatomic location of the inflammatory process, and risk factors for a particular uveitic syndrome.
History and ophthalmic examination
The clinical ophthalmologic evaluation includes a careful history, ophthalmic exam, and review of systems with attention to other potential systemic inflammatory symptoms (e.g., back pain in ankylosing spondylitis, fevers, productive cough, or night sweats suggestive of pulmonary tuberculosis). Past medical history including history of autoimmune diseases, cancer, and systemic immunodeficiency, travel history to areas that may be endemic for certain uveitic syndromes (e.g., West Nile chorioretinitis, onchocerciasis in sub-Saharan Africa), and family history of ocular inflammatory disease (e.g., juvenile systemic granulomatosis) should be elicited. Medications, medication allergies, and social history including use of illicit drugs should also be documented, as these may influence the therapeutic options available to a patient.
The history taking should be directed and relate to the major ophthalmic complaint (e.g., visual acuity loss, peripheral visual field loss, photophobia, floaters). The rapidity of the symptom onset should be ascertained, as some uveitic syndromes such as HLA-B27-associated anterior uveitis are associated with sudden onset of symptoms, while other syndromes may be more indolent in nature. The duration of the uveitis is described as limited if it is less than 3 months or persistent for conditions lasting greater than 3 months. The course of the uveitic syndrome may be characterized as acute, if the condition is a sudden onset and with a limited duration, recurrent if a relapse occurs greater than 3 months after discontinuing therapy, and chronic, if a relapse occurs less than 3 months after discontinuing therapy [14]. These descriptors have relevance to the differential diagnosis of an ophthalmic condition, as well as tailoring therapies for the inflammatory process. In addition, history of prior therapies, a patient’s response to therapy, and past ophthalmic history should be thoroughly investigated.
The ophthalmic exam should be thorough, but emphasis should be placed on anatomic structures likely to be implicated based on the patient’s symptoms (e.g., photophobia associated with anterior chamber inflammation in anterior uveitis; floaters associated with vitritis in intermediate uveitis). Key elements of the ophthalmic exam include visual acuity, pupillary examination, visual field testing, ocular motility testing, slit lamp biomicroscopic exam, and dilated funduscopic exam. Visual acuity loss may be due to a variety of etiologies including anterior segment disease involving the cornea (e.g., corneal edema) or lens (e.g., cataract), as well as vitreous debris or cystoid macular edema (CME), a complication seen in a number of uveitic entities. Pupils should be assessed for a relative afferent pupillary defect, which may implicate widespread retinal dysfunction or an optic nerve process. Slit lamp examination may reveal perilimbal conjunctival inflammation (ciliary flush), which is suggestive of ciliary body inflammation, granulomatous or nongranulomatous keratic precipitates on the corneal endothelium, and anterior chamber cell and flare resulting from anterior uveitis. Examination of the iris may demonstrate granulomatous nodules on the surface of the iris suggestive of sarcoidosis or posterior synechiae, adhesions forming from the iris to the lens. Patients with uveitis may also develop cataracts from corticosteroid therapy or from long-standing inflammation. The anterior vitreous should also be assessed for vitreous inflammation and the presence of pigment. Dilated funduscopic examination may reveal optic disc edema, macular edema, retinal vascular sheathing from white blood cells enveloping retinal vessels, or retinal or choroidal lesions (e.g., birdshot retinochoroidopathy and serpiginous choroidopathy). A thorough ophthalmic exam is essential for accurate anatomic classification of the disease process, identification of the secondary complications of ocular inflammation (e.g., CME), construction of a broad differential diagnosis, and directed ancillary clinical and laboratory testing.
Ancillary testing
Ancillary testing routinely performed in the clinic for the evaluation of uveitis now includes FA and OCT in cases of intermediate, posterior, and panuveitis. FAF has been increasingly utilized in the evaluation of inflammatory processes involving the retina, retinal pigment epithelium (RPE), and choroid. Three-dimensional OCT has also been used in the evaluation of posterior segment processes, and its indications in ocular inflammatory disease will likely continue to increase. B-scan ultrasound continues to play a role in the evaluation of vitreous disorders and retinal and choroidal processes. In addition, laboratory testing including PCR-based testing, cytokine fluid analysis, and flow cytometry may contribute valuable information in the diagnostic workup of a uveitis patient.
Fluorescein angiography
FA remains a mainstay in the clinical evaluation of uveitis for the evaluation of optic disc leakage, macular edema, and retinal vascular leakage. Sodium fluorescein dye is given via intravenous injection and is visualized as hyper- or hypofluorescence within choroidal and retinal structures with an absorption wavelength of 460–495 nm and emission wavelength of 520–530 nm. Hyperfluorescence of the optic nerve may represent optic disc edema either as a primary process (i.e., optic neuritis) or as a secondary sequela of intraocular inflammation (Fig. 1). CME is visualized on FA as a petalloid accumulation of fluorescein dye extravasates into the outer plexiform layer of the retina (Fig. 2). Leakage of fluorescein in a perivascular location may also be a primary process (i.e., retinal vasculitis) or be secondary to intraocular inflammation (e.g., periphlebitis secondary to sarcoid-associated panuveitis). Decreased fluorescence or hypofluorescence may occur due to the blockage of underlying choroidal fluorescence due to retinal pigment epithelial alteration or scarring or may result from retinal ischemia, observed in some cases of retinal vasculitis. For example, in lupus vasculitis, decreased fluorescence is seen due to small-vessel capillary dropout, which may predispose patients to retinal neovascularization and visual loss from foveal ischemia (Fig. 3). The advent of digital FA has provided a method of rapid transfer of data, so that consultation with another physician from a remote location regarding a complex patient is possible.

Fluorescein angiogram in late venous phase shows hyperfluorescence of optic nerve due to inflammation

In this patient, with birdshot retinochoroidopathy, fluorescein dye extravasates into cystoid spaces inferior, nasal, and temporal to the fovea due to incompetence of perifoveal vessels. Hyperfluorescent regions surrounding the optic nerve are due to areas of retinal pigment epithelial dropout

In lupus vasculitis, numerous capillary vessel dropouts are observed on angiography, resulting in retinal nonperfusion during an episode of active inflammation
Fundus autofluorescence
FAF has also been valuable in the evaluation of uveitic syndromes, and its use will likely increase with standardization of FAF photography and improved understanding of this diagnostic modality [16]. FAF relies on the property of autofluorescence, an intrinsic property of some cellular structures due to intracellular photoreactive components [17]. When excited at a particular wavelength, the autofluorescent signal may be detected with the correct barrier filter. The FAF signal from posterior segment structures is derived primarily from lipofuscin, which normally accumulates in aged RPE [18]. A number of filter sets (excitation and barrier filters) have been described for use in the clinical setting. Spaide described the clinical use of an excitation wavelength of 585 nm and barrier filter of 690 nm [19]. Indocyanine green filters (excitation wavelength 790 nm, barrier filter 810 nm) have also been used. In pathologic conditions such as ARMD and central serous chorioretinopathy, distinct FAF patterns have been identified and, in some cases, may be correlated with ongoing and possibly future injury [20, 21]. FAF abnormalities have been reported in a number of ocular inflammatory conditions including acute posterior multifocal pigment placoid epitheliopathy [22], acute syphilitic chorioretinopathy [23], and acute zonal occult outer retinopathy [24]. The FAF signal has also been evaluated in a number of degenerative conditions including retinitis pigmentosa [25], Stargardt’s disease (fundus flavimaculatus) [26], Best vitelliform macular dystrophy [27], and pseudoxanthoma elasticum [28]. In uveitic entities involving the retina, RPE, and choroid, we have observed abnormalities of the FAF signal (unpublished data), and this technique will likely continue to provide valuable information about structural changes found in posterior uveitis (Fig. 4).

a Fundus photo of patient with VKH showing nummular areas of RPE hyperpigmentation and RPE loss. b Corresponding FAF photo of VKH patient shows mottled areas of hyperfluorescent (bright) signal amid areas of reticulated hypofluorescent (dark) signal, demonstrating widespread RPE abnormalities and accumulation of lipofuscin deposits
Optical coherence tomography
The use of OCT in the clinical evaluation of patients with ophthalmic disease was first reported in 1991 by Huang et al. [29]. This noninvasive imaging modality initially demonstrated 10-μm resolution and the ability to visualize cross-sectional anatomy of anterior segment and posterior segment tissues. OCT relies on optical reflectivity to image various planes of retinal tissue similar to ultrasound technology, and its use in the evaluation of uveitis has been previously reported [30]. Macular edema, a secondary complication of a number of uveitic syndromes, is responsible for a significant proportion of uveitis patients with visual loss. Several different patterns of macular edema have been identified in patients with uveitis, including serous neurosensory retinal detachment, diffuse macular edema, and CME [31, 32]. In one study, distance visual acuity was negatively correlated with retinal thickness, CME, and the presence of a serous retinal detachment [31]. In another study, retinal thickness as measured by OCT appeared to be better correlated to reading (near) acuity and reading speed than distance visual acuity [33]. Quantitative OCT measures of retinal thickness and qualitative interpretation of OCT images have been useful in following the response of patients to immunotherapy. In a study of 50 patients with uveitis, Sivaprasad et al. observed that patients with diffuse macular edema, serous retinal detachments, and outer retinal edema appeared to respond the best to anti-inflammatory medication; cysts of the inner retina did not fare as well and were more resistant to treatment [34]. Complications of uveitis, including vitreomacular traction, epiretinal membrane formation, and posterior vitreous abnormalities, are readily visualized and monitored using OCT (Fig. 5).

OCT demonstrates zone of vitreo-macular traction with tenting of the fovea and resultant visual loss
The introduction of spectral domain/Fourier transform three-dimensional OCT into clinical practice has improved the ability to resolve retinal microstructures including inner segment and outer segment portions of photoreceptors. Three-dimensional OCT with spectral/Fourier domain technology has improved axial resolution from 10-μm to 2–3-μm resolution. Its faster acquisition time when compared to conventional OCT and ability to record three-dimensional images of retina and optic nerve pathology will likely complement the clinical exam in detecting and following ocular inflammatory disease involving posterior segment structures. Utilizing high-speed ultrahigh-resolution three-dimensional OCT, Srinivasan et al. examined 588 eyes of 327 patients with macular pathology including macular holes, ARMD, epiretinal membranes, diabetic retinopathy, and central serous retinopathy. They described improved image quality, coverage of macular area, and anatomic registration with high-definition 3D OCT when compared to standard OCT [35]. High-resolution three-dimensional OCT has been particularly useful in the volumetric analysis of uveitic entities and in visualizing pathology in three-dimensional space. Sophisticated software analysis has also provided a means to visualize topographic features of individual layers including the RPE, internal limiting membrane, and difference maps between the RPE and internal limiting membrane (Fig. 6).

a High-resolution three-dimensional OCT demonstrates the focal epiretinal membrane (arrows) leading to mild metamorphopsia. b Segmentation of retinal layers on high-resolution 3D OCT allows visualization of the epiretinal membrane in three-dimensional space. The difference in elevation between RPE and the internal limiting membrane pictured here allows improved visualization of the focal epiretinal membrane formation (red/orange color). The foveal contour is normal (central blue depression) with normal visual acuity
B-Scan ultrasound
B-scan ultrasound is a safe, noninvasive method of visualizing the anatomic status of the vitreous, retina, and choroid. Ultrasound examination has been most useful in cases of uveitis in which the posterior pole cannot be visualized, usually due to media opacity (e.g., diffuse corneal edema, severe cataract, diffuse vitreous hemorrhage, or severe vitreous haze). In cases of VKH and posterior scleritis, characteristic findings have also been seen that may be useful to follow during treatment [36, 37]. In VKH for example, ultrasound features include low to medium reflectivity of the choroid, serous retinal detachment, and thickening of the sclera.
Higher-frequency ultrasound in the 40- to 60-MHz range is utilized in ultrasound biomicroscopy (UBM) and may be utilized for the detection of anterior segment pathology with improved visualization of the iris, ciliary body, and pars plana structures. For example, ciliary body effusions and anterior fibrovascular proliferation of the pars plana may be visualized using UBM. UBM has been particularly useful in cases of chronic hypotony due to uveitis and in patients with intermediate uveitis in several reports [38, 39]. In two independent reports, ciliary body abnormalities were observed in approximately 80% of eyes evaluated for ocular hypotony [40, 41]. Uveitis patients with ocular hypotony appeared to derive the greatest benefit from UBM evaluation, and in one report, therapeutic intervention for this condition resulted in the restoration of normal intraocular pressure in 50% of cases [40].
Laboratory studies
The role of laboratory studies in the evaluation of patients with uveitis requires careful consideration because of the significant expenditure on unnecessary laboratory testing, as well as problems with false-positive results including errant diagnosis and unnecessary therapy at times. Serologic and laboratory investigations should be guided by a limited differential diagnosis. If the pretest likelihood of a disease is high, a positive test may serve to confirm clinical suspicions. However, if the pretest likelihood of a disease is extremely low (e.g., systemic lupus erythematosus in a patient with isolated anterior uveitis), the likelihood of a patient having lupus with a positive antinuclear antibody test result still remains very low [42]. The choice of laboratory investigations for anterior, intermediate, posterior, and panuveitis should be tailored according to the diagnostic possibilities, as opposed to a “shotgun” approach to laboratory testing. Patients who have bilateral, granulomatous anterior uveitis from an area endemic for tuberculosis may require PPD testing and a chest X-ray; in other patients with unilateral, acute anterior uveitis responsive to a short course of topical corticosteroid, no evaluation may be necessary.
Polymerase chain reaction-based testing
The emergence of PCR testing of ocular fluid for uveitic syndromes has improved our ability to more accurately diagnose herpetic viral retinitides, toxoplasmosis, toxocariasis, and Mycobacteria infections in some cases [43, 44]. The list of etiologies that PCR allows us to identify will likely increase as PCR techniques continue to improve. PCR testing has been valuable in the confirmation of atypical cases of cytomegalovirus (CMV) retinitis [45], as well as in the identification of viral etiologies of progressive outer retinal necrosis and acute retinal necrosis (ARN) [46, 47]. Pathogenic viruses identified in ARN thus far have included varicella–zoster virus (VZV), herpes simplex (HSV)-1, HSV-2, and CMV [46]. It is interesting to note that one child with an atypical case of ARN who was serology negative for HSV was diagnosed with HSV-2 retinitis via PCR of an aqueous humor specimen [48]. Real-time quantitative PCR may also have a role in the monitoring of VZV deoxyribonucleic acid (DNA) levels and clinical response during the treatment of progressive outer retinal necrosis and ARN [49, 50].
Besides its use in the diagnosis of the herpetic viral retinitides, PCR diagnostics have also been useful in the diagnosis of serpiginous-like choroiodopathy [51], retinal vasculitis [52], and retinochoroiditis associated with tuberculosis [53]. The use of nested PCR has also been used clinically for the detection of Mycobacterial DNA with promising results [54, 55].
PCR-based testing has also been performed for T. gondii retinochoroiditis [56]. However, in cases in which PCR for Toxoplasmosis DNA is negative, an elevation in the local antibody titer to T. gondii relative to serum antibody titer levels may contribute to the diagnosis of ocular toxoplasmosis. In one study, calculation of the Goldmann–Witmer coefficient (GWC) was complementary to PCR for DNA of herpetic viruses and Toxoplasmosis. If PCR was the only modality used for diagnosis, herpetic viruses would have been missed in 34% of cases, and Toxoplasmosis would have been missed in 64% of cases [57]. In another retrospective series comparing the utility of PCR and GWC for the diagnosis of 56 patients with uveitis, viral infections were detected by PCR in 16 of 17 cases (94%), while GWC identified T. gondii as the cause of an infectious uveitis in nine of ten cases (90%) [58].
Cytokine fluid analysis
Cytokine testing of patient samples obtained from serum and aqueous and vitreous fluids has contributed to our understanding of the pathogenic mechanisms underlying ocular inflammatory disease [59]. Their clinical utility will likely increase, as disease-specific cytokine profiles are characterized.
The ability to distinguish different ocular inflammatory pathologies from cytokine analysis is exemplified in work by Chan et al., which reported an elevation of interleukin (IL)-10 in the vitreous fluids of patients with PIOL. In vitrectomy specimens from patients with PIOL, IL-10 levels were correlated with clinical activity and the number of malignant cells [60]. In clinical practice, an elevation of the vitreous IL-10 level relative to the IL-6 level has been helpful in the diagnosis of intraocular B cell lymphoma [61]. An IL-10 to IL-6 ratio greater than 1.0 has been more frequently observed in vitritis secondary to intraocular lymphoma than in vitritis from other inflammatory etiologies (IL-10/IL-6 less than 1.0) [62].
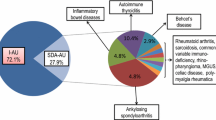
A number of cytokine-profiling studies have been performed to examine disease-specific cytokine profiles, and their findings may help to guide future immunotherapy. Ahn et al. compared the aqueous and serum cytokine profiles of Behçet’s patients with active uveitis to those who were inactive [63]. Interferon (IFN)-γ and tumor necrosis factor (TNF)-α levels were higher in the aqueous fluids of patients with active Behçet’s uveitis when compared to patients without uveitis. In addition, the immunoregulatory cytokine IL-10 was expressed in both the aqueous fluids and serum of patients with quiescent Behçet’s uveitis but was not observed in patients with active disease. Their findings suggested that a T-helper 1 cell polarization and proinflammatory state were present in Behçet’s uveitis. Several studies have also recently supported the use of infliximab, a TNF-α antagonist, in the treatment of active Behçet’s disease-associated uveitis (discussed below).
Studies in VKH patients have demonstrated elevated aqueous levels of IL-6 [64], as well as elevated IFN-γ messenger ribonucleic acid (mRNA) from peripheral blood mononuclear cells [65]. Imai et al. also demonstrated higher IFN-γ and IL-2 levels in cell culture supernatant obtained from VKH patients and a higher proportion of CD4+ T-helper cells, which produced IFN-γ and IL-2. The propensity of VKH patients toward a T-helper 1 cell-type cytokine profile may explain, in part, the efficacy of medications such as daclizumab (directed against IL-2 receptor) for this indication (discussed below).
Besides studies examining specific disease entities, a number of observations have been made on cytokine profiles in various classes of uveitis. Takase et al. examined cytokine profiles in serum and aqueous humor samples in infectious and noninfectious uveitis. IFN-γ and IL-10 levels were higher in both aqueous humor and sera samples of both infectious and noninfectious uveitis. It is interesting to note that IFN-γ was elevated in both aqueous fluids and sera of patients with noninfectious uveitic conditions, possibly indicating a systemic inflammatory process, rather than solely local inflammation, as seen in some cases of isolated infectious uveitis [66]. In a study by Curnow et al., multiplex bead immunoassay technology was used to analyze aqueous humor cytokine profiles in noninfectious uveitis versus controls. In their study, IL-6, IL-8, and IFN-γ were elevated in the aqueous humor of patients with idiopathic uveitis; the immunoregulatory cytokine transforming growth factor-β2 was decreased in idiopathic uveitis [67]. In another study comparing multiplex cytokine detection versus enzyme-linked immunosorbent assay for uveitis patients, aqueous humor IFN-γ was elevated in both anterior uveitis and panuveitis patients when compared to controls. IL-10 was notably elevated in patients treated with corticosteroid, implicating glucocorticoid-mediated upregulation of IL-10 as a possible mechanism for its immunosuppressive effect [68].
Advances in immunotherapy
Corticosteroids remain a mainstay for the treatment of active, noninfectious uveitis. However, the myriad of side effects (i.e., hypertension, osteoporosis, hyperglycemia, and growth retardation in children) associated with their long-term use requires the consideration of other corticosteroid-sparing agents including traditional agents such as the antimetabolites, T cell calcineurin inhibitors, and alkylating agents. A newer class of medications, the biologics, targets specific cytokines, cytokine receptors, and other cellular or soluble targets implicated in inflammation, and their use for ophthalmic inflammatory disease continues to increase. Agents in this class include the TNF-α inhibitors (infliximab, adalimumab, etanercept) and the IL-2 receptor daclizumab. Advances in corticosteroid delivery systems have also contributed to the therapeutic options for the ophthalmologist.
The efficacy of the antimetabolites, calcineurin inhibitors, and alkylating agents has been demonstrated for a number of uveitic entities [2, 3]. The antimetabolite class of medications includes mycophenolate mofetil, methotrexate, and azathioprine. Mycophenolate mofetil, the newest medication in this class, inhibits inosine monophosphate dehydrogenase, inhibiting a pathway of guanosine nucleotide synthesis preferentially used by T and B cells. Initially used for the prevention of renal transplant rejection, the efficacy of mycophenolate mofetil for a number of ophthalmic inflammatory diseases has been reported [69]. Some of these ophthalmic diseases include HLA-B27-associated anterior uveitis, juvenile idiopathic arthritis (JIA), scleritis, and several posterior uveitic syndromes (birdshot retinochoroidopathy, VKH, sympathetic ophthalmia) [70, 71]. Azathioprine has also been successfully used for a variety of posterior uveitic syndromes including serpiginous choroidopathy [72, 73], VKH [74], and sympathetic ophthalmia [3]. “Triple therapy” or the combination of cyclosporine, azathioprine, and prednisone has also been valuable in the treatment VKH [75], sympathetic ophthalmia [76], and serpiginous choroidopathy [77]. Methotrexate has demonstrated efficacy for the treatment of sarcoidosis-associated uveitis, intermediate uveitis, rheumatoid arthritis, and JIA [78, 79]. Primary intraocular lymphoma has also been treated with intravenous methotrexate previously [80]. Each medication requires monitoring of symptoms and laboratory tests because of their associated side effects. Azathioprine may be acutely associated with cytopenias and gastrointestinal disturbances. Mycophenolate may also be associated with gastrointestinal disturbances, but myalgias, fatigue, and headache may also be experienced by patients on this medication. Nonmelanoma skin cancer, leukopenia, and opportunistic infections have also been associated with mycophenolate mofetil use; however, these complications have frequently been observed in heavily immunosuppressed individuals. Side effects of methotrexate include leukopenias, hepatotoxicity, and impairment of renal function. In addition, approximately 1–5% of patients experience an acute pneumonitis due to a drug hypersensitivity reaction [1].
The T cell calcineurin inhibitors cyclosporine [81, 82] and tacrolimus [83] have also been used as immunotherapy for noninfectious, endogenous uveitis. Cyclosporine appears to interfere with T cell activation and recruitment, but the precise mechanism is debatable. Following binding to its immunophilin, cyclosporine is transported to the nucleus where it interferes with mRNA and protein synthesis. Cyclosporine also binds to proteins that bind to an IL-2 enhancer to prevent transcriptional activation of the IL-2 gene [1]. The efficacy of cyclosporine was first demonstrated in animal models of uveitis [84, 85] and, subsequently, in several prospective clinical trials for uveitis [81, 86]. The successful use of cyclosporine for the treatment of uveitis has been reported in a number of posterior uveitic entities including Behçet’s disease, sympathetic ophthalmia, serpiginous choroidopathy, birdshot retinochoroidopathy, intermediate uveitis, and pediatric uveitis [1]. Tacrolimus (FK506) binds to its specific immunophilin (FK506-binding protein) and also suppresses the transcription of IL-2 mRNA. Its efficacy has been demonstrated in Behçet’s disease, intermediate uveitis, sarcoidosis-associated uveitis, and uveitis refractory to cyclosporine in one case report [87, 88]. Hypertension and renal toxicity are the most common adverse effects that require vigilant monitoring in patients treated with cyclosporine. Alteration in renal function was seen more often when cyclosporine was used at a much higher dose (10 mg kg−1 day−1) than the current dose range used for uveitis patients currently (2–5 mg kg−1 day−1). Tacrolimus has been associated with nephrotoxicity, tremors, gastrointestinal symptoms, and hyperglycemia [1].
The alkylating agents, cyclophosphamide and chlorambucil, are the least commonly used immunomodulatory agents for uveitis but may be required in cases of severe, bilateral refractory uveitis not amenable to other therapies when visual acuity is salvageable [89, 90]. The efficacy of cyclophosphamide for Wegener’s granulomatosis-associated ocular and orbital inflammatory disease has been reported previously [91, 92]. In addition, cyclophosphamide has been used in the treatment of serpiginous choroidopathy and Behçet’s disease [93, 94]. Chlorambucil has been used for the treatment of sympathetic ophthalmia [95] and Behçet’s disease in several reports [96, 97]. A range of side effects associated with alkylating agents has been reported, but leukopenia and serious opportunistic infections requiring hospitalization and intravenous antibiotics have been reported. Secondary malignancy including bladder cancer in patients who have had hemorrhagic cystitis has been observed with cyclophosphamide use. Other complications of cyclophosphamide include interstitial pulmonary fibrosis and testicular atrophy. Sterility may also be seen in patients on either cyclophosphamide or chlorambucil, and cryopreservation should be considered in any patient considering alkylating agent therapy [1].
Biologic therapy for ocular inflammatory disease
A newer class of medications, the biologics, targets specific cytokines, cytokine receptors, and soluble mediators of inflammation. The use of biologics may allow the treating physician to eventually tailor the immunotherapy against a specific immune target. This will be especially relevant as the specific immune mechanisms of each disease entity are elucidated. Biologic agents have been used for a number of rheumatologic, dermatologic, hematologic, and neurologic indications. In ophthalmic practice, biologic agent therapy is currently considered an off-label use of Food and Drug Administration (FDA)-approved medications. Without FDA approval and the benefit of large prospective, controlled trial data, caution is recommended when considering the use of these medications. Three groups of biologic medications that have been used for ophthalmic indications include the TNF-α antagonists, IL-2 receptor antagonist daclizumab, and the IL-1 receptor antagonist anakinra.
Tumor necrosis factor-α antagonists
In EAU, uveal infiltration of macrophages and T cells has been observed, resulting in the elaboration of proinflammatory cytokines and subsequent loss of tissue architecture and function. TNF-α has also been detected in the aqueous humor of patients with intraocular inflammation and serves as an attractive target for anti-inflammatory therapy [98]. Three TNF-α antagonists are FDA approved for systemic administration—infliximab, adalimumab, and etanercept.
Infliximab (Remicade)
Infliximab is a mouse-derived chimeric monoclonal antibody directed against soluble and transmembrane TNF-α, preventing the binding of TNF-α with its receptor. Several clinical trials have supported the efficacy of infliximab for a number of rheumatologic diseases including rheumatoid arthritis [99, 100], Crohn’s disease [101], anklosing spondylitis [102], and psoriatic arthritis [103]. The successful use of infliximab for uveitis associated with Behçet’s disease, JIA-associated uveitis, HLA-B27-associated uveitis, and anklyosing spondylitis has been reported in a number of retrospective case series.
Suhler et al. reported their experience in a phase II, open-label study using infliximab for refractory autoimmune uveitis. Diagnoses of patients enrolled in their series included sarcoidosis, pars planitis, Behçet’s disease, and Crohn’s disease. Infliximab was given in doses of 3 mg/kg or 5 mg/kg via intravenous infusion at weeks 0, 2, and 6, and clinical efficacy was assessed at week 10. Of the 23 patients treated, 18 (78%) of the subjects met the clinical criteria for success at the week 10 time point (i.e., improved visual acuity, two-step decrease in intraocular inflammation, ability to taper immunosuppression, or decrease in inflammatory signs by OCT or FA). Seven (50%) of 14 patients maintained on infliximab for a full year maintained successful grading. Of concern, however, were a number of adverse events observed including congestive heart failure, lupus-like reaction, pulmonary embolus, and vitreous hemorrhage in two patients. Antinuclear antibodies were also found in 15 of 20 patients who received more than three infusions. Thus, while some patients achieved benefit from the medication, the rate of adverse events was unexpectedly high, and the authors recommended additional long-term studies to assess the safety and efficacy infliximab for ocular inflammatory diseases [104].
Niccoli et al. recently reported their experience with infliximab for Behçet’s disease-associated uveitis in a 24-month prospective, open-label, multicenter trial. In their study, 9 of 12 (75%) of patients treated with infliximab demonstrated complete remission with no relapses at 12-month follow-up. At the 24-month follow-up, seven of nine (78%) remained in remission. Visual acuity improved, and the number of ocular attacks was decreased compared to the year prior to infliximab therapy [105]. Abu El-Asrar et al. also reported a smaller prospective trial of six patients treated with infliximab. Follow-up varied from 16 to 36 months, but all six patients achieved remission at 2-month follow-up. Two patients experienced uveitic relapses, which resolved following a subsequent infusion of infliximab, and one patient required more frequent infusions (every 6 weeks) because of a relapse while undergoing infusion at 8-week intervals [106]. Tugal-Tutkun et al. described their experience with infliximab in Behçet’s disease-associated uveitis in patients who were resistant to combination therapy with corticosteroids, azathioprine, and cyclosporine. Infliximab infusions (5 mg/kg) given at weeks 0, 2, 6, and 14 resulted in remission during the infusion period (weeks 0–22) in 4 (30.8%) of 13 patients. One patient demonstrated a sustained remission without any attacks of uveitis at 54-week follow-up, and 36 total uveitic attacks were documented. However, the number of attacks occurring during the observation period was less than the number during the period prior to infliximab therapy [107].
The treatment of JIA-associated uveitis with infliximab has been reported in several retrospective case series [108, 109]. Richards et al. described the efficacy of infliximab in five of six JIA patients treated with a drug-free remission observed in three patients. Low-dose immunosuppression was continued in combination with infliximab therapy, supporting the role of infliximab as an adjunctive therapy for this ophthalmic indication [109].
The use of infliximab for pediatric uveitis of other etiologies has also been reported. Successful tapering of immunosuppression was reported in six patients treated with infliximab for refractory uveitis by Rajaraman et al. Diagnoses in their series included idiopathic pars planitis, juvenile rheumatoid arthritis, and retinal vasculitis [110]. Kahn et al. reported their experience with high-dose infliximab (10–20 mg/kg) in a series of 17 pediatric patients with refractory uveitis. All 17 patients demonstrated rapid responses with no observed inflammation. This was achieved in 13 of 17 patients after two infusions and in the other four patients after three infliximab infusions [111].
One prospective, noncomparative case series evaluated infliximab (10 mg/kg) as monotherapy for HLA-B27-associated anterior uveitis. In this report, six of seven patients demonstrated total resolution of inflammation, and all patients experienced an improvement of clinical symptoms and decrease in anterior chamber cells. However, relapses were observed in four patients after 5 ± 6.4 months [112].
Besides its efficacy in intraocular inflammation, infliximab has also been successfully used for the treatment of scleritis and peripheral ulcerative keratitis [113]. Murphy et al. retrospectively reviewed their experience with infliximab for three patients with scleritis, one patient with a combination of peripheral ulcerative keratitis, and three other patients with intraocular inflammation. In their series, infliximab given at a dose of 200 mg at 4- or 8-week intervals resulted in clinical improvement in six patients with five achieving remission and one patient experiencing a significant reduction in immunosuppression [114]. Of the patients described, one patient discontinued treatment because of a hypersensitivity reaction. Several case reports and case series have described the efficacy of infliximab for peripheral ulcerative keratitis and scleritis due to Wegener’s granulomatosis [115], rheumatoid arthritis [116], and relapsing polychondritis [117].
Reported adverse effects associated with infliximab therapy include reactivation of latent tuberculosis, the unmasking of demyelinating disease, congestive heart failure exacerbations, the development of antinuclear antibodies, and the formation of anti-infliximab antibodies [104, 106]. Herpes zoster and optic neuritis have also been rarely reported following infliximab therapy [118, 119]. Reports of hepatosplenic lymphoma in young patients receiving infliximab therapy for inflammatory bowel disease have also been worrisome, given the increasing use of this medication in the pediatric population [120]. The potential long-term risk of lymphoma in patients treated with infliximab is of concern and requires further study. A definite causal relationship between anti-TNF therapy and lymphoma has not been established [121], and reports in the literature differ with respect to whether anti-TNF therapy is associated with a higher incidence of lymphoma [122, 123].
Adalimumab (Humira)
Adalimumab is a humanized, recombinant TNF-α-specific immunoglobulin G1 monoclonal antibody, blocking the interaction of TNF-α with the p55 and p75 cell surface receptors. Adalimumab binds to cell-bound TNF-α and may theoretically be more effective in immunosuppression than etanercept, which is a soluble TNF-α receptor.
The use of adalimumab for uveitis has been reported in both adults and pediatric patients. In one retrospective series examining the efficacy of adalimumab in 14 children with uveitis (nine JIA-associated and five idiopathic), decreased inflammation was observed in 21 of 26 eyes (80.8%) with four eyes (15.4%) remaining stable and only one eye worsening (3.8%). Use of adalimumab allowed a decrease in topical corticosteroid usage in 11 of 14 patients (78.5%) with 4 of 14 (28.5%) completely discontinuing drops. Corticosteroid-sparing medications were also discontinued or decreased in a number of patients in this series [124]. Biester et al. reviewed their experience in the use of adalimumab for pediatric uveitis (17 JIA-associated, one idiopathic uveitis) and observed efficacy in 16 of 18 patients (88%). Of note, adalimumab was effective or mildly effective for arthritis in 13 of 16 patients (81%) [125]. Thus, while the efficacy of adalimumab was comparable to etanercept for arthritis, it appeared to be more effective for the treatment of uveitis than etanercept [125].
Two retrospective case series examining the efficacy of adalimumab for uveitis secondary to Behçet’s disease have shown promising results. In a report by Mushtaq et al., three patients with bilateral panuveitis due to Behçet’s disease were switched successfully from infliximab to adalimumab during a period of clinical remission. At variable follow-up intervals ranging from 11 to 24 months, the intraocular inflammation in all three patients remained well controlled [126, 127].
Adalimumab is given either as a 20- or 40-mg subcutaneous injection on a weekly or biweekly dosing schedule and may be advantageous versus infliximab, which requires an intravenous infusion. The most commonly reported adverse events associated with adalimumab were injection site reactions, occurring in up to 10% of patients. In one trial of adalimumab for ankylosing spondylitis, adverse events were higher in patients treated with adalimumab versus controls; it is interesting to note that the incidence of infections did not appear to be higher in the adalimumab group. No reports of reactivation of latent tuberculosis, demyelinating disorders, drug-induced lupus, congestive heart failure, or secondary malignancies were observed [128]. In a postmarketing surveillance study of rheumatoid arthritis population, adalimumab also appeared to be safe and well tolerated, without an increase in the incidence of lymphoma greater than that observed in rheumatoid arthritis patients [129]. Adverse events reported in the ophthalmic literature include injection site reactions, HSV keratitis, and elevation of liver enzymes requiring the cessation of adalimumab [125].
Etanercept (Enbrel)
Etanercept is a fusion protein composed of the extracellular ligand-binding portion of TNF receptor p75 and the Fc portion of human IgG1. Etanercept has demonstrated efficacy in joint inflammation associated with rheumatoid arthritis [130], JIA [131], psoriatic arthritis [132], and ankylosing spondylitis [133]. However, its efficacy in the treatment of ocular inflammatory disease has been variable.
Schmeling and Horneff reported the findings from questionnaires completed by pediatric rheumatologists to document the incidence of chronic anterior uveitis and associated complications in 310 JIA patients. From the 229 questionnaires returned (74%), 31 patients (13.5%) with 102 uveitic flares were identified in patients who had a history of uveitis prior to etanercept treatment. Following etanercept treatment, 32 uveitis exacerbations were reported in 19 patients; it is interesting to note that two patients experienced their first episode of uveitis. Arthritis significantly or completely responded in 87% of uveitis patients, and etanercept did not appear to influence the frequency or severity of uveitis episodes [134]. A randomized, masked trial of etanercept for JIA-associated anterior uveitis conducted at the National Eye Institute (NEI) demonstrated no difference between etanercept and placebo. Three of seven patients randomized to etanercept and two of five patients randomized to placebo were considered ophthalmic successes. However, this pilot study was limited by its small sample size [135]. In one case report of a patient with idiopathic panuveitis who only partially responded to multiple immunosuppressive medications (methotrexate, cyclosporine, corticosteroid), etanercept resulted in complete control of his intraocular inflammation [136].
Galor et al. recently compared the efficacy of etanercept versus infliximab in a retrospective analysis. Fifty-nine percent of patients treated with infliximab demonstrated a greater than or equal to 50% reduction in uveitis recurrences on therapy; conversely, none of the patients on etanercept therapy experienced a similar reduction. Seventeen of 18 (94%) patients on infliximab showed a reduction in intraocular inflammation at their final follow-up, whereas zero of four patients on etanercept experienced a reduction in intraocular inflammation [137].
Another recent report by Foeldvari et al. examined the use of the three TNF-α inhibitors for the treatment of JIA-associated uveitis in a cross-sectional survey of 33 pediatric rheumatology centers. In this report, a total of 47 patients on anti-TNF therapy were identified with a mean age of 12.5 years. Etanercept was used in 34 cases, infliximab in 25 cases, and adalimumab in three cases. In 12 of 34 patients in which etanercept was used, the treatment was inefficacious, and patients were subsequently switched to infliximab therapy. In their report, infliximab was more efficacious than etanercept for the treatment of JIA-associated uveitis. Adalimumab was efficacious in all three patients treated [138].
Etanercept is given via subcutaneous injection at a dose of 25–50 mg per dose, twice per week. Of concern are reports of ocular inflammatory exacerbations (scleritis, uveitis, myositis) associated with etanercept therapy [139]. In one case report, acute exacerbations of ankylosing spondylitis-associated uveitis were temporally associated with etanercept injections [140]. Optic neuritis has also been observed in four patients receiving etanercept injections, with three of four patients requiring discontinuation of therapy [141]. A retrospective cohort study, which reviewed 70 patients on etanercept, found no statistically significant increased risk in the development of new-onset uveitis. However, treatment with etanercept was unable to prevent uveitis onset in two patients in their cohort of patients (2.9%) [142].
Daclizumab (Zenapax)
Daclizumab is a humanized monoclonal recombinant immunoglobulin targeting Tac, a 55-kDa IL-2 receptor subunit expressed by most T, B, and natural killer cells. The IL-2 receptor system is a lymphokine receptor system, which plays a key role in the induction of the immune response. Expression of the Tac subunit is considered a critical step in the activation of all T cells that are contributors to autoimmune disease.
In animal models of uveitis, the presence of high-affinity IL-2 receptors has been demonstrated previously [85]. In uveitis patients, levels of soluble IL-2 receptor in both patient serum [143]and aqueous fluids [144] were elevated versus control subjects.
A number of studies have reported the efficacy of daclizumab for the treatment of noninfectious intermediate and posterior uveitis and panuveitis. A nonrandomized, open-label pilot study showed that the use of intravenous daclizumab therapy in up to 4-week intervals allowed patients to successfully taper other immunosuppressive medications. In addition, daclizumab was effective in preventing the expression of sight-threatening inflammatory disease in eight of ten patients treated over a 12-month period. The various diagnoses treated included sarcoidosis, idiopathic intermediate uveitis, VKH, idiopathic panuveitis, and multifocal choroiditis [145]. A longer-term phase I/II interventional study of intravenous daclizumab and a short-term phase II study using subcutaneous daclizumab also has demonstrated encouraging results. Seven of ten enrolled patients in the long-term intravenous daclizumab study were tapered off their original immunosuppressive medications. It is interesting to note that the use of 6-week intervals of daclizumab infusion resulted in recurrent uveitic disease, whereas 2- to 4-week dosing intervals did not. All five patients in the short-term subcutaneous daclizumab study met endpoints for success by 26 weeks of therapy; four of five patients met endpoints for success within the first 12 weeks [146].
A retrospective review by Papaliodis et al. documented the efficacy of daclizumab for a variety of ocular inflammatory diseases including scleritis, ocular cicatricial pemphigoid, and panuveitis [147]. Visual acuity improvement was reported in 12 of 27 (44%) eyes and in 5 of 14 (36%) patients. Intraocular inflammation improved in 16 of 27 eyes (59%), remained stable in 3 of 27 (11%) eyes, and worsened in 8 of 27 (30%) eyes.
Several other retrospective studies have reported the use of daclizumab for a variety of other ocular inflammatory conditions including birdshot retinochoroidopathy and childhood uveitis of varying etiologies [148, 149]. Gallagher et al. reported decreased ocular inflammation in five pediatric patients following the use of daclizumab. Diagnoses in their cohort included sarcoidosis, panuveitis, keratouveitis, and anterior uveitis. Four of ten eyes improved vision, while five of ten eyes remained stable. One eye demonstrated a deterioration of vision in this series [149].
For the uveitis protocol treatments conducted at the NEI, two induction treatments are typically completed within 14 days. A higher-dose induction treatment of intravenous daclizumab at 4–8 mg/kg is given on day 0 followed by another intravenous dose of 2–4 mg/kg at day 14. Maintenance therapy is then continued at 1–2-mg/kg doses in 4-week intervals.
In several studies of daclizumab for the prevention of renal allograft rejection, no difference in serious infectious complications or cancer has been observed when comparing patients receiving daclizumab or placebo [150, 151]. One patient from the NEI was diagnosed with low-grade renal cell carcinoma after 4 years of monthly intravenous daclizumab therapy. The renal cell carcinoma was surgically removed, but it was not clear whether this event preceded intravenous daclizumab therapy. Following surgical removal of the renal cell carcinoma, the participant was restarted on intravenous daclizumab and converted to subcutaneous daclizumab without any further untoward effects.
Anakinra (Kineret)
IL-1 receptor antagonist (IL-1RA) is a naturally occurring protein that inhibits IL-1 activity, downregulating the proinflammatory functions of IL-1 including chemotaxis, activation of antigen-presenting cells, and the upregulation of cell surface adhesion molecules involved in leukocyte trafficking. IL-1RA binds to IL-1 type I receptor competitively with an affinity comparable to IL-1α and IL-1β. During experimentally induced inflammation, IL-1RA limits IL-1 activity presumably through a negative feedback mechanism. Anakinra is a recombinant, human, nonglycosylated IL-1RA. Anakinra competes with the IL-1 ligand-binding site but does not induce downstream signaling pathways after it binds to its target.
In several rodent models for uveitis, increased IL-1α [152152], IL-1β [152, 153], and IL1-RA [154] have been observed. In one therapeutic trial for murine EAU, mice treated with anakinra demonstrated lower levels of clinical and histological inflammation compared to untreated mice and decreased cellular immune response and inflammatory cytokine expression (i.e., IFN-γ, TNF-α, IL-1α, IL-1β, IL-1RA, and IL-6) [155].
Benezra et al. examined the IL-1RA levels of patients with pars planitis, Behçet’s disease, and normal volunteers who were naïve to therapy and following immunosuppressive therapy (i.e., corticosteroid and cyclosporine). Prior to immunosuppressive treatment, no difference was observed in IL-1RA levels between uveitis patients and controls. Following immunosuppression, a statistically significant increase in IL-1RA was observed in uveitis patients compared to their baseline mean IL-1RA levels. This study suggests a possible mechanism involved in the immunomodulatory properties of the medications used and a potential immune target for the treatment of uveitis [156].
The efficacy of anakinra in rheumatoid arthritis patients has been reported by Cohen et al. In their report, patients treated with 1.0–2.0 mg/kg anakinra demonstrated significant improvement by the American College of Rheumatology 20% improvement criteria (i.e., ACR20 response) following 12 weeks of therapy when compared with patients receiving placebo. All patients were also on concomitant methotrexate therapy [157]. A noncomparative, open-label study of anakinra for polyarticular JIA also demonstrated encouraging results with 46 of 82 (58%) of patients experiencing clinical improvement [158]. Injection site reactions were the most commonly observed adverse event, occurring in 70% of patients in this series; however, most injection reactions were mild [158].
Thus far, the use of anakinra for ocular inflammatory disease has been limited to few case reports and small case series. Teoh et al. reported the efficacy of anakinra for ocular inflammation associated with chronic infantile neurological cutaneous articular (CINCA) syndrome. In their report, a 4-year-old patient with CINCA syndrome demonstrated resolution of both posterior uveitis and arthritis with anakinra treatment after failing other immunosuppressive medications (i.e., prednisolone, methotrexate, and etanercept) [159]. In another report by Aróstegui et al., anakinra combined with mycophenolate mofetil was successfully used to treat severe uveitis in a 15-year-old female patient with NOD2 gene-associated pediatric granulomatous arthritis. IL-1β, IL-6, and TNF-α had been elevated in the patient’s plasma prior to anakinra therapy compared to controls. Following treatment with anakinra, IL-1β, IL-6, and TNF-α normalized, correlating the plasma inflammatory cytokine levels to clinical ophthalmic findings [160].
Botsios et al. reported the use of anakinra for rheumatoid arthritis-associated anterior scleritis in two patients, both of whom were refractory to other immunosuppressive medications. One patient was initially treated with infliximab and methotrexate for a total of six infusions for her scleritis but experienced a decrease in visual acuity while on therapy. Anakinra, given at a dose of 100 mg/day combined with methotrexate, led to remission 8 weeks following initiation of therapy. The other patient reported had been on etanercept therapy for rheumatoid arthritis for 15 months prior to the onset of scleritis. Anakinra treatment (100 mg/day) resulted in quiescence of the scleritis following 6 weeks of therapy, with a sustained remission after 12-month follow-up [161].
Doses of anakinra range from 1–2 mg kg−1 day−1 via subcutaneous injection. In the ophthalmic literature, the dose of 100 mg/day has been used. Injection site reactions have been the most common adverse event in long-term follow-up of rheumatoid arthritis patients [157]. In studies of anakinra in pediatric patients, injection site reactions also appear to be the most commonly reported adverse event [158, 162].
Other biologic agents
Several other biologic agents used by other subspecialty disciplines have also been described in limited case series and require further investigation.
Alemtuzumab, a humanized monoclonal antibody, which recognizes the pan-lymphocyte antigen CD52, is lymphocytotoxic and has been reported to be effective for lymphoproliferative malignancies including peripheral T cell lymphoma. Because of its ability to deplete T cells with its subsequent immunosuppressive effects, alemtuzumab has also been used for stem cell transplantation and autoimmune conditions as well [163]. Its use for severe, refractory noninfectious ocular inflammatory disease was recently described. Dick et al. reported its use in a series of ten patients with a variety of ocular inflammatory diseases including corneal graft rejection, retinal vasculitis, sympathetic ophthalmia, Wegener’s granulomatosis, and Behçet’s disease-associated uveitis. Eight of ten patients achieved clinical remission, allowing a decrease in their immunosuppressive medications. In the two patients with Behçet’s disease and sympathetic ophthalmia, their clinical disease stabilized and became easier to control [164]. The successful use of alemtuzumab for the prevention of corneal graft rejection in a patient who had failed nine previous graft attempts despite corticosteroid, cyclophosphamide, azathioprine, and cyclosporine A has also been reported [165]. Adverse effects associated with alemtuzumab include hematologic toxicity (pancytopenia, neutropenia, thrombocytopenia, anemia) and rare cases of autoimmune hemolytic anemia. Opportunistic infection secondary to leukopenia and infusion-related illnesses consisting of fevers, rigors, and hypotension have also been reported [166].
Efalizumab, a humanized form of murine IgG1 against CD11a, the alpha subunit of lymphocyte function-associated antigen (LFA)-1, inhibits the interaction of LFA-1 with intercellular adhesion molecule (ICAM)-1, which is found on endothelial cells. LFA-1 is important in T cell trafficking into sites of inflammation via ICAM-1. Efalizumab has been approved by the FDA for the treatment of adults with moderate to severe plaque psoriasis [167, 168] and has been studied in a pilot study in patients with atopic dermatitis [169].
In EAU, expression of ICAM-1 was associated with blood–retinal barrier breakdown, immune cell adhesion, and extravasation [170]. In a mouse model of uveitis, ICAM-1 was observed on the ciliary body 6 h following induction, and LFA-1 have been observed on infiltrating lymphocytes [171]. Subsequent experiments showed that monoclonal antibodies against LFA-1 (CD11a) and ICAM-1 (CD54) inhibited the development of uveitis in a rat model and showed efficacy in the treatment of ocular inflammation [172].
The efficacy for efalizumab in the treatment of macular edema associated with noninfectious intermediate and posterior uveitis and panuveitis is currently being studied at the NEI. In the NEI treatment protocol, uveitis patients receive efalizumab at a dose of 0.7 mg/kg initially and 1-mg/kg weekly maintenance doses for a 16-week duration.
Adverse effects associated with efalizumab therapy observed during the clinical studies for psoriasis included injection reactions (i.e., headache, fever, chills, myalgias) within 48 h following the first two doses of medication. Adverse effects resulting in therapy discontinuation included pain, arthritis, and psoriasis [168]. A review of phase III clinical trials of the use of efalizumab for plaque psoriasis found no significant increase in the incidence of malignant melanoma, nonmelanoma skin cancers, and lymphoproliferative disorders in patients treated with efalizumab when compared to placebo [173]. However, in this study, the incidence of nonmelanoma skin cancers was elevated in both efalizumab and placebo-treated groups compared to external databases.
As the number of biologic immunosuppressive agents continues to increase, their use for ophthalmic inflammatory disease will likely continue. Caution is certainly warranted, as the long-term adverse effects are still unknown for many of the therapies currently being developed. Scrutiny of the basic science evidence with regard to efficacy for ocular inflammatory disease, preclinical testing, and postmarketing surveillance are also required for the treating physician. Before initiation of biologic therapy, a thorough medical evaluation is warranted, as well as a full discussion of the potential risks of immunosuppressive therapy including serious infections and secondary malignancies. The reported adverse effects as well as unknown long-term risks are also discussed so that the patients are aware of the implications of long-term systemic immunosuppressive therapy. While the TNF-α inhibitors infliximab and adalimumab and IL-2 receptor antagonist daclizumab have demonstrated promise in the treatment of ocular inflammatory disease, judicious use of these agents and future biologic therapies is necessary.
Corticosteroid delivery systems
Jaffe et al. initially reported their data from a prospective, interventional pilot trial of the fluocinolone acetonide-sustained drug delivery device for the treatment of severe, noninfectious uveitis. The fluocinolone-sustained-release implant was designed to release corticosteroid for at least 2.5 years following implantation into the vitreous cavity. Patients treated in the initial pilot trial included Behçet’s disease, idiopathic panuveitis, and intermediate uveitis. They reported stabilization or improvement in visual acuity after implantation of the device and four of seven eyes improved by three lines or more following a mean follow-up of 10 months [174].
The results of a historically controlled, multicenter trial for patients with unilateral or bilateral uveitis showed a reduction in uveitis recurrence rate from 51.4% in the 34 weeks preceding device implantation to 6.1% in the 34-week period following surgery. In addition, 87% of patients maintained or improved visual acuity with a decreased number of eyes that required topical medications, periocular injections, and systemic medications. The development of glaucoma and cataract were the most frequent complications observed with 51.1% of patients requiring topical ocular antihypertensive agents and 5.8% required glaucoma filtration surgery. In addition, 19.8% of patients showed an increase in their lens opacity, and 9.9% of the patients required cataract surgery at final follow-up [175].
The Multicenter Uveitis Steroid Treatment Trial is currently underway to compare the fluocinolone acetonide implant to standard systemic therapy for patients with noninfectious intermediate uveitis, posterior uveitis, or panuveitis. Results from this study will better enable us to determine where the corticosteroid implant may fall in our current therapeutic paradigm.
A recent study evaluating the efficacy of a novel intravitreous dexamethasone drug delivery system for persistent macular edema was performed. In this study, patients with macular edema due to diabetic retinopathy, venous occlusive disease, and uveitis demonstrated similar improvements in visual acuity. At the primary endpoint assessment at 90-day follow-up, improvement in visual acuity by at least ten letters was achieved by a greater proportion of patients treated with dexamethasone 700 μg (35%) compared to patients receiving dexamethasone 350 μg (24%) and observation (13%; P < 0.001 vs. 700-μg group; P = 0.04 vs. 350-μg group). Injections were well tolerated in most patients; however, 11% of treated patients experienced intraocular pressure elevations of 10 mmHg or higher. Further studies evaluating this drug delivery system are underway [176].
Conclusion
In summary, a significant number of advances have been made to expand the diagnostic capabilities and therapeutic armentarium of the uveitis and ocular inflammatory specialist. The development of a standardized anatomic classification scheme for uveitis has provided a framework for future clinical outcome research. In addition, this scheme may aid the clinician in honing their differential diagnoses based on the anatomic sites of uveal inflammation for directed laboratory and radiographic testing. Ancillary testing including traditional tests such as FA and OCT continue to play a major role in the evaluation of ocular inflammatory disease. However, the advent of FAF and three-dimensional spectral OCT in clinical practice will likely assist our evaluation of ocular inflammatory diseases. Laboratory-based testing including PCR, cytokine, and chemokine evaluation has improved our ability to identify infectious etiologies and immunologic features of these conditions. As the pathogenic mechanisms that cause uveitis become apparent, medications such as the biologic agents will allow us to target specific soluble mediators of inflammation and provide improved care for patients with uveitis and other related immune-mediated ophthalmic diseases.
References
Nussenblatt RB, Whitcup SC (2004) Uveitis: Fundamentals and clinical practice, 3rd edn. Elsevier, Philadelphia
Jabs DA, Akpek EK (2005) Immunosuppression for posterior uveitis. Retina 25(1):1–18
Jabs DA, Rosenbaum JT, Foster CS, Holland GN, Jaffe GJ, Louie JS, Nussenblatt RB, Stiehm ER, Tessler H, Van Gelder RN, Whitcup SM, Yocum D (2000) Guidelines for the use of immunosuppressive drugs in patients with ocular inflammatory disorders: recommendations of an expert panel. Am J Ophthalmol 130(4):492–513
Lim L, Suhler EB, Smith JR (2006) Biologic therapies for inflammatory eye disease. Clin Experiment Ophthalmol 34(4):365–374
Yeh S, Nussenblatt RB, Levy-Clarke GA (2007) Exp Rev Clin Immunol 3(5):781–796
Nussenblatt RB, Caspi RR, Dinning WJ, Palestine AG, Hiestand P, Borel J (1986) A comparison of the effectiveness of cyclosporine A, D, and G in the treatment of experimental autoimmune uveitis in rats. J Immunopharmacol 8(3):427–435
Caspi RR, Roberge FG, Chan CC, Wiggert B, Chader GJ, Rozenszajn LA, Lando Z, Nussenblatt RB (1988) A new model of autoimmune disease. Experimental autoimmune uveoretinitis induced in mice with two different retinal antigens. J Immunol 140(5):1490–1495
Nussenblatt RB, Ferris F 3rd (2007) Age-related macular degeneration and the immune response: implications for therapy. Am J Ophthalmol 144(4):618–626
Joussen AM, Poulaki V, Le ML, Koizumi K, Esser C, Janicki H, Schraermeyer U, Kociok N, Fauser S, Kirchhof B, Kern TS, Adamis AP (2004) A central role for inflammation in the pathogenesis of diabetic retinopathy. FASEB J 18(12):1450–1452
Klein RJ, Zeiss C, Chew EY, Tsai JY, Sackler RS, Haynes C, Henning AK, SanGiovanni JP, Mane SM, Mayne ST, Bracken MB, Ferris FL, Ott J, Barnstable C, Hoh J (2005) Complement factor H polymorphism in age-related macular degeneration. Science 308(5720):385–389
Edwards AO, Ritter R 3rd, Abel KJ, Manning A, Panhuysen C, Farrer LA (2005) Complement factor H polymorphism and age-related macular degeneration. Science 308(5720):421–424
Dastgheib K, Green WR (1994) Granulomatous reaction to Bruch’s membrane in age-related macular degeneration. Arch Ophthalmol 112(6):813–818
Sarks JP, Sarks SH, Killingsworth MC (1997) Morphology of early choroidal neovascularisation in age-related macular degeneration: correlation with activity. Eye 11(Pt 4):515–522
Jabs DA, Nussenblatt RB, Rosenbaum JT, Standardization of Uveitis Nomenclature (SUN) Working Group (2005) Standardization of uveitis nomenclature for reporting clinical data. Results of the First International Workshop. Am J Ophthalmol 140(3):509–516
Moorthy RS, Valluri S, Jampol LM (1998) Drug-induced uveitis. Surv Ophthalmol 42(6):557–570
Hopkins J, Walsh A, Chakravarthy U (2006) Fundus autofluorescence in age-related macular degeneration: an epiphenomenon? Invest Ophthalmol Vis Sci 47(6):2269–2271
Delori FC, Dorey CK, Staurenghi G, Arend O, Goger DG, Weiter JJ (1995) In vivo fluorescence of the ocular fundus exhibits retinal pigment epithelium lipofuscin characteristics. Invest Ophthalmol Vis Sci 36(3):718–729
Delori FC, Goger DG, Dorey CK (2001) Age-related accumulation and spatial distribution of lipofuscin in RPE of normal subjects. Invest Ophthalmol Vis Sci 42(8):1855–1866
Spaide RF (2003) Fundus autofluorescence and age-related macular degeneration. Ophthalmology 110(2):392–399
Spaide RF, Klancnik JM Jr (2005) Fundus autofluorescence and central serous chorioretinopathy. Ophthalmology 112(5):825–33
Yannuzzi LA, Ober MD, Slakter JS, Spaide RF, Fisher YL, Flower RW, Rosen R (2004) Ophthalmic fundus imaging: today and beyond. Am J Ophthalmol 137(3):511–524
Souka AA, Hillenkamp J, Gora F, Gabel VP, Framme C (2006) Correlation between optical coherence tomography and autofluorescence in acute posterior multifocal placoid pigment epitheliopathy. Graefes Arch Clin Exp Ophthalmol 244(10):1219–1223
Bellmann C, Holz FG, Breitbart A, Völcker HE (1999) Bilateral acute syphilitic posterior placoid chorioretinopathy–angiographic and autofluorescence characteristics. Ophthalmologe 96(8):522–528
Spaide RF (2004) Collateral damage in acute zonal occult outer retinopathy. Am J Ophthalmol 138(5):887–889
Popović P, Jarc-Vidmar M, Hawlina M (2005) Abnormal fundus autofluorescence in relation to retinal function in patients with retinitis pigmentosa. Graefes Arch Clin Exp Ophthalmol 243(10):1018–1027
Ergun E, Hermann B, Wirtitsch M, Unterhuber A, Ko TH, Sattmann H, Scholda C, Fujimoto JG, Stur M, Drexler W (2005) Assessment of central visual function in Stargardt’s disease/fundus flavimaculatus with ultrahigh-resolution optical coherence tomography. Invest Ophthalmol Vis Sci 46(1):310–316
Spaide RF, Noble K, Morgan A, Freund KB (2006) Vitelliform macular dystrophy. Ophthalmology 113(8):1392–1400
Sawa M, Ober MD, Freund KB, Spaide RF (2006) Fundus autofluorescence in patients with pseudoxanthoma elasticum. Ophthalmology 113(5):814–820 e2
Huang D, Swanson EA, Lin CP, Schuman JS, Stinson WG, Chang W, Hee MR, Flotte T, Gregory K, Puliafito CA et al (1991) Optical coherence tomography. Science 254(5035):1178–1181 (Nov 22)
Gallagher MJ, Yilmaz T, Cervantes-Castañeda RA, Foster CS (2007) The characteristic features of optical coherence tomography in posterior uveitis. Br J Ophthalmol 91(12):1680–1685
Markomichelakis NN, Halkiadakis I, Pantelia E, Peponis V, Patelis A, Theodossiadis P, Theodossiadis G (2004) Patterns of macular edema in patients with uveitis: qualitative and quantitative assessment using optical coherence tomography. Ophthalmology 111(5):946–953
Catier A, Tadayoni R, Paques M, Erginay A, Haouchine B, Gaudric A, Massin P (2005) Characterization of macular edema from various etiologies by optical coherence tomography. Am J Ophthalmol 140(2):200–206
Kiss CG, Barisani-Asenbauer T, Maca S, Richter-Mueksch S, Radner W (2006) Reading performance of patients with uveitis-associated cystoid macular edema. Am J Ophthalmol 142(4):620–624
Sivaprasad S, Ikeji F, Xing W, Lightman S (2007) Tomographic assessment of therapeutic response to uveitic macular oedema. Clin Exp Ophthalmol 35(8):719–723
Srinivasan VJ, Wojtkowski M, Witkin AJ, Duker JS, Ko TH, Carvalho M, Schuman JS, Kowalczyk A, Fujimoto JG (2006) High-definition and 3-dimensional imaging of macular pathologies with high-speed ultrahigh-resolution optical coherence tomography. Ophthalmology 113(11):2054 e1–14
Forster DJ, Cano MR, Green RL, Rao NA (1990) Echographic features of the Vogt–Koyanagi–Harada syndrome. Arch Ophthalmol 108(10):1421–1426
McCluskey PJ, Watson PG, Lightman S, Haybittle J, Restori M, Branley M (1999) Posterior scleritis: clinical features, systemic associations, and outcome in a large series of patients. Ophthalmology 106(12):2380–2386
Greiner KH, Kilmartin DJ, Forrester JV, Atta HR (2002) Grading of pars planitis by ultrasound biomicroscopy–echographic and clinical study. Eur J Ultrasound 15(3):139–144
Häring G, Nölle B, Wiechens B (1998) Ultrasound biomicroscopic imaging in intermediate uveitis. Br J Ophthalmol 82(6):625–629
Roters S, Szurman P, Engels BF, Bartz-Schmidt KU, Krieglstein GK (2002) Ultrasound biomicroscopy in chronic ocular hypotony: its impact on diagnosis and management. Retina 22(5):581–588
Tran VT, LeHoang P, Herbort CP (2001) Value of high-frequency ultrasound biomicroscopy in uveitis. Eye 15(Pt 1):23–30
Rosenbaum JT, Wernick R (1990) The utility of routine screening of patients with uveitis for systemic lupus erythematosus or tuberculosis. A Bayesian analysis. Arch Ophthalmol 108(9):1291–1293
Van Gelder RN (2001) Applications of the polymerase chain reaction to diagnosis of ophthalmic disease. Surv Ophthalmol 46(3):248–258
Van Gelder RN (2003) Cme review: polymerase chain reaction diagnostics for posterior segment disease. Retina 23(4):445–452
Doornenbal P, Seerp Baarsma G, Quint WG, Kijlstra A, Rothbarth PH, Niesters HG (1996) Diagnostic assays in cytomegalovirus retinitis: detection of herpesvirus by simultaneous application of the polymerase chain reaction and local antibody analysis on ocular fluid. Br J Ophthalmol 80(3):235–240
Tran TH, Rozenberg F, Cassoux N, Rao NA, LeHoang P, Bodaghi B (2003) Polymerase chain reaction analysis of aqueous humour samples in necrotising retinitis. Br J Ophthalmol 87(1):79–83
Knox CM, Chandler D, Short GA, Margolis TP (1998) Polymerase chain reaction-based assays of vitreous samples for the diagnosis of viral retinitis. Use in diagnostic dilemmas. Ophthalmology 105(1):37–44
Chiquet C, Bodaghi B, Mouqin C, Najioullah F (2006) Acute retinal necrosis diagnosed in a child with chronic panuveitis. Graefes Arch Clin Exp Ophthalmol 244(9):1206–1208
Yin PD, Kurup SK, Fischer SH, Rhee HH, Byrnes GA, Levy-Clarke GA, Buggage RR, Nussenblatt RB, Mican JM, Wright ME (2007) Progressive outer retinal necrosis in the era of highly active antiretroviral therapy: successful management with intravitreal injections and monitoring with quantitative PCR. J Clin Virol 38(3):254–259
Asano S, Yoshikawa T, Kimura H, Enomoto Y, Ohashi M, Terasaki H, Nishiyama Y (2004) Monitoring herpesvirus DNA in three cases of acute retinal necrosis by real-time PCR. J Clin Virol 29(3):206–209
Gupta V, Gupta A, Arora S, Bambery P, Dogra MR, Agarwal A (2003) Presumed tubercular serpiginouslike choroiditis: clinical presentations and management. Ophthalmology 110(9):1744–1749
Gupta A, Gupta V, Arora S, Dogra MR, Bambery P (2001) PCR-positive tubercular retinal vasculitis: clinical characteristics and management. Retina 21(5):435–444
Arora SK, Gupta V, Gupta A, Bambery P, Kapoor GS, Sehgal S (1999) Diagnostic efficacy of polymerase chain reaction in granulomatous uveitis. Tuber Lung Dis 79(4):229–233
Rao NA, Saraswathy S, Smith RE (2006) Tuberculous uveitis: distribution of Mycobacterium tuberculosis in the retinal pigment epithelium. Arch Ophthalmol 124(12):1777–1779
Ortega-Larrocea G, Bobadilla-del-Valle M, Ponce-de-León A, Sifuentes-Osornio J (2003) Nested polymerase chain reaction for Mycobacterium tuberculosis DNA detection in aqueous. and vitreous of patients with uveitis. Arch Med Res 34(2):116–119
Aouizerate F, Cazenave J, Poirier L, Verin P, Cheyrou A, Begueret J, Lagoutte F (1993) Detection of Toxoplasma gondii in aqueous humour by the polymerase chain reaction. Br J Ophthalmol 77(2):107–109
De Groot-Mijnes JD, Rothova A, Van Loon AM, Schuller M, Ten Dam-Van Loon NH, De Boer JH, Schuurman R, Weersink AJ (2006) Polymerase chain reaction and Goldmann–Witmer coefficient analysis are complimentary for the diagnosis of infectious uveitis. Am J Ophthalmol 141(2):313–318
Westeneng AC, Rothova A, de Boer JH, de Groot-Mijnes JD (2007) Infectious uveitis in immunocompromised patients and the diagnostic value of polymerase chain reaction and Goldmann–Witmer coefficient in aqueous analysis. Am J Ophthalmol 144(5):781–785
Curnow SJ, Murray PI (2006) Inflammatory mediators of uveitis: cytokines and chemokines. Curr Opin Ophthalmol 17(6):532–537
Chan CC, Whitcup SM, Solomon D, Nussenblatt RB (1995) Interleukin-10 in the vitreous of patients with primary intraocular lymphoma. Am J Ophthalmol 120(5):671–673
Chan CC (2003) Molecular pathology of primary intraocular lymphoma. Trans Am Ophthalmol Soc 101:275–292
Buggage RR, Whitcup SM, Nussenblatt RB, Chan CC (1999) Using interleukin 10 to interleukin 6 ratio to distinguish primary intraocular lymphoma and uveitis. Invest Ophthalmol Vis Sci 40(10):2462–2463
Ahn JK, Yu HG, Chung H, Park YG (2006) Intraocular cytokine environment in active Behçet uveitis. Am J Ophthalmol 142(3):429–434
Norose K, Yano A, Wang XC, Tokushima T, Umihira J, Seki A, Nohara M, Segawa K (1994) Dominance of activated T cells and interleukin-6 in aqueous humor in Vogt–Koyanagi–Harada disease. Invest Ophthalmol Vis Sci 35(1):33–39
Imai Y, Sugita M, Nakamura S, Toriyama S, Ohno S (2001) Cytokine production and helper T cell subsets in Vogt–Koyanagi–Harada’s disease. Curr Eye Res 22(4):312–318
Takase H, Futagami Y, Yoshida T, Kamoi K, Sugita S, Imai Y, Mochizuki M (2006) Cytokine profile in aqueous humor and sera of patients with infectious or noninfectious uveitis. Invest Ophthalmol Vis Sci 47(4):1557–1561
Curnow SJ, Falciani F, Durrani OM, Cheung CM, Ross EJ, Wloka K, Rauz S, Wallace GR, Salmon M, Murray PI (2005) Multiplex bead immunoassay analysis of aqueous humor reveals distinct cytokine profiles in uveitis. Invest Ophthalmol Vis Sci 46(11):4251–4259
Ooi KG, Galatowicz G, Towler HM, Lightman SL, Calder VL (2006) Multiplex cytokine detection versus ELISA for aqueous humor: IL-5, IL-10, and IFNgamma profiles in uveitis. Invest Ophthalmol Vis Sci 47(1):272–277
Deierhoi MH, Kauffman RS, Hudson SL, Barber WH, Curtis JJ, Julian BA, Gaston RS, Laskow DA, Diethelm AG (1993) Experience with mycophenolate mofetil (RS61443) in renal transplantation at a single center. Ann Surg 217(5):476–482
Doycheva D, Deuter C, Stuebiger N, Biester S, Zierhut M (2007) Mycophenolate mofetil in the treatment of uveitis in children. Br J Ophthalmol 91(2):180–184
Choudhary A, Harding SP, Bucknall RC, Pearce IA (2006) Mycophenolate mofetil as an immunosuppressive agent in refractory inflammatory eye disease. J Ocul Pharmacol Ther 22(3):168–175
Vianna RN, Ozdal PC, Deschênes J, Burnier MN Jr (2006) Combination of azathioprine and corticosteroids in the treatment of serpiginous choroiditis. Can J Ophthalmol 41(2):183–189
Akpek EK, Baltatzis S, Yang J, Foster CS (2001) Long-term immunosuppressive treatment of serpiginous choroiditis. Ocul Immunol Inflamm 9(3):153–167
Kim SJ, Yu HG (2007) The use of low-dose azathioprine in patients with Vogt–Koyanagi–Harada disease. Ocul Immunol Inflamm 15(5):381–387
Berker N, Ozdamar Y, Soykan E, Ozdal P, Ozkan SS (2007) Vogt–Koyanagi–Harada syndrome in children: report of a case and review of the literature. Ocul Immunol Inflamm 15(4):351–357
Hakin KN, Pearson RV, Lightman SL (1992) Sympathetic ophthalmia: visual results with modern immunosuppressive therapy. Eye 6(Pt 5):453–455
Hooper PL, Kaplan HJ (1991) Triple agent immunosuppression in serpiginous choroiditis. Ophthalmology 98(6):944–951
Kaplan-Messas A, Barkana Y, Avni I, Neumann R (2003) Methotrexate as a first-line corticosteroid-sparing therapy in a cohort of uveitis and scleritis. Ocul Immunol Inflamm 11(2):131–139
Samson CM, Waheed N, Baltatzis S, Foster CS (2001) Methotrexate therapy for chronic noninfectious uveitis: analysis of a case series of 160 patients. Ophthalmology 108(6):1134–1139
Kim SK, Chan CC, Wallace DJ (2005) Management of primary intraocular lymphoma. Curr Oncol Rep 7(1):74–79
Nussenblatt RB, Palestine AG, Chan CC, Stevens G Jr, Mellow SD, Green SB (1991) Randomized, double-masked study of cyclosporine compared to prednisolone in the treatment of endogenous uveitis. Am J Ophthalmol 112(2):138–146
Walton RC, Nussenblatt RB, Whitcup SM (1998) Cyclosporine therapy for severe sight-threatening uveitis in children and adolescents. Ophthalmology 105(11):2028–2034
Murphy CC, Greiner K, Plskova J, Duncan L, Frost NA, Forrester JV, Dick AD (2005) Cyclosporine vs. tacrolimus therapy for posterior and intermediate uveitis. Arch Ophthalmol 123(5):634–641
Fujino Y, Okumura A, Nussenblatt RB, Gery I, Mochizuki M (1988) Cyclosporine-induced specific unresponsiveness to retinal soluble antigen in experimental autoimmune uveoretinitis. Clin Immunol Immunopathol 46(2):234–248
Caspi RR, McAllister CG, Gery I, Nussenblatt RB (1988) Differential effects of cyclosporins A and G on functional activation of a T-helper-lymphocyte line mediating experimental autoimmune uveoretinitis. Cell Immunol 113(2):350–360
Nussenblatt RB, de Smet MD, Rubin B, Freidlin V, Whitcup SM, Davis J, Herman D, Bloom JN, Sran PK, Whitcher S et al (1993) A masked, randomized, dose-response study between cyclosporine A and G in the treatment of sight-threatening uveitis of noninfectious origin. Am J Ophthalmol 115(5):583–591
Hogan AC, McAvoy CE, Dick AD, Lee RW (2007) Long-term efficacy and tolerance of tacrolimus for the treatment of uveitis. Ophthalmology 114(5):1000–1006
Sloper CM, Powell RJ, Dua HS (1999) Tacrolimus (FK506) in the treatment of posterior uveitis refractory to cyclosporine. Ophthalmology 106(4):723–728
Durrani K, Papaliodis GN, Foster CS (2004) Pulse IV cyclophosphamide in ocular inflammatory disease: efficacy and short-term safety. Ophthalmology 111(5):960–965
Goldstein DA, Fontanilla FA, Kaul S, Sahin O, Tessler HH (2002) Long-term follow-up of patients treated with short-term high-dose chlorambucil for sight-threatening ocular inflammation. Ophthalmology 109(2):370–377
Valmaggia C, Neuweiler J (2001) Orbital involvement as the first manifestation in classic Wegener’s granulomatosis. Orbit 20(3):231–237
Florine CW, Dwyer M, Holland EJ (1993) Wegener’s granulomatosis presenting with sclerokeratitis diagnosed by antineutrophil cytoplasmic autoantibodies (ANCA). Surv Ophthalmol 37(5):373–376
Akpek EK, Jabs DA, Tessler HH, Joondeph BC, Foster CS (2002) Successful treatment of serpiginous choroiditis with alkylating agents. Ophthalmology 109(8):1506–1513
Davatchi F, Baygan F, Chams H, Chams C (1984) Cyclophosphamide in the treatment of the ocular manifestations of Behçet’s disease. J Rheumatol 11(3):404–405
Yang CS, Liu JH (1995) Chlorambucil therapy in sympathetic ophthalmia. Am J Ophthalmol 119(4):482–488
Tessler HH, Jennings T (1990) High-dose short-term chlorambucil for intractable sympathetic ophthalmia and Behçet’s disease. Br J Ophthalmol 74(6):353–357
Mudun BA, Ergen A, Ipcioglu SU, Burumcek EY, Durlu Y, Arslan MO (2001) Short-term chlorambucil for refractory uveitis in Behcet’s disease. Ocul Immunol Inflamm 9(4):219–229
Hale S, Lightman S (2006) Anti-TNF therapies in the management of acute and chronic uveitis. Cytokine 33(4):231–237
Maini R, St Clair EW, Breedveld F, Furst D, Kalden J, Weisman M, Smolen J, Emery P, Harriman G, Feldmann M, Lipsky P (1999) Infliximab (chimeric anti-tumour necrosis factor alpha monoclonal antibody) versus placebo in rheumatoid arthritis patients receiving concomitant methotrexate: a randomised phase III trial. ATTRACT Study Group. Lancet 354(9194):1932–1939
Lipsky PE, van der Heijde DM, St Clair EW, Furst DE, Breedveld FC, Kalden JR, Smolen JS, Weisman M, Emery P, Feldmann M, Harriman GR, Maini RN, Anti-Tumor Necrosis Factor Trial in Rheumatoid Arthritis with Concomitant Therapy Study Group (2000) Infliximab and methotrexate in the treatment of rheumatoid arthritis. Anti-Tumor Necrosis Factor Trial in Rheumatoid Arthritis with Concomitant Therapy Study Group. N Engl J Med 343(22):1594–1602
Hanauer SB, Feagan BG, Lichtenstein GR, Mayer LF, Schreiber S, Colombel JF, Rachmilewitz D, Wolf DC, Olson A, Bao W, Rutgeerts P, ACCENT I Study Group (2002) Maintenance infliximab for Crohn’s disease: the ACCENT I randomised trial. Lancet 359(9317):1541–1549
Braun J, Brandt J, Listing J, Zink A, Alten R, Golder W, Gromnica-Ihle E, Kellner H, Krause A, Schneider M, Sorensen H, Zeidler H, Thriene W, Sieper J (2002) Treatment of active ankylosing spondylitis with infliximab: a randomised controlled multicentre trial. Lancet 359(9313):1187–1193
Cauza E, Spak M, Cauza K, Hanusch-Enserer U, Dunky A, Wagner E (2002) Treatment of psoriatic arthritis and psoriasis vulgaris with the tumor necrosis factor inhibitor infliximab. Rheumatol Int 22(6):227–232
Suhler EB, Smith JR, Wertheim MS, Lauer AK, Kurz DE, Pickard TD, Rosenbaum JT (2005) A prospective trial of infliximab therapy for refractory uveitis: preliminary safety and efficacy outcomes. Arch Ophthalmol 123(7):903–912
Niccoli L, Nannini C, Benucci M, Chindamo D, Cassarà E, Salvarani C, Cimino L, Gini G, Lenzetti I, Cantini F (2007) Long-term efficacy of infliximab in refractory posterior uveitis of Behcet’s disease: a 24-month follow-up study. Rheumatology (Oxford) 46(7):1161–1164
Abu El-Asrar AM, Abboud EB, Aldibhi H, Al-Arfaj A (2005) Long-term safety and efficacy of infliximab therapy in refractory uveitis due to Behçet’s disease. Int Ophthalmol 26(3):83–92
Tugal-Tutkun I, Mudun A, Urgancioglu M, Kamali S, Kasapoglu E, Inanc M, Gul A (2005) Efficacy of infliximab in the treatment of uveitis that is resistant to treatment with the combination of azathioprine, cyclosporine, and corticosteroids in Behcet’s disease: an open-label trial. Arthritis Rheum 52(8):2478–2484
Sharma SM, Ramanan AV, Riley P, Dick AD (2007) Use of infliximab in juvenile onset rheumatological disease associated refractory uveitis: Efficacy in joint and ocular disease. Ann Rheum Dis 66(6):840–841
Richards JC, Tay-Kearney ML, Murray K, Manners P (2005) Infliximab for juvenile idiopathic arthritis-associated uveitis. Clin Exp Ophthalmol 33(5):461–468
Rajaraman RT, Kimura Y, Li S, Haines K, Chu DS (2006) Retrospective case review of pediatric patients with uveitis treated with infliximab. Ophthalmology 113(2):308–314
Kahn P, Weiss M, Imundo LF, Levy DM (2006) Favorable response to high-dose infliximab for refractory childhood uveitis. Ophthalmology 113(5):864 e1–2
El-Shabrawi Y, Hermann J (2002) Anti-tumor necrosis factor-alpha therapy with infliximab as an alternative to corticosteroids in the treatment of human leukocyte antigen B27-associated acute anterior uveitis. Ophthalmology 109(12):2342–2346
Thomas JW, Pflugfelder SC (2005) Therapy of progressive rheumatoid arthritis-associated corneal ulceration with infliximab. Cornea 24(6):742–744
Murphy CC, Ayliffe WH, Booth A, Makanjuola D, Andrews PA, Jayne D (2004) Tumor necrosis factor alpha blockade with infliximab for refractory uveitis and scleritis. Ophthalmology 111(2):352–356
El-Shabrawi Y, Hermann J (2005) Anti-TNF alpha therapy in chronic necrotizing scleritis resistant to standard immunomodulatory therapy in a patient with Wegener’s granulomatosis. Eye 19(9):1017–1018
Atchia II, Kidd CE, Bell RW (2006) Rheumatoid arthritis-associated necrotizing scleritis and peripheral ulcerative keratitis treated successfully with infliximab. J Clin Rheumatol 12(6):291–293
Cazabon S, Over K, Butcher J (2005) The successful use of infliximab in resistant relapsing polychondritis and associated scleritis. Eye 19(2):222–224 (Feb)
Mejico LJ, Infliximab-associated retrobulbar optic neuritis (2004) Arch Ophthalmol 122(5):793–794
Lanthier N, Parc C, Scavennec R, Dhôte R, Brézin AP, Guillevi L (2005) Infliximab in the treatment of posterior uveitis in Behçet’s disease. Long term follow up in four patients. Presse Med 34(13):916–918
Mackey AC, Green L, Liang LC, Dinndorf P, Avigan M (2007) Hepatosplenic T cell lymphoma associated with infliximab use in young patients treated for inflammatory bowel disease. J Pediatr Gastroenterol Nutr 44(2):265–267
Geborek P, Bladström A, Turesson C, Gulfe A, Petersson IF, Saxne T, Olsson H, Jacobsson LT (2005) Tumour necrosis factor blockers do not increase overall tumour risk in patients with rheumatoid arthritis, but may be associated with an increased risk of lymphomas. Ann Rheum Dis 64(5):699–703
Wolfe F, Michaud K (2007) The effect of methotrexate and anti-tumor necrosis factor therapy on the risk of lymphoma in rheumatoid arthritis in 19,562 patients during 89,710 person-years of observation. Arthritis Rheum 56(5):1433–1439
Askling J, Fored CM, Baecklund E, Brandt L, Backlin C, Ekbom A, Sundström C, Bertilsson L, Cöster L, Geborek P, Jacobsson LT, Lindblad S, Lysholm J, Rantapää-Dahlqvist S, Saxne T, Klareskog L, Feltelius N (2005) Haematopoietic malignancies in rheumatoid arthritis: lymphoma risk and characteristics after exposure to tumour necrosis factor antagonists. Ann Rheum Dis 64(10):1414–1420
Vazquez-Cobian LB, Flynn T, Lehman TJ (2006) Adalimumab therapy for childhood uveitis. J Pediatr 149(4):572–575
Biester S, Deuter C, Michels H, Haefner R, Kuemmerle-Deschner J, Doycheva D, Zierhut M (2007) Adalimumab in the therapy of uveitis in childhood. Br J Opthalmol 91(3):319–324
van Laar JA, Missotten T, van Daele PL, Jamnitski A, Baarsma GS, van Hagen PM (2007) Adalimumab: a new modality for Behçet’s disease? Ann Rheum Dis 66(4):565–566
Mushtaq B, Saeed T, Situnayake RD, Murray PI (2007) Adalimumab for sight-threatening uveitis in Behcet’s disease. Eye 21(6):824–825
van der Heijde D, Kivitz A, Schiff MH, Sieper J, Dijkmans BA, Braun J, Dougados M, Reveille JD, Wong RL, Kupper H, Davis JC Jr, ATLAS Study Group (2006) Efficacy and safety of adalimumab in patients with ankylosing spondylitis: results of a multicenter, randomized, double-blind, placebo-controlled trial. Arthritis Rheum 54(7):2136–2146
Schiff MH, Burmester GR, Kent JD, Pangan AL, Kupper H, Fitzpatrick SB, Donovan C (2006) Safety analyses of adalimumab (HUMIRA) in global clinical trials and US postmarketing surveillance of patients with rheumatoid arthritis. Ann Rheum Dis 65(7):889–894
Klareskog L, van der Heijde D, de Jager JP, Gough A, Kalden J, Malaise M, Martin Mola E, Pavelka K, Sany J, Settas L, Wajdula J, Pedersen R, Fatenejad S, Sanda M, TEMPO (Trial of Etanercept and Methotrexate with Radiographic Patient Outcomes) study investigators (2004) Therapeutic effect of the combination of etanercept and methotrexate compared with each treatment alone in patients with rheumatoid arthritis: double-blind randomised controlled trial. Lancet 363(9410):675–681
Lovell DJ, Giannini EH, Reiff A, Cawkwell GD, Silverman ED, Nocton JJ, Stein LD, Gedalia A, Ilowite NT, Wallace CA, Whitmore J, Finck BK (2000) Etanercept in children with polyarticular juvenile rheumatoid arthritis. Pediatric Rheumatology Collaborative Study Group. N Engl J Med 342(11):763–769
Mease PJ, Kivitz AJ, Burch FX, Siegel EL, Cohen SB, Ory P, Salonen D, Rubenstein J, Sharp JT, Tsuji W (2004) Etanercept treatment of psoriatic arthritis: safety, efficacy, and effect on disease progression. Arthritis Rheum 50(7):2264–2272
Calin A, Dijkmans BA, Emery P, Hakala M, Kalden J, Leirisalo-Repo M, Mola EM, Salvarani C, Sanmarti R, Sany J, Sibilia J, Sieper J, van der Linden S, Veys E, Appel AM, Fatenejad S (2004) Outcomes of a multicentre randomised clinical trial of etanercept to treat ankylosing spondylitis. Ann Rheum Dis 63(12):1594–1600
Schmeling H, Horneff G (2005) Etanercept and uveitis in patients with juvenile idiopathic arthritis. Rheumatology (Oxford) 44(8):1008–1011
Smith JA, Thompson DJ, Whitcup SM, Suhler E, Clarke G, Smith S, Robinson M, Kim J, Barron KS (2005) A randomized, placebo-controlled, double-masked clinical trial of etanercept for the treatment of uveitis associated with juvenile idiopathic arthritis. Arthritis Rheum 53(1):18–23
Smith BJ, McMillan VM, Newton JS (2001) Corticosteroid-sparing effect of etanercept in idiopathic panuveitis resistant to immunosuppressive therapy. J Clin Rheumatol 7(3):175–178
Galor A, Perez VL, Hammel JP, Lowder CY (2006) Differential effectiveness of etanercept and infliximab in the treatment of ocular inflammation. Ophthalmology 113(12):2317–2323
Foeldvari I, Nielsen S, Kümmerle-Deschner J, Espada G, Horneff G, Bica B, Olivieri AN, Wierk A, Saurenmann RK (2007) Tumor necrosis factor-alpha blocker in treatment of juvenile idiopathic arthritis-associated uveitis refractory to second-line agents: results of a multinational survey. J Rheumatol 34(5):1146–1150
Reddy AR, Backhouse OC (2003) Does etanercept induce uveitis? Br J Ophthalmol 87(7):925
Taban M, Dupps WJ, Mandell B, Perez VL (2006) Etanercept (Enbrel)-associated inflammatory eye disease: case report and review of the literature. Ocul Immunol Inflamm 14(3):145–150
Tauber T, Turetz J, Barash J, Avni I, Morad Y (2006) Optic neuritis associated with etanercept therapy for juvenile arthritis. J AAPOS 10(1):26–29
Saurenmann RK, Levin AV, Feldman BM, Laxer RM, Schneider R, Silverman ED (2006) Risk of new-onset uveitis in patients with juvenile idiopathic arthritis treated with anti-TNFalpha agents. J Pediatr 149(6):833–836
Torun N, Callizo J, Orlic N, Scherer M, Hartmann C, Pleyer U (2005) Serum cytokine receptor levels in noninfectious uveitis. Ophthalmic Res 37(2):112–116
Martin CM, Lacomba MS, Molina CI, Chamond RR, Galera JM, Estevez EC (2000) Levels of soluble ICAM-1 and soluble IL-2R in the serum and aqueous humor of uveitis patients. Curr Eye Res 20(4):287–292
Nussenblatt RB, Fortin E, Schiffman R, Rizzo L, Smith J, Van Veldhuisen P, Sran P, Yaffe A, Goldman CK, Waldmann TA, Whitcup SM (1999) Treatment of noninfectious intermediate and posterior uveitis with the humanized anti-Tac mAb: a phase I/II clinical trial. Proc Natl Acad Sci USA 96(13):7462–7466
Nussenblatt RB, Thompson DJ, Li Z, Chan CC, Peterson JS, Robinson RR, Shames RS, Nagarajan S, Tang MT, Mailman M, Velez G, Roy C, Levy-Clarke GA, Suhler EB, Djalilian A, Sen HN, Al-Khatib S, Ursea R, Srivastava S, Bamji A, Mellow S, Sran P, Waldmann TA, Buggage RR (2003) Humanized anti-interleukin-2 (IL-2) receptor alpha therapy: long-term results in uveitis patients and preliminary safety and activity data for establishing parameters for subcutaneous administration. J Autoimmun 21(3):283–93
Papaliodis GN, Chu D, Foster CS (2003) Treatment of ocular inflammatory disorders with daclizumab. Ophthalmology 110(4):786–789
Kiss S, Ahmed M, Letko E, Foster CS (2005) Long-term follow-up of patients with birdshot retinochoroidopathy treated with corticosteroid-sparing systemic immunomodulatory therapy. Ophthalmology 112(6):1066–1071
Gallagher M, Quinones K, Cervantes-Castañeda RA, Yilmaz T, Foster CS (2007) Biological response modifier therapy for refractory childhood uveitis. Br J Ophthalmol 91(10):1341–1344
Bumgardner GL, Hardie I, Johnson RW, Lin A, Nashan B, Pescovitz MD, Ramos E, Vincenti F, Phase III Daclizumab Study Group (2001) Results of 3-year phase III clinical trials with daclizumab prophylaxis for prevention of acute rejection after renal transplantation. Transplantation 72(5):839–845
Vincenti F, Kirkman R, Light S, Bumgardner G, Pescovitz M, Halloran P, Neylan J, Wilkinson A, Ekberg H, Gaston R, Backman L, Burdick J (1998) Interleukin-2-receptor blockade with daclizumab to prevent acute rejection in renal transplantation. Daclizumab Triple Therapy Study Group. N Engl J Med 338(3):161–165
Thomas PB, Albini T, Giri RK, See RF, Evans M, Rao NA (2005) The effects of atorvastatin in experimental autoimmune uveitis. Br J Ophthalmol 89(3):275–279
Luna JD, Chan CC, Derevjanik NL, Mahlow J, Chiu C, Peng B, Tobe T, Campochiaro PA, Vinores SA (1997) Blood-retinal barrier (BRB) breakdown in experimental autoimmune uveoretinitis: comparison with vascular endothelial growth factor, tumor necrosis factor alpha, and interleukin-1beta-mediated breakdown. J Neurosci Res 49(3):268–280
de Vos AF, Klaren VN, Kijlstra A (1994) Expression of multiple cytokines and IL-1RA in the uvea and retina during endotoxin-induced uveitis in the rat. Invest Ophthalmol Vis Sci 35(11):3873–3883
Lim WK, Fujimoto C, Ursea R, Mahesh SP, Silver P, Chan CC, Gery I, Nussenblatt RB (2005) Suppression of immune-mediated ocular inflammation in mice by interleukin 1 receptor antagonist administration. Arch Ophthalmol 123(7):957–963
Benezra D, Maftzir G, Barak V (1997) Blood serum interleukin-1 receptor antagonist in pars planitis and ocular Behcet disease. Am J Ophthalmol 123(5):593–598
Cohen S, Hurd E, Cush J, Schiff M, Weinblatt ME, Moreland LW, Kremer J, Bear MB, Rich WJ, McCabe D (2002) Treatment of rheumatoid arthritis with anakinra, a recombinant human interleukin-1 receptor antagonist, in combination with methotrexate: results of a twenty-four-week, multicenter, randomized, double-blind, placebo-controlled trial. Arthritis Rheum 46(3):614–624
Reiff A (2005) The use of anakinra in juvenile arthritis. Curr Rheumatol Rep 7(6):434–440
Teoh SC, Sharma S, Hogan A, Lee R, Ramanan AV, Dick AD (2007) Tailoring biological treatment: anakinra treatment of posterior uveitis associated with the CINCA syndrome. Br J Ophthalmol 91(2):263–264
Aróstegui JI, Arnal C, Merino R, Modesto C, Antonia Carballo M, Moreno P, García-Consuegra J, Naranjo A, Ramos E, de Paz P, Rius J, Plaza S, Yagüe J (2007) NOD2 gene-associated pediatric granulomatous arthritis: clinical diversity, novel and recurrent mutations, and evidence of clinical improvement with interleukin-1 blockade in a Spanish cohort. Arthritis Rheum 56(11):3805–3813
Botsios C, Sfriso P, Ostuni PA, Todesco S, Punzi L (2007) Efficacy of the IL-1 receptor antagonist, anakinra, for the treatment of diffuse anterior scleritis in rheumatoid arthritis. Report of two cases. Rheumatology (Oxford) 46(6):1042–1043
Carrasco R, Smith JA, Lovell D (2004) Biologic agents for the treatment of juvenile rheumatoid arthritis: current status. Paediatr Drugs 6(3):137–146
Flynn JM, Byrd JC (2000) Campath-1H monoclonal antibody therapy. Curr Opin Oncol 12(6):574–581
Dick AD, Meyer P, James T, Forrester JV, Hale G, Waldmann H, Isaacs JD (2000) Campath-1H therapy in refractory ocular inflammatory disease. Br J Ophthalmol 84(1):107–109
Isaacs JD, Hale G, Waldmann H, Dick AD, Haynes R, Forrester JV, Watson P, Meyer PA (1995) Monoclonal antibody therapy of chronic intraocular inflammation using Campath-1H. Br J Ophthalmol 79(11):1054–1055
Klastersky J (2006) Adverse effects of the humanized antibodies used as cancer therapeutics. Curr Opin Oncol 18(4):316–3120
Dubertret L, Sterry W, Bos JD, Chimenti S, Shumack S, Larsen CG, Shear NH, Papp KA, CLEAR Multinational Study Group (2006) Clinical experience acquired with the efalizumab (Raptiva) (CLEAR) trial in patients with moderate-to-severe plaque psoriasis: results from a phase III international randomized, placebo-controlled trial. Br J Dermatol 155(1):170–181
Menter A, Gordon K, Carey W, Hamilton T, Glazer S, Caro I, Li N, Gulliver W (2005) Efficacy and safety observed during 24 weeks of efalizumab therapy in patients with moderate to severe plaque psoriasis. Arch Dermatol 141(1):31–38
Takiguchi R, Tofte S, Simpson B, Harper E, Blauvelt A, Hanifin J, Simpson E (2007) Efalizumab for severe atopic dermatitis: a pilot study in adults. J Am Acad Dermatol 56(2):222–227
Xu H, Forrester JV, Liversidge J, Crane IJ (2003) Leukocyte trafficking in experimental autoimmune uveitis: breakdown of blood-retinal barrier and upregulation of cellular adhesion molecules. Invest Ophthalmol Vis Sci 44(1):226–234
Whitcup SM, DeBarge LR, Rosen H, Nussenblatt RB, Chan CC (1993) Monoclonal antibody against CD11b/CD18 inhibits endotoxin-induced uveitis. Invest Ophthalmol Vis Sci 34(3):673–681
Whitcup SM, Hikita N, Shirao M, Miyasaka M, Tamatani T, Mochizuki M, Nussenblatt RB, Chan CC (1995) Monoclonal antibodies against CD54 (ICAM-1) and CD11a (LFA-1) prevent and inhibit endotoxin-induced uveitis. Exp Eye Res 60(6):597–601
Leonardi CL, Toth D, Cather JC, Langley RG, Werther W, Compton P, Kwon P, Wetherill G, Curtin F, Menter A (2006) A review of malignancies observed during efalizumab (Raptiva) clinical trials for plaque psoriasis. Dermatology 213(3):204–214
Jaffe GJ, McCallum RM, Branchaud B, Skalak C, Butuner Z, Ashton P (2005) Long-term follow-up results of a pilot trial of a fluocinolone acetonide implant to treat posterior uveitis. Ophthalmology 112(7):1192–1198
Jaffe GJ, Martin D, Callanan D, Pearson PA, Levy B, Comstock T (2006) Fluocinolone acetonide implant (Retisert) for noninfectious posterior uveitis: thirty-four-week results of a multicenter randomized clinical study. Ophthalmology 113(6):1020–1027
Kuppermann BD, Blumenkranz MS, Haller JA, Williams GA, Weinberg DV, Chou C, Whitcup SM, Dexamethasone DDS Phase II Study Group (2007) Randomized controlled study of an intravitreous dexamethasone drug delivery system in patients with persistent macular edema. Arch Ophthalmol 125(3):309–317
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Yeh, S., Faia, L.J. & Nussenblatt, R.B. Advances in the diagnosis and immunotherapy for ocular inflammatory disease. Semin Immunopathol 30, 145–164 (2008). https://doi.org/10.1007/s00281-008-0109-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00281-008-0109-4