
Abstract
The coagulation, fibrinolytic, anticoagulation, and complement systems are in delicate balance with the vessel wall endothelium ensuring appropriate hemostasis. Coagulopathy in coronavirus disease 2019 (COVID-19) is not a simple disorder of one hemostatic component but a complicated process affecting most of the hemostasis system. COVID-19 disturbs the balance between the procoagulant systems and the regulatory mechanisms. Here, we investigate the effect of COVID-19 on key hemostatic components, including platelets, endothelial cells, coagulation factors, fibrinolytic system, anticoagulant protein system, and complement system, to improve our understanding of the pathophysiological processes underlying COVID-19 coagulopathy based on evidence.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
From the early days of the severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) outbreak to the present, clinical and basic studies have indicated that coronavirus disease 2019 (COVID-19) may be associated with coagulopathy (CAC), which is involved in its related morbidity and mortality [1]. Deep vein thrombosis and pulmonary embolism are common in COVID-19 patients and are remarkably high in the intensive care unit (ICU)–admitted patients [2]. CAC can lead to the formation of circulating microthrombi and macrothrombi which can involve multiple sites, including the lungs, brain, heart, and visceral organs like kidneys and spleen [3, 4]. Hematological findings show that CAC is distinct from consumption coagulopathy including conventional sepsis–induced coagulopathy and disseminated intravascular coagulation (DIC). Coagulopathy of COVID-19 is characterized by high plasma levels of D-dimer, fibrin split products, C-reactive protein, and P-selectin but less frequently with change in activated partial thromboplastin time, prothrombin time, and platelet (Plt) count and with elevation of fibrinogen [5, 6]. However, DIC can occur in COVID-19 which is an ominous and late sign of the disease [6].
There is a close relationship between the immune system and coagulation. The components of the hemostatic system play a role in the body’s immunity, and the activation of the immune system strongly influences the hemostatic system. Abnormal activation of the immune system may promote the growth of pathologies associated with thrombosis [7,8,9]. COVID-19 is accompanied by an immune-cell hyperactivation and excessive production of proinflammatory cytokines, known as “cytokine storm” [9]. CAC is theorized to result from dysregulated interactions between the immune and coagulation systems [9, 10].
COVID-19 can affect both cellular and non-cellular components of the hemostatic system [11, 12]. Here, we review the effects of COVID-19 on the major hemostatic components, including Plts and endothelial cells (ECs), as well as Von Willebrand factor (VWF), ADAMTS13 (a disintegrin-like metalloproteinase with a thrombospondin type 1 motif, member 13), clot-forming (coagulation) protein system, clot lysing (fibrinolytic) protein system, anticoagulant protein system, and complement system to improve the understanding of the pathophysiological processes underlying COVID-19 coagulopathy. Further, we also discuss the relationship of changes in hemostatic components to COVID-19 morbidity and mortality.
Effects of COVID-19 on Plts, VWF, and ADAMTS13
Plt is the main effector of hemostasis and has several roles, such as monitoring the continuity of blood vessels, forming a hemostatic plug through Plt-Plts and Plt-coagulation proteins interactions, and assisting the healing of injured tissue. In vascular injury, Plts adhere to exposed collagen at the injury site, followed by activation and aggregation of Plts to those already adhering, forming a surface that supports activation of coagulation factors [13]. SARS-CoV-2 directly and indirectly can affect Plts, which is important in the pathogenesis of COVID-19 associated coagulopathy.
Zhang et al. [14] reported that SARS-CoV-2 can directly interact with Plts and enhance their activation by binding spike to angiotensin-converting enzyme 2 (ACE2). They found that blood Plts show ACE2, the receptor for SARS-CoV-2, and TMPRSS2, a protease for spike priming, on their surface, allowing the virus to directly stimulate Plts via its spike protein, possibly through the MAPK pathway, and subsequently promote the release of clot forming factors and inflammatory mediators and the development of leukocyte-Plt aggregates [14]. In contrast to this report, Manne et al. did not detect ACE2 as either mRNA or protein in Plts. Astonishingly, however, SARS-CoV-2 N1 gene mRNA was found in Plts of some COVID-19 patients. This suggests that Plts are able to absorb SARS-COV-2 mRNA independently of ACE2 [15]. The presence of SARS-CoV-2 RNA in Plts from patients with COVID-19 has also been demonstrated in other studies [16].
Plt adhesion and aggregation have been shown to be increased in COVID-19, especially in severe disease [17]. Plts exhibit altered gene expression and functional responses in COVID-19 and are hyperactivated [15]. In COVID-19 patients, Plts show increased P-selectin expression compared to healthy donors and increased circulating Plt-leukocyte aggregates, and Plts aggregate more rapidly and show increased spread to fibrinogen and collagen [15, 17]. Calderon-Lopez et al. [18] tested Plt function by closure time in COVID-19 and found that closure time was shorten 20% and 40% in response to collagen and ADP and collagen and epinephrine agonists, respectively. In addition, Plts in COVID-19 are thought to release their alpha and dense granule contents and contribute directly to the plasmatic cytokine load [16]. COVID-19 Plts are able to disseminate pro-inflammatory and pro-coagulant process in the systemic circulation because they release greater amounts of cytokines, chemokines, growth factors, and procoagulant factors upon stimulation than the control group [17].
Evidences show that Plt activation is involved in the severity of COVID-19; in other words, the severity of the disease is directly related to Plt activation [19]. Hottz et al. in their report [19] demonstrate an augmented Plt stimulation and increased formation of Plt-monocyte aggregate in COVID-19 patients with severe illness compared with patients with mild COVID-19. Likewise, Nicolai et al. [20] provided evidence that Plt activation changes with disease severity. While patients intermediately affected with COVID-19 demonstrate a depleted Plt and hyporeactive neutrophil phenotype, cases with severe illness show an excessive activation of Plts and neutrophils, compared to healthy controls and patients with non-COVID-19 pneumonia [20]. Accordingly, Viecca et al. [21] showed that the anti-Plt therapy could be capable to make the ventilation/perfusion ratio better in COVID-19 patients with severe respiratory failure, which would be indirect evidence that Plts are involved in the severity of the disease.
COVID-19 in most cases does not reduce Plt counts below the normal range or reduces slightly, but about 5% of patients met severe thrombocytopenia [22]. As with Plt activation, thrombocytopenia seems to be connected to disease progression and death. Patients with mild COVID-19 infection have been shown to have higher Plt values compared to those with severe disease, and thrombocytopenia is related to a higher risk of severe illness and death, and more than half of fatal COVID-19 had a severe thrombocytopenia [23]. Therefore, the severity of thrombocytopenia and its worsening can be indicative of COVID-19 patients’ clinical status and its changing to an inferior state. Worsening thrombocytopenia in COVID-19 patients could be due to development of DIC as COVID-19 non-survivors met significantly higher the criteria of DIC compared to survivors [24]. In addition to DIC as a pre-terminal event, alternative causes may also contribute to the development of severe thrombocytopenia in COVID-19. Development of secondary infections [25], drug-induced thrombocytopenia such as heparin [26], and anti-phospholipid antibodies [27] are among the factors that have been reported so far. In general, it seems that Plt counts can differentiate between severe and non-severe COVID-19 infections, so that a progressive and considerable reduction in Plt counts may be a signal for disease deterioration and severity, of course, if there is no other reasonable explanation for worsening thrombocytopenia.
Plts derive from megakaryocytes (MKs), which developed from the hematopoietic stem cell (HSCs), and then from a hematopoietic committed progenitor cells (HPCs). MK proliferation and Plt production is mainly controlled by thrombopoietin (TPO) [28]. COVID-19 may affect MK and Plt production through different mechanisms. It has been proposed that SARS-CoV2 may directly attach to the HSCs/HPCs and MKs and internalize into them, and induces thrombocytopenia by induction of apoptosis in these cells [29]. In addition, the excessive secretion of inflammatory cytokines in COVID-19 could alter the BM microenvironment that leads to inhibition of HSC differentiation to MK progenitors and also suppress the MK maturation process [30]. Another proposed mechanism is the effect on TPO production or expression of its receptor. SARS-CoV-2 may reduce TPO production or attenuate TPO effects on MKs by inducing impairment of the liver, which is the major site of TPO production, or by inhibiting expression of the c-MPL gene, which encodes the TPO receptor on MKs [15, 29]. Furthermore, based on the theory that mature MKs can settle in lung capillary beds and release circulating PLTs, it was hypothesized that SARS-CoV-2-induced lung damage could affect the settlement of MKs in the pulmonary capillary beds and subsequently impair PLT production [22]. In contrast to the above statements about the possible effects of COVID-19 on megakaryocytes, Roncati et al. [31] reported in postmortem and biopsy report that naked megakaryocyte nuclei in BM and lungs of severe COVID-19 were increased more than 10-fold, which was attributed to high serum interleukin-6 levels that stimulate megakaryopoiesis and Plt production. In addition, their electron microscopic studies excluded the presence of virions in megakaryocytes and did not support this hypothesis that SARS-CoV-2 infect megakaryocytes. Other studies also did not find evidence of direct infection of megakaryocytes by SARS-CoV-2; nonetheless, the presence of a large number of these cells in the lungs is probably linked to the large quantity of small thrombi, and foci of hemorrhage [3].
VWF is a multimeric adhesion protein that helps Plts adhere to the subendothelium of the harmed vessel walls. ECs and MKs produce VWF and store it in Weibel Palade bodies and alpha granules, respectively. VWF in plasma is derived from the secretion of ECs, and released vWF upon Plt activation is generally not capable of appreciably increasing the total plasma levels of VWF. VWF is an acute-phase response protein that plays a role in vascular inflammation. Activated ECs in response to inflammatory stimuli release VWF [32]. COVID-19 is associated with the activation and damage of ECs. An increase in VWF has been observed in COVID-19 patients. Goshua et al. [33] evaluated activation indicators of EC and Plt in severe and non-severe patients with COVID-19 and observed that VWF antigen was meaningfully high in ICU patients compared with non-ICU patients and that VWF level correlated with mortality rate. In addition, VWF antigen was increased above the normal range in 80% of non-ICU patients [33]. A marked increase in plasma levels of VWF antigen and activity and its correlation with mortality in COVID-19 patients was also observed in other studies [34]. Wibowo et al. [35] in a meta-analysis compared VWF antigen levels between poor outcome and good outcome COVID-19 patients, and found that the VWF antigen levels were higher in poor outcome COVID-19 patients. Since the increased VWF levels are considered a risk factor for coagulopathies in different diseases [36], the increased VWF levels in COVID-19 patients, due to activation of ECs or endotheliopathy and activation of Plts, could also be involved in the increased thrombotic risk of these patients.
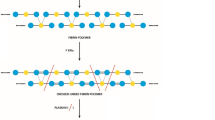
Prothrombotic ultra-large VWF multimers (UL-VWFM), in an important physiological process of coagulation in the circulation, are cleaved by ADAMTS13 into smaller and less procoagulant forms. Deficiency of ADAMTS13 results in the circulation of UL-VWFM in the plasma. UL-VWFM can spontaneously aggregate Plts along with founding large strands attached to the endothelial surface leading to uncontrolled thrombosis [32]. There is a reciprocal relationship between VWF and ADAMTS-13 levels; the VWF-cleaving protease tends to be low in plasma when VWF is high. An abnormal VWF-ADAMTS-13 ratio could be associated with an increased risk of thrombosis, stroke, and myocardial infarction [37, 38]. In COVID-19, the increased level of VWF also seems to be associated with concomitant reduction in the level and activity of ADAMTS13. Different studies assessed both ADAMTS-13 and VWF levels in COVID-19 and observed decreased ADAMTS-13 and elevated VWF levels which worsen in parallel with disease severity [39, 40]. Furthermore, there is a significant association between higher risk of severe COVID-19 or mortality with high VWF antigen concentrations or decreased ADAMTS13 activity [39, 40]. Alternatively, Escher et al. [41] observed normal ADAMTS13 activity together with a persistently and excessively elevated level of VWF antigen and activity in 3 ICU-admitted patients. Nevertheless, it should be known that “normal” levels of ADAMTS13, every time reported, may still recognize a relative decrease in individual cases. However, in general, ADAMTS13 appears to be reduced in COVID-19 which along with an increase in VWF levels, i.e., the VWF/ADAMTS13 imbalance, can be effective in COVID-19 coagulopathy through enhancement of Plt-Plt and Plt-endothelial interactions (Fig. 1).

Overview of hypercoagulability in COVID-19. COVID-19 can upregulate prothrombogenic state and downregulate anti-thrombogenic state which predispose patients to fibrin clot formation and platelet aggregation. The complement system and coagulation are functionally dependent, and complement activation is among the main drivers of COVID-19-associated coagulopathy. Neutrophils contribute to this process through the release of NETs, which activate platelets and the extrinsic and intrinsic coagulation pathways and inactivate natural anticoagulants. Monocytes activate the extrinsic pathway of coagulation by expressing and releasing of activated TFs. AT, antithrombin; EPCR, endothelial protein C receptor; PC, protein C; PS, protein S; TF, tissue factor; TFPI, tissue factor pathway inhibitor; TM, thrombomodulin; t-PA, tissue plasminogen activator, is profibrinolytic and serve to activate plasminogen; PAI-1, plasminogen activator inhibitor 1, inhibits t-PA and is antifibrinolytic; TAFI, thrombin-activatable fibrinolysis inhibitor; vWF, von Willebrand factor, helping platelets to attach to each other and to the surface of the vessel; NO, nitric oxide; CD39, a membrane-associated ectoADPase; NETs, neutrophil extracellular traps; MDMs, monocyte-derived microvesicles
In addition to consumption of ADAMTS13 owing to increased release of large VWF multimers from ECs, which could be the most common reason, other factors can also be involved in reducing the amount and activity of ADAMTS13 in COVID-19. Significantly increased plasma levels of the ADAMTS-13 inhibitors, interleukin-6, thrombospondin-1, and platelet factor 4 have also been suggested to be involved in the reduction of ADAMTS13 activity [42]. Another potential involved factor in ADAMTS-13 deficiency could be related to the formation of antiphospholipid antibodies during COVID-19 [43]. It is speculated that these antibodies can interfere with the recognition and proteolysis of VWF by ADAMTS-13 similar to the autoantibodies against ADAMTS-13 present in thrombotic thrombocytopenic purpura causing clinical thrombosis [44]. Another factor which can be interesting to mention is about tobacco smoking which has been shown to be associated with decreased plasma levels of ADAMTS13 [45] and is also almost certainly associated with adverse course and outcome of COVID-19 [46].
Effects of COVID-19 on coagulation protein systems
Physiologic clotting is started when adequate tissue factor (TF) and factor (F) VIIa complex (TF/FVIIa) becomes available to activate coagulation cascade. Control of TF expression is an important regulator of the onset of hemostasis. TF is found in most tissues and cells, and its expression can be upregulated in inflammation [47]. In the COVID-19, it seems that the severe inflammatory environment within the lungs triggers an aberrant expression of TF on ECs, monocytes/macrophages, neutrophils, and Plts, which causes hypercoagulable state [48]. In addition, entry of SARS-CoV-2 into the ECs by ACE2 receptor reduces the expression and enzymatic activity of ACE2, which lead to increased vascular permeability and TF expression in subendothelial cells [49]. Further, reciprocal action with Plts is also important in TF expression by monocyte and neutrophil [50, 51]. Hottz et al. showed that during COVID-19, Plt-monocyte interaction stimulates both Plts and monocytes via TF-mediated signaling and makes hyperinflammation and hypercoagulability stronger in a mutual augmentation loop [50]. Besides that, development of local hypoxia in COVID-19 patients may progressively exacerbate the aberrant TF expression through stimulation of alveolar epithelial cells and monocytes-macrophages [52]. In agreement, Rosell et al. [53] in measurement of TF activity of plasma extracellular vesicles (EVs) in patients with COVID-19 observed increased levels of EVs TF activity which were also associated with D-dimer levels, disease severity, and mortality. On the contrary, examination of mRNA profiles of bronchoalveolar lavage fluid (BALF) in COVID-19 patients by Mast et al. showed that while mRNA for several clotting factors were amplified, TF transcripts were not increased in BALF [54]. Nevertheless, FitzGerald et al. re-analyzed the data sets of later examination and explained that the used data sets were not comparable, and that the COVID-19 sample set was not appropriate for the analysis of transcriptome [55].
Following inflammation and thereby cytokines secretion, activated monocytes and macrophages express TF [47]. These cells can also be activated by direct interaction with COVID-19 virus, so that after detection of a pathogen by pattern recognition receptors, TF can be presented by monocyte and monocyte-derived microvesicles [9] (Fig. 1). According to the high activation of these cells in COVID-19, the markedly release of TF can be expected. In this regard, hyperexpressed-TF of monocytes has been found in the severe form of COVID-19 [56]. In addition to monocyte activation, extensive neutrophil activation has been detected in prothrombotic state of COVID-19 patients. These cells can be activated directly by viruses or even by other cells such as ECs, monocytes/macrophages, and Plts [56]. Coordinately, Plt overactivation and Plt-neutrophil and Plt-monocyte aggregation along with P-selectin expression has been found in severe form of COVID-19 [15]. All above, high levels of TF correlate with the presence of circulating Plt-monocyte and Plt-neutrophil aggregation, constituting the occlusive thrombus [57].
TF/FVIIa activates the zymogen FIX to enzymatically active FIXa. FIXa converts FX to FXa in the presence of FVIIIa. Subsequently, FXa with FVa activates prothrombin (FII) to thrombin (FIIa). FIIa then leads to the coagulation of fibrinogen, and additionally activates FXI to FXIa, rendering other pathway for the activation of FIX. FVIIIa and FVa are cofactors and accelerate the activation of FX and FII, respectively [47, 58, 59]. In COVID-19 patients, a substantial increase in FVIII and FV antigen levels and activity has been observed in several studies [18, 34, 60,61,62,63]. FVIII is an acute-phase protein that responds in transcription to interleukin-6 and increases under inflammatory conditions [64]. With respect to FV, Wang et al. [61] identified leukocytes, neutrophil granulocytes, monocytes, and regulatory T cells, as causes of high FV in hospitalized COVID-19 patients and showed FV in the leukocytes infiltrated in the lungs of these patients. In addition, increased FV activity in COVID-19 was found to have the strongest association with COVID-19 of parameters such as FVIII, fibrinogen, and D-dimer [60]. However, as the disease progresses and after the initial elevation, FV levels decrease [65], and the activity of FV and also the activity of FVII are significantly lower in end-stage patients [63]. In FXI following a similar pattern, higher FII and FXI levels were observed in patients with asymptomatic/mild and moderate COVID-19 compared with healthy individuals, whereas lower FII and FXI levels were observed in patients with thromboembolism and in patients who developed severe disease [66, 67].
Fibrinogen (FI), synthesized by the liver, is the substrate of coagulation pathway and the important protein involved in aggregation of Plts. COVID-19 is uniformly associated with high fibrinogen levels (hyperfibrinogenemia), which are correlated with disease severity [5, 18, 34, 49, 63, 68,69,70]. Inflammatory stimuli in the acute phase affect fibrinogen transcription, and fibrinogen increases with inflammation [71]. In COVID-19 patients, increased levels of inflammatory cytokines such as IL-6 are involved in increasing the expression of this protein [72]. In COVID-19 patients, Plts, in addition to the liver, are predisposed to engage in procoagulant activities in the circulation and appear to be involved in the elevation of fibrinogen [17]. In rare cases, decreased fibrinogen levels have been observed in patients with severe COVID-19 due to the occurrence of DIC [69], which is associated with a poor prognosis [73].
FXII (Hageman) is a proenzyme of the contact system. This system includes FXII, prekallikrein (Fletcher factor), and FXI. FXII is not important for in vivo hemostasis and its deficiency is not associated with bleeding. However, it seems that FXII is essential for thrombus formation, so that mice deficient in FXII are protected from arterial thrombosis [74]. In the setting of inflammation, FXII autoactivates when associated with polysomes released by stimulated Plts, extracellular RNA, protein aggregations, or exposed collagen in the blood circulation and participates in in vivo FIIa production and potentiate the formation of Plt-fibrin thrombi [74, 75]. In COVID-19, activation of FXII and, thus, the contact system of coagulation through the polyphosphate release from the activated Plts and release of neutrophil extracellular traps (NETs) from recruited neutrophils can be involved in thrombotic process [76, 77] (Fig. 1). Englert et al. found increased FXII expression and activity in lung tissue and plasma of patients with COVID-19. Activated FXIIs in the lung tissues were colocalized with NET accumulation suggesting that NETs make a platform available for activation of FXII in COVID-19 [78]. Taus et al. [17] showed that contribution of circulating Plts in hypercoagulability of COVID-19 is FXII dependent and COVID-19 patients have accelerated FXII-dependent coagulation. Plts, in addition to increasing the fibrinogen and VWF, also contribute to increasing FXII in COVID-19 patients; an increased FXII activity was found when washed Plts from patients were suspended in control plasma. In addition, Ceballos et al. [66] evaluated the association of antithrombin (AT), prothrombin, FXI, FXII, and FXIII with COVID-19 severity and mortality and observed higher levels of FXII in asymptomatic/mild and moderate patients compared to healthy individuals. This observation was also associated with higher levels of AT, prothrombin, FXI, and FXIII. However, decreased levels of FXII, and also AT and FXI, and FXIII, were found in those patients who in the end developed severe illness. They also observed that patients with lower levels of these coagulation proteins had a higher risk of mortality. On the other hand, regarding to FXII and COVID-19, Bowles et al. [43] in exploring the causes of APTT prolongation in COVID-19 patients found that the most patients with a prolonged APTT had lupus anticoagulants (91%) which were often in association with FXII deficiency. In their investigation, coagulation screening of 216 patients admitted to the hospital showed that 44 (20%) have a prolonged APTT that the FXII level was 50 U/dl or lower in 16 patients. It is worthy to note that, the prolonged APTT in the patients was present even with considerable elevations in FVIII, which shortens the length of APTT. In agreement, Calderon-Lopez et al. also observed the reduction of FXII in a proportion of COVID-19 patients (25.3%) (median: 90.3 U/dl) [18].
FXIII is a transglutaminase that cross-links fibrin strands and forms an insoluble fibrin clot. In addition, FXIIIa through cross-linking α2-antiplasmin to the fibrin increases resistance to fibrinolysis [79]. Ceballos et al. [66] found increased FXIII in asymptomatic/mild and moderate COVID-19 patients and, interestingly, reduced FXIII in those patients who finally developed severe illness. Von Meijenfeldt et al. [80], in an evaluation of FXIII activity in 97 COVID-19 patients, detected a substantial decrease in plasma FXIII activity. The majority (57) of their patients was with “mild” disease and the decline in FXIII levels was more noticeable in high care facility admitted patients. Due to increased levels of D-dimer in COVID-19 patients and as D-dimers are the product of fibrinolytic action on cross-linked fibrin generated by FXIIIa, they considered a consumptive mechanism possible for the acquired FXIII deficiency in COVID-19 patients.
In accord, Gupta et al., to define association of COVID-19-induced thromboembolism with FXIII, performed the correlation analyses between FXIII and D-dimer, a biomarker of thromboembolism, in moderate and severe COVID-19 patients, and observed a negative correlation between plasma FXIII and D-dimer levels [81]. Al-Tamimi et al. [68] assessed the levels of FXIII at moderate and severe stages of COVID-19 and observed a marked decreased in plasma FXIII levels particularly in the severe COVID-19. They also found that the decrease in FXIII was in reverse correlation with fibrinogen levels when COVID-19 becomes more severe. In general, it can be concluded that COVID-19 can cause an early increase in some coagulation factors, including FVII, FV, FII, FXI, FXII, and FXIII, and then a decrease in these factors after onset of coagulation and exacerbation of the disease.
Effects of COVID-19 on fibrinolytic protein systems
Plasminogen and its activators are the main components of the clot lysing system. The plasminogen activators, including tissue plasminogen activator (TPA), single-chain urokinase plasminogen activator (ScuPA), and two-chain urokinase plasminogen activator (TcuPA), convert the zymogen plasminogen to its active form, plasmin. Wright et al. [82] observed a complete lack of fibrinolysis in 57% of severe COVID-19 patients through high D-dimer and unsuccessful clot lysis at 30 min by means of thromboelastography. In agreement with this, Heinz et al. [83] found more resistance to fibrinolysis in critically ill COVID-19 patients than in healthy controls. They compared thromboelastometry results and found substantial differences in peak clot strength and lysis time. Hypofibrinolysis in COVID-19 patients has also been observed in other studies, which has also been associated with the severity and poor outcomes [34, 84]. The contribution of hypofibrinolysis or suppression of fibrinolysis to COVID-19-associated coagulopathy has also been hypothesized [85], but the detailed mechanisms is not yet clear.
Henry et al. [86] investigated plasminogen levels in patients with COVID-19 and found no difference in the values between COVID-19 patients and COVID-19-negative sick controls, and the plasminogen concentrations for all patients were within the normal range. In this context, Hammer et al. [87] also observed no difference in plasminogen concentration between patients with COVID-19 (non-ICU and ICU) and healthy controls. However, in the study by Henry et al., hospitalized COVID-19 patients and patients with COVID-19 requiring ICU admission had slightly lower plasma plasminogen concentrations than COVID-19 patients discharged from the emergency department. Although the decrease was small, the trend of decrease was significantly associated with the disease severity and suggested plasminogen consumption for fibrinolysis activation (as suggested by elevated D-dimer in hospitalized COVID-19 patients). Consistent with this, to describe the relationship between plasminogen and thromboembolism in COVID-19, Gupta et al. [81] performed correlation analyses between plasminogen and D-dimer in moderate and severe COVID-19 patients and found a reverse correlation of plasminogen and D-dimer in these patients. In addition, Juneja et al. [84] observed in a prospective longitudinal single-center study that lower plasminogen levels were among the biomarkers that characterized the high-mortality cluster. It can be concluded that disease progression and the associated increase in coagulability and clotting lead to an increase in fibrinolysis and consumption and a decrease in plasminogen, and that the decrease in plasminogen and hypofibrinolysis contributes to the worsening of the disease and the increase in mortality in a positive feedback loop.
With respect to plasminogen activators and plasminogen activator inhibitors, Nougier et al. [88] examined FIIa production and fibrinolytic activity in patients with COVID-19 who have significant hypercoagulability and found an altered equilibrium between clot formation and lysis which was associated with elevated levels of plasminogen activator inhibitor-1 (PAI-1), increased activation of thrombin-activatable fibrinolysis inhibitor (TAFI), and increased levels of TPA. Given the continuous deposit of fibrin in the lung of COVID-19 patients, they hypothesized that high levels of PAI-1, a major inhibitor of plasminogen activators, may offset elevated levels of TPA [88]. The hypothesis of an impaired fibrinolytic system in COVID-19 patients was also investigated by Hamme et al. [87]. They observed significantly higher activity of PAI-1 in ICU COVID-19 patients, which also showed a positive correlation with resistance to clot dissolution. They also found significantly higher levels of TPA in non-ICU and ICU COVID-19 patients compared with healthy donors. Increased PAI-1 and TPA concentrations were also found in several other studies [68, 70, 81, 84, 89]. White et al. [70] observed that TPA was significantly increased in critical versus non-critical COVID-19 patients. Consistent with this, Francischetti et al. found in a cohort that association of D-dimers with high mortality in COVID-19 were correlated with TPA [89]. Gupta et al. [81] found a strong positive correlation between PAI-1 and D-dimers in addition to TPA, and Juneja et al. [84] identified antigen levels of PAI-1 as biomarkers associated with death. Given the hypofibrinolytic state in SARS-CoV-2 infection despite elevated TPA levels, it appears that the PAI-1 and TAFI elevation is stronger than the TPA increase.
About the urokinase plasminogen activator, Mast et al. [54] by examination of transcriptional profiles of BALF samples showed that mRNA levels for urokinase and urokinase receptor were meaningfully decreased in COVID-19 patients. About α2-antiplasmin, the major inhibitor of plasmin, Goshua et al. [33] observed unaltered activity of α2-antiplasmin in both ICU and non-ICU COVID-19 patients compared to standard reference range. In agreement, measurement of α2-antiplasmin levels by Nougier et al. [88] showed no significant difference between ICU and non-ICU COVID-19 patients. In contrast, Hammer et al. [87] found that α2-antiplasmin levels were significantly lower in ICU COVID-19 patients compared with healthy donors.
Based on the information on the effects of COVID-19 on the fibrinolytic system, it appears that in the early stage of the disease, the balance between activators and inhibitors of fibrinolysis is sufficient for adequate activation of clot lysing. But, progression of COVID-19 and consumption of plasminogen and elevation of PAI-1 eventually lead to the hypofibrinolysis state. However, given the elevated D-dimer in COVID-19 patients, even in the hypofibrinolytic condition, the general processes of coagulation and fibrinolysis is increased in these patients [49].
Effects of COVID-19 on the anticoagulation systems
The protein C/protein S system, AT, and tissue factor pathway inhibitor (TFPI) are three major inhibitors of coagulation. Activated protein C (APC) is an enzyme that acts as an inhibitor of FVa and FVIIIa to reduce thrombin formation. Protein S is a cofactor for APC on the cell surfaces. Regarding the protein C/protein S system in COVID-19 patients, Calderon-Lopez et al. [18] found in a study of 80 consecutive COVID-19 patients with different degrees of disease that 7% of the patients had a reduced protein C level and 22% of the women and 62% of the men had a reduced protein S level. Tabatabai et al. [90] studied a case series of ten patients with severe COVID-19 and observed that they have low normal activity of protein C associated with high activity of FVIII. Zhang et al. [63] found mean protein C and protein S activities below the normal range in a study of 19 patients with COVID-19 requiring ICU admission. However, conflicting data were also found in a study of 24 patients admitted to the ICU, with protein C elevated in 11 patients [34]. Regarding associations with disease severity, Corrêa et al. [91] performed a longitudinal analysis of the coagulation profile of 30 patients admitted to the ICU and found that patients with a Sequential Organ Failure Assessment (SOFA) score > 10 had lower protein C levels and activity than patients with a SOFA score ≤ 10. But plasma free protein S levels were low in both the SOFA ≤ 10 and SOFA > 10 groups increasing with time at baseline. Sehgal et al. [92] identified a significant association of protein C decrease with a high SOFA score among the natural anticoagulants, protein C, protein S, and AT. Furthermore, Gerotziafas et al. [93] found in the prospective observational cohort study COMPASS-COVID-19 that protein C activity in ICU patients was significantly lower than that in patients with mild or moderate disease. Finally, Stanne et al. [94], while characterizing uptake plasma levels of 12 hemostatic proteins in hospitalized COVID-19 patients, observed that low uptake levels of protein C are a risk factor for worsening disease and mortality in moderate or severe patients.
Thrombomodulin (TM), a protein of ECs, is a site of protein C zymogen activation by thrombin to APC. TM-bound thrombin, together with protein C receptor on the endothelial cells (EPCR), activates protein C. Won et al. [95] observed decreased TM and EPCR on pulmonary ECs from deceased COVID-19 patients. Consistent with this, several studies described increased plasma levels of soluble TM (sTM) in COVID-19 patients [33, 84, 96] which were associated with a lower rate of hospital discharge [33] and a higher probability of mortality [33, 84, 89, 96]. The elevated sTM concentrations in hyperinflammatory states are thought to be the result of endothelial disruption or injury [97]. Vassiliou et al. [98] measured soluble form of EPCR (sEPCR) in hospitalized and non-hospitalized COVID-19 patients and found that sEPCR levels in hospitalized patients are considerably higher than outpatients. When sEPCR binds to APC, it not only reduces APC anticoagulant function, but also makes a functional alteration in APC rendering it pro-coagulant [99]. Therefore, elevated levels of sEPCR could contribute to the procoagulant phenotype reported in hospitalized patients by altering APC activities [98]. In agreement with this, Bayrakci et al. [100] have shown in a study that sEPCR levels are higher in RT-qPCR-positive COVID-19 patients than in clinically diagnosed COVID-19 patients and in patients with ground-glass opacity or bilateral involvement on the thorax CT regardless of the PCR result. Therefore, protein C levels and protein C function may generally decrease in COVID-19 patients, and this decrease may contribute to a high risk of thrombosis in these patients and may be related to disease severity and mortality. Moreover, in SARS-CoV2 infection, the decreased amount of TM and EPCR on the surface of pulmonary ECs and their increased plasma levels may also be involved in reducing the inhibition of coagulation by the protein C/protein S system at the site of endothelial injury.
AT primarily inhibits thrombin and FXa to prevent thrombosis. Several studies investigated the activity of AT in COVID-19 patients [18, 34, 63]. Calderon-Lopez et al. [18] analyzed 80 patients with COVID-19 and observed decreased AT activity in 21%. Ranucci et al. [72] observed in a prospective study that the mean activity of AT in 16 patients with COVID-19 ARDS on admission to the ICU was 85% [65–91%], with 25% of patients having an AT activity of <80%. Zhang et al. [63] in a study of 19 patients with COVID-19, who were admitted to ICU, found that the median activity of AT was below the normal range. In the study by Panigada et al. [34], 24 patients admitted to the ICU for COVID-19 were found to have slightly decreased AT activity in approximately half of the patients. Han et al. prospectively gathered coagulation tests results of 94 SARS-CoV-2-infected patients and found that the patients had significantly lower AT values compared to healthy controls (85% vs. 99%) [101]. Gross et al. [102] in Germany found even lower AT concentrations in COVID-19 patients before their ICU admission, ranging from 26 to 62% (reference > 70%). AT concentrations in patients with COVID-19 treated for mild symptoms in the intermediate care unit were also low but within the normal range. However, conflicting results have also been reported. Cani et al. [96], in a study to distinguish between COVID-19 patients and septic patients with pneumonia who did not have coronavirus disease, found that AT had significant powers along with fibrinogen, sEPCR, and cell-free DNA, such that AT levels were persistently decreased in patients who did not have COVID, but AT levels were within the normal range in patients with COVID-19. However, the results of most studies support a decrease AT in COVID-19 patients. In addition, Buijsers et al. measured heparanase activity and heparan sulfate levels in the plasma of 48 COVID-19 patients in a cross-sectional study and found that these parameters were increased in COVID-19 patients. Heparanase is an enzyme that degrades endothelial glycocalyx heparan sulfate. The increased activity of heparanase in COVID-19 patients probably impairs endothelial glycocalyx heparan sulfate. Because heparan sulfate enhances the anticoagulant effect of AT, the loss of heparan sulfate in COVID-19 may weaken the anticoagulant effect of AT [103].
The association between AT reduction and clinical outcomes has already been noted in studies of sepsis [104]. Studies on COVID-19 generally suggest that low AT levels are associated with poor treatment outcomes. For example, Gazzaruso et al. [105] recruited 49 consecutive hospitalized COVID-19 patients and found that amounts of AT were lower in nonsurvivors than in survivors and that low AT amounts (below 80%) were a mortality predictor. They also documented a negative relationship between AT levels and BMI and hypothesized that AT might be the link between obesity and a worse prognosis in COVID-19 patients. In addition, retrospectively analyzing the coagulation test findings and outcomes of 183 successive COVID-19 patients, Tang et al. [24] observed that there was a statistically significant difference between nonsurvivors and survivors in AT after day 7 of admission. In addition, Anaklı et al. retrospectively analyzed the prognostic values of AT activity scores of 104 consecutive critically ill COVID-19 patients and reported that AT activity scores at admission were significantly lower in nonsurvivors compared with survivors [106]. In addition, Boknaes et al. evaluated hemostatic function in 217 consecutive patients (96 SARS-CoV-2-positive patients and 121 SARS-CoV-2-negative patients) admitted to Linkoeping College Hospital in Sweden and observed increased venous thromboembolism and mortality in COVID-19 patients with the following abnormalities in hemostatic markers: high D-dimers, low AT, or low plasmin-antiplasmin complexation (PAP) [107]. Indeed, Lippi et al. [108] and Rostami et al. [109] found in meta-analyses that low plasma AT levels were significantly associated with severe disease in COVID-19 patients. Altogether, these observations propose that AT is increasingly depleted during the progression of COVID-19, and low AT levels may be associated with exacerbation of COVID-19-induced coagulopathy and poor prognosis.
TFPI is the key regulator of TF-induced coagulation. TFPI achieves its inhibitory effect via a quaternary complexation with FVIIa, TF, and FXa [47, 58]. It typically is found on the endothelial surface. In baboon, it has been shown that sepsis-induced coagulation in the lung is associated with decreased TFPI mRNA and antigen in the lung endothelium [110]. It has also been reported that sepsis can lead to marked release of TFPI from ECs and that plasma levels of TFPI are markedly increased in patients with sepsis [111]. Several studies have examined TFPI levels in COVID-19 patients and have reached conflicting results. For example, Mast et al. compared data from RNA sequencing (RNA-seq) of BALF samples from COVID-19 patients with data from BALF RNA-seq samples from obese and asthmatic adults and found an 8-fold upregulation of TFPI transcripts, whereas TF expression remained unchanged [54]. These results differ from those obtained in the baboon because of sepsis-induced coagulation in the lung. As mentioned previously, in a commentary on the work of Mast et al., FitzGerald et al. [55] reanalyzed their data sets and showed that the two data sets used were not comparable and that the COVID-19 sample set was not suitable for transcriptome analysis. However, in agreement with Mast et al., when TFPI plasma levels were determined in moderate (n=40) and severe (n=26) COVID-19 patients, Francischetti et al. found that TFPI levels were significantly elevated in patients compared with 9 individuals who were negative for COVID-19 and had no significant pathology [89]. Nevertheless, they found no difference in TFPI levels between moderate and severe COVID-19 patients. In contradiction to the later results, Al-Tamimi et al. [68] noted a tendency for plasma TFPI to decrease in patients with moderate to severe stages of COVID-19, which was particularly low in the severe COVID-19 patients compared with healthy controls. In contrast, when Plt-depleted plasma from 34 patients with noncritical COVID-19 and 75 patients with critical COVID-19 was compared, White et al. [70] observed significantly increased plasma levels of TFPI in the critical patients. Consistent with this, Cacciola et al. observed increased TFPI levels in moderately severe COVID-19 compared with mild/asymptomatic COVID-19 and controls. Because TFPI can interfere with blood coagulation, Cacciola et al. also compared diluted prothrombin time (dPT) in moderate-severe COVID-19 patients with mild/asymptomatic COVID-19 and controls and found high dPT levels in moderate-severe COVID-19 patients and a correlation between TFPI and dPT [112]. Consistent with this, Kelliher et al. [113] observed that the delay time to thrombin generation was prolonged in patients with moderately severe COVID-19 compared with non-COVID-19-hospitalized patients. In the presence of an inhibitory antibody directed against TFPI, the difference in delay time between the groups was abolished; in other words, the delay time to thrombin formation in COVID-19 patients was more sensitive to neutralization of TFPI activity. The contradictory point in their observations was that TFPI levels were similar in both COVID-19 and non-COVID-19-hospitalized patients.
To describe the relationship between TFPI and thromboembolism in COVID-19, Gupta et al. [81] performed correlation analyzes between TFPI and D-dimer, a biomarker of thromboembolism, in moderate (non-ICU) and severe COVID-19 (ICU) patients and found an inverse correlation between plasma TFPI and D-dimer levels. Contrary to this finding, Francischetti et al. showed that TFPI correlated with high D-dimer levels, which are associated with high mortality in SARS-CoV-2 infections [89]. Those who observed the lower TFPI level, particularly in severe COVID-19 patients, stated that TFPI in these patients is probably insufficient to inhibit the procoagulant response induced by SARS-CoV-2 and suggested a contribution of TF pathway activation to COVID-19 coagulopathy [68]. Those who observed the increased plasma TFPI in COVID-19 patients attributed it to the loss of TFPI from the endothelium due to endothelial damage and suggested a role of the uncontrolled TF pathway in the hypercoagulable state at the site of endothelial damage [89, 112]. It is important to point out that endothelial glycocalyx damage is a characteristic feature of critically ill patients and that TFPI is primarily associated with ECs through binding to the glycocalyx at the EC surface [114]. In addition, a contribution of heparin to the release of TFPI from the endothelium has also been suggested [70]. Because of the disparity in the results of these studies, the status of TFPI in plasma and on the surface of ECs at the site of injury and the relationship between TFPI and disease severity require further investigation.
Effects of COVID-19 on the vessel wall endothelium
Under normal conditions, ECs prevent Plt aggregation and coagulation and trigger fibrinolysis, whereas in pathophysiologic states after activation or injury, they activate the coagulation system and promote thrombus formation [115]. ECs contribute to their constitutive anticoagulant action through the following mechanisms: binding of glycosaminoglycans of ECs to AT; presence of TM on EC membranes, which is a site for protein C activation by low levels of thrombin; degradation of adenosine diphosphate by intact ECs by secretion of an ectonucleotidase, CD39; secreting of prostacyclin and nitric oxide, which leads to preventing Plt activity and thus preventing arterial thrombosis; and involved in fibrinolysis and maintaining an anticoagulant state by binding to plasminogen, TPA, and ScuPA [116]. Some of these pathways may interfere with SARS-CoV-2 infection, which are discussed below.
There is evidence that SARS-CoV infection can activate and/or destroy ECs, leading to increase in PAI-1 [117] and VWF levels and increased Plt activation, which causes hypercoagulable state that can result in venous, arterial, and microvascular thrombosis. In this setting, the release of TF, VWF, and adhesion molecules such as P-selectin, E-selectin, and intercellular adhesion molecule from impaired ECs can collectively trigger thrombosis. The exact mechanism of endotheliopathy is not yet known, but several factors may be involved, including the direct action of the virus (may cause damage to ECs leading to stimulation of the coagulation cascade) [118] the infiltration of immune cells (Plts, neutrophils and/or macrophages) [14], hypoxemia, complement-mediated injury, or the sudden and remarkable release of pro-inflammatory cytokines (such as IL-1β and IL-6) as a result of direct cellular damage [9]. As soon as cells expressing ACE-2 are infected by SARS-CoV-2, active replication and then viral release occurs. This causes pyroptosis, releases damage-associated molecular patterns and oxidant stress activation of host cells, and triggers the production of chemokines and pro-inflammatory cytokines by adjacent epithelial cells, ECs, and alveolar macrophages. By attracting inflammatory cells to the infection site, these proteins trigger a proinflammatory feedback loop. The exposure of hidden TFs of subendothelium during the inflammation results in the upregulation of Plts, leukocytes, and ECs, thereby leading to thrombin generation following the activation of both the extrinsic and intrinsic coagulation pathways [34, 119]. There is an increasing recognition that endothelial activation is the primary cause of thrombosis. The presence of viral inclusion bodies in ECs of various organs including the lung emphasizes this point [118]. It is shown that the intense inflammation of ECs of the lungs due to activation by high levels of pro-inflammatory cytokines (IL-1, IL-6, and TNF) and ferritin may result in “in situ” microvascular thrombosis in patients with COVID-19 [9, 119]. Interestingly, reversely direct infection of vascular ECs by the virus, and thus cellular damage and apoptosis, leads to continuous decreases in the antithrombotic activity of the normal endothelium [9].
Endothelial injury and dysfunction is a commonplace finding in patients with COVID-19. The presence of viral particles in the ECs of the lungs as the primary site of viral infection and resulting occurrence of endotheliitis, cell degeneration, and necrosis underscore this problem. However, this process may later spread to other organs via the blood vessels [56]. By and large, the decrease in the antithrombotic activity of the endothelium results in thrombosis formation. Further details of EC injury have been studied in patients with COVID-19, which are as follows: (1) increased levels of soluble P-selectin in ICU patients compared with non-ICU patients; (2) increased levels of TM which is generally released during EC damage and its association with higher mortality rate; (3) increased numbers of circulating ECs, particularly in ICU patients; and (4) consequently, increased plasma levels of soluble vascular cell adhesion molecule 1 (sVCAM1; as a classical endothelial marker) [120]. The above results are corroborated by the fact that a high number of ECs has a positive correlation with lymphocyte and Plt counts and with high levels of soluble sICAM1 and sVCAM1, all of which support EC activation in COVID-19 [9]. In this context, direct multi-organ damage associated with COVID-19 infection has been observed in postmortem studies, accompanied by diffuse inflammation implicating endotheliitis. Therefore, apoptosis and pyroptosis have been considered possible mechanisms of endothelial damage. It is worth mentioning that in addition to the direct disruption of microvascular function and occlusion by the inflammatory endothelial cascade, a hypercoagulable state can also be induced, which in turn results in microvascular thrombosis. In addition to a likely driver of vasoconstriction, and inflammation, profound hypoxemia especially in severe COVID-19 has been shown to stimulate thrombosis through the expression of hypoxia-inducible transcription factors, which in turn can target several genes involved in thrombosis [119, 121]. Interestingly, the release of TM in plasma which represents lung endothelial injury and disease severity may be associated with reduced generation of APC. Concomitantly, elevated levels of soluble EPCR may also cause protein C pathway dysfunction (due to reduced APC generation as well as the reduced ability of APC to induce cytoprotective signaling) [122].
In patients with severe COVID-19, the presence of profoundly high levels of D-dimer and fibrin split products in absence of DIC suggests pulmonary microvascular thrombosis. In this regard, extensive microthrombosis that promotes and aggravates due to endothelial dysfunction could elucidate the role of inflamed endothelial in the elevation of D-dimers and thrombocytopenia in severe forms of COVID-19 [25]. Moreover, the small-vessel hyperplasia, wall thickening, focal pulmonary hemorrhage, and vascular hyaline thrombosis possibly due to venous congestion have been observed [121, 123]. Relying on the Johnson et al. study on heart autopsy tissue of 7 succumb patients, frequent and extensive thrombosis in small and large heart vessels in the context of no evidence of viral involvement or lymphocytic infiltration has been observed. However, they showed that endothelial activation may not be the primary cause of increased thrombotic diathesis in COVID-19 patients’ hearts and alterations in circulating neutrophils may have more roles in this phenomenon. In this setting, they do not observe any evidence of cytokine- or inflammatory-mediated endothelial activation with little evidence of cell death or desquamation while in contrary manner, they indicated the frequent markers of neutrophil activation as well as evidence of NET formation [124]. However, Varga et al. in a postmortem study on 3 cases with multiorgan failure showed viral inclusion structure beside lymphocytic endotheliitis [118]. In another autopsy study, Copin et al. identified cytoplasmic vacuolation in EC and its detachment in small- and medium-sized pulmonary arteries [125]. In large postmortem study by Su et al., the presence of variable foamy-like degeneration of renal ECs along with fibrin thrombus in glomerular capillary has revealed in some of patients with COVID-19 [126]. Of note, endothelial dysregulation may occur by loss of EC barrier integrity, leading to a procoagulant state. Interestingly, not only elevated circulating EC was observed in critically ill COVID-19 patients, but this biomarker was identified to be significantly increased even in the convalescent patients in comparison with healthy groups [120, 127]. In this context, direct viral infection, endotheliitis, and occasionally other coinfections seem to contribute to thrombotic events in different patients [118, 128]. All these data reflect the role of altered ECs injury and dysfunction in patients with COVID-19 especially in those with severe conditions, resulting in thrombotic events [129]. Currently, abnormal coagulation and thrombotic complication have obviously been perceived. Generally, the vascular endothelium (EC injury and/or dysfunction), the innate immune response (hyper-inflammatory immune response), and the coagulation and fibrinolytic pathways (hypercoagulability) are considered to be the main causes of the procoagulant condition [122].
Effects of COVID-19 on the complement system
The proteins of the complement system, which are liver-derived and function in plasma and interstitial fluids, play an important role in the innate immunity by recognizing invading pathogens, inducing lysis, and causing inflammatory cells to come to the infection site [8, 130]. The complement system and the coagulation system may have a common origin and are closely related to each other. Both systems rely on an amplifying enzyme waterfall to function. Proteins of the complement system, such as the membrane attack complex (MAC) and mannose-binding lectin (MBL)-associated serine proteases (MASPs), can also promote coagulation. Additionally, C5a can upregulate TF and PAI-1 expression of ECs, while the serine protease inhibitor C1-inhibitor inhibits coagulation. The concentration of C4b-binding protein bound protein S is also related to the level of free protein S. Conversely, coagulation proteins can participate in the activity of the complement system, with multiple coagulation factors (e.g., FXIa, FIXa, FXa, and FIIa) able to cleave C3. Surface-bound factors such as FXII and Kallikrein and anticoagulant components like TFPI and TM support activation and inactivation of complement, respectively. Several clinical disorders, including paroxysmal nocturnal hemoglobinuria, atypical hemolytic uremic syndrome, and antiphospholipid syndrome, exhibit combined engagement of both complement and coagulation [7, 131]. SARS-CoV-2 can directly trigger the complement cascade through the classical, alternative, and MBL pathways [8, 9]. Additionally, SARS-CoV-2 can indirectly activate the complement system due to EC damage, coagulation activation, and thrombosis [8, 130]. Histopathological, preclinical, transcriptomic, proteomic, multiomics, bioinformatic, and observational studies identified that over-activation of the complement system is involved in the pathogenesis, disease severity, and mortality of COVID-19 [130]. SARS-CoV-2-infection of airway epithelial cells causes overexpression of complement genes, particularly C3 and complement factor B. Factor B induces an intracellular convertase formation, which leads to conversion of C3 to C3a in the infected cells [132]. Patients with severe COVID-19 show increased circulating values of C3 fragments, C5a, and C5b–9 MAC, which associate with advanced disease severity and death [133,134,135]. Histopathological studies of tissues from patients with COVID-19 showed widespread microvascular deposition of complement components, such as C5b-9, C4d, and MASP2 throughout the skin, lungs, and kidneys [130, 136], and distinctive cytomorphological alterations of ECs in these patients are specifically characteristic of injury induced by MAC formation [8].
Complement activation can contribute to COVID-19-induced coagulopathy by affecting various components of homeostasis, such as ECs, leukocytes, Plts, and coagulation proteins. When SARS-CoV-2 affects ECs, it can increase the expression of C5a receptor 1 (C5aR1), which makes them more susceptible to pathological activation of C5a and formation of MAC. This can lead to EC death and a loss of resistance to thrombosis [8]. Microvasculature thrombosis associated with damaged EC is associated with deposits of complement activation products within injured microvasculature (positive fibrin and MAC immunostaining) [137]. In the site, where SARS-CoV-2 has infected capillaries, the increase in C3a and C5a production can cause the recruitment of neutrophils and monocytes, which can then adhere to ECs and participate in the thrombosis formation by the releasing NETs and MDMs [51, 138]. Complement activation can also stimulate Plts and coagulation factors, mainly through the action of MAC and MASPs, respectively, which supports the construction of thrombosis [7, 8]. Increase of coagulation factors in COVID-19, including FXa, FIIa, and FXII, and decrease of anti-thrombogenic components, including TM and TFPI, may also support activation of complement. Therefore, SARS-CoV-2-induced complement activation can amplify coagulation activation, while SARS-CoV-2-induced upregulation and downregulation of the coagulation and anticoagulant factors, respectively, as positive feedback loop enhance complement activation. Overall, the complement system and the coagulation system are functionally dependent and play important roles against SARS-CoV-2 infection. Complement activation is one of the main drivers of COVID-19-associated coagulopathy, and its overactivation is involved in severity and mortality of the disease.
Conclusion
COVID-19 patients have a hypercoagulability state, and thrombosis is a life-threatening complication of them. A process leading to thrombus formation involves a combination of imbalanced factors and is not as simple as a sole factor imbalance to the other factors [139]. It seems that COVID-19 by disturbing the balance between the procoagulant systems and the regulatory mechanisms causes this predisposition to thrombus formation. In other words, an endotheliopathy, enhanced Plt activation, complement activation, and increased procoagulant factors in combination with impaired endogenous anticoagulation and decreased fibrinolysis will cooperatively lead to the increased risk of thrombus formation. It is possible that the changes in the components involved in homeostasis be different at distinct stages of the disease and one or more of these changes predominate in different stages. The degree of coagulation abnormalities in COVID-19 seems to connect with the severity of disease. It seems that, to discover the overall risk potential for thrombosis in a COVID-19 patient, a complete evaluation of the recognized risk factors is required.
References
Luo H-c, You C-y, Lu S-w, Fu Y-q (2021) Characteristics of coagulation alteration in patients with COVID-19. Ann Hematol 100(1):45–52. https://doi.org/10.1007/s00277-020-04305-x
Hanif A, Khan S, Mantri N et al (2020) Thrombotic complications and anticoagulation in COVID-19 pneumonia: a New York City hospital experience. Ann Hematol 99(10):2323–2328. https://doi.org/10.1007/s00277-020-04216-x
Fox SE, Akmatbekov A, Harbert JL, Li G, Quincy Brown J, Vander Heide RS (2020) Pulmonary and cardiac pathology in African American patients with COVID-19: an autopsy series from New Orleans. Lancet Respir Med 8(7):681–686. https://doi.org/10.1016/s2213-2600(20)30243-5
Sarmadian R, Ghasemikhah R, Sarmadian H, Khosravi M, Hassani S (2022) Post-COVID-19 splenic infarction in a patient with chronic atrial fibrillation: a case report. Clin Case Rep 10(7):e6011. https://doi.org/10.1002/ccr3.6011
Sayyadi M, Khosravi M, Ghaznavi-Rad E (2021) Contribution value of coagulation abnormalities in COVID-19 prognosis: a bright perspective on the laboratory pattern of patients with coronavirus disease 2019. Eur Rev Med Pharmacol Sci 25(1):518–522. https://doi.org/10.26355/eurrev_202101_24423
Allegra A, Innao V, Allegra AG, Musolino C (2020) Coagulopathy and thromboembolic events in patients withSARS-CoV-2 infection: pathogenesis and management strategies. Ann Hematol 99(9):1953–1965. https://doi.org/10.1007/s00277-020-04182-4
Dzik S (2019) Complement and coagulation: cross talk through time. Transfus Med Rev 33(4):199–206. https://doi.org/10.1016/j.tmrv.2019.08.004
Afzali B, Noris M (2022) The state of complement in COVID-19. Nat Rev Immunol 22(2):77–84. https://doi.org/10.1038/s41577-021-00665-1
Bonaventura A, Vecchie A, Dagna L et al (2021) Endothelial dysfunction and immunothrombosis as key pathogenic mechanisms in COVID-19. Nat Rev Immunol 21(5):319–329. https://doi.org/10.1038/s41577-021-00536-9
Rad F, Dabbagh A, Dorgalaleh A, Biswas A (2021) The relationship between inflammatory cytokines and coagulopathy in patients with COVID-19. J Clin Med 10(9). https://doi.org/10.3390/jcm10092020
Bahraini M, Dorgalaleh A (2021) The impact of SARS-CoV-2 infection on blood coagulation and fibrinolytic pathways: a review of prothrombotic changes caused by COVID-19. Semin Thromb Hemost 48(01):19–30
Dorgalaleh A (2020) Bleeding and bleeding risk in COVID-19. Semin Thromb Hemost 46(07):815–818
Sang Y, Roest M, de Laat B, de Groot PG, Huskens D (2021) Interplay between platelets and coagulation. Blood Rev 46:100733. https://doi.org/10.1016/j.blre.2020.100733
Zhang S, Liu Y, Wang X et al (2020) SARS-CoV-2 binds platelet ACE2 to enhance thrombosis in COVID-19. J Hematol Oncol 13(1):120. https://doi.org/10.1186/s13045-020-00954-7
Manne BK, Denorme F, Middleton EA et al (2020) Platelet gene expression and function in patients with COVID-19. Blood 136(11):1317–1329. https://doi.org/10.1182/blood.2020007214
Zaid Y, Puhm F, Allaeys I et al (2020) Platelets can associate with SARS-Cov-2 RNA and are hyperactivated in COVID-19. Circ Res 127(11):1404–1418. https://doi.org/10.1161/circresaha.120.317703
Taus F, Salvagno G, Canè S et al (2020) Platelets promote thromboinflammation in SARS-CoV-2 pneumonia. Arterioscler Thromb Vasc Biol 40(12):2975–2989. https://doi.org/10.1161/atvbaha.120.315175
Calderon-Lopez MT, Garcia-Leon N, Gomez-Arevalillo S, Martin-Serrano P, Matilla-Garcia A (2021) Coronavirus disease 2019 and coagulopathy: other prothrombotic coagulation factors. Blood Coagul Fibrinolysis 32(1):44–49. https://doi.org/10.1097/mbc.0000000000000996
Hottz ED, Azevedo-Quintanilha IG, Palhinha L et al (2020) Platelet activation and platelet-monocyte aggregate formation trigger tissue factor expression in patients with severe COVID-19. Blood 136(11):1330–1341. https://doi.org/10.1182/blood.2020007252
Nicolai L, Leunig A, Brambs S et al (2020) Immunothrombotic dysregulation in COVID-19 pneumonia is associated with respiratory failure and coagulopathy. Circulation 142(12):1176–1189. https://doi.org/10.1161/circulationaha.120.048488
Viecca M, Radovanovic D, Forleo GB, Santus P (2020) Enhanced platelet inhibition treatment improves hypoxemia in patients with severe Covid-19 and hypercoagulability. A case control, proof of concept study. Pharmacol Res 158:104950. https://doi.org/10.1016/j.phrs.2020.104950
Xu P, Zhou Q, Xu J (2020) Mechanism of thrombocytopenia in COVID-19 patients. Ann Hematol 99(6):1205–1208. https://doi.org/10.1007/s00277-020-04019-0
Lippi G, Plebani M, Henry BM (2020) Thrombocytopenia is associated with severe coronavirus disease 2019 (COVID-19) infections: a meta-analysis. Clin Chim Acta 506:145–148. https://doi.org/10.1016/j.cca.2020.03.022
Tang N, Li D, Wang X, Sun Z (2020) Abnormal coagulation parameters are associated with poor prognosis in patients with novel coronavirus pneumonia. J Thrombo Haemost 18(4):844–847. https://doi.org/10.1111/jth.14768
Zhou F, Yu T, Du R et al (2020) Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet 395(10229):1054–1062. https://doi.org/10.1016/S0140-6736(20)30566-3
Liu X, Zhang X, Xiao Y et al (2020) Heparin-induced thrombocytopenia is associated with a high risk of mortality in critical COVID-19 patients receiving heparin-involved treatment. MedRxiv
Zhang Y, Xiao M, Zhang S et al (2020) Coagulopathy and antiphospholipid antibodies in patients with Covid-19. New Eng J Med 382(17):e38. https://doi.org/10.1056/NEJMc2007575
Kaushansky K, Broudy VC, Lin N et al (1995) Thrombopoietin, the Mp1 ligand, is essential for full megakaryocyte development. Proc Natl Acad Sci U S A 92(8):3234–3238. https://doi.org/10.1073/pnas.92.8.3234
Delshad M, Safaroghli-Azar A, Pourbagheri-Sigaroodi A, Poopak B, Shokouhi S, Bashash D (2021) Platelets in the perspective of COVID-19; pathophysiology of thrombocytopenia and its implication as prognostic and therapeutic opportunity. Int Immunopharmacol 99:107995. https://doi.org/10.1016/j.intimp.2021.107995
Chen G, Wu D, Guo W et al (2020) Clinical and immunological features of severe and moderate coronavirus disease 2019. J Clin Invest 130(5):2620–2629. https://doi.org/10.1172/jci137244
Roncati L, Ligabue G, Nasillo V et al (2020) A proof of evidence supporting abnormal immunothrombosis in severe COVID-19: naked megakaryocyte nuclei increase in the bone marrow and lungs of critically ill patients. Platelets 31(8):1085–1089. https://doi.org/10.1080/09537104.2020.1810224
Chen J, Chung DW (2018) Inflammation, von Willebrand factor, and ADAMTS13. Blood 132(2):141–147. https://doi.org/10.1182/blood-2018-02-769000
Goshua G, Pine AB, Meizlish ML et al (2020) Endotheliopathy in COVID-19-associated coagulopathy: evidence from a single-centre, cross-sectional study. Lancet Haematol 7(8):e575–e582. https://doi.org/10.1016/s2352-3026(20)30216-7
Panigada M, Bottino N, Tagliabue P et al (2020) Hypercoagulability of COVID-19 patients in intensive care unit: a report of thromboelastography findings and other parameters of hemostasis. J Thromb Haemost 18(7):1738–1742. https://doi.org/10.1111/jth.14850
Wibowo A, Pranata R, Lim MA, Akbara MR, Martha JW (2022) Endotheliopathy marked by high von Willebrand factor (vWF) antigen in COVID-19 is associated with poor outcome: a systematic review and meta-analysis. Int J Infect Dis 117:267–273. https://doi.org/10.1016/j.ijid.2021.06.051
Patmore S, Dhami SPS, O'Sullivan JM (2020) Von Willebrand factor and cancer; metastasis and coagulopathies. J Thromb Haemost 18(10):2444–2456. https://doi.org/10.1111/jth.14976
Sonneveld MA, de Maat MP, Leebeek FW (2014) Von Willebrand factor and ADAMTS13 in arterial thrombosis: a systematic review and meta-analysis. Blood Rev 28(4):167–178. https://doi.org/10.1016/j.blre.2014.04.003
Sonneveld MA, de Maat MP, Portegies ML et al (2015) Low ADAMTS13 activity is associated with an increased risk of ischemic stroke. Blood 126(25):2739–2746. https://doi.org/10.1182/blood-2015-05-643338
Marco A, Marco P (2021) Von Willebrand factor and ADAMTS13 activity as clinical severity markers in patients with COVID-19. J Thromb Thromb 52(2):497–503. https://doi.org/10.1007/s11239-021-02457-9
Rodríguez Rodríguez M, Castro Quismondo N, Zafra Torres D, Gil Alos D, Ayala R, Martinez-Lopez J (2021) Increased von Willebrand factor antigen and low ADAMTS13 activity are related to poor prognosis in covid-19 patients. Int J Lab Hematol 43(4):152–155. https://doi.org/10.1111/ijlh.13476
Escher R, Breakey N, Lammle B (2020) ADAMTS13 activity, von Willebrand factor, factor VIII and D-dimers in COVID-19 inpatients. Thromb Res 192:174–175. https://doi.org/10.1016/j.thromres.2020.05.032
Ward SE, Fogarty H, Karampini E et al (2021) ADAMTS13 regulation of VWF multimer distribution in severe COVID-19. J Thromb Haemost 19(8):1914–1921. https://doi.org/10.1111/jth.15409
Bowles L, Platton S, Yartey N et al (2020) Lupus anticoagulant and abnormal coagulation tests in patients with Covid-19. New Eng J Med 383(3):288–290. https://doi.org/10.1056/NEJMc2013656
Katneni UK, Alexaki A, Hunt RC et al (2020) Coagulopathy and thrombosis as a result of severe COVID-19 infection: a microvascular focus. Thromb Haemost 120(12):1668–1679. https://doi.org/10.1055/s-0040-1715841
Ma Q, Jacobi PM, Emmer BT et al (2017) Genetic variants in ADAMTS13 as well as smoking are major determinants of plasma ADAMTS13 levels. Blood Adv 1(15):1037–1046. https://doi.org/10.1182/bloodadvances.2017005629
Vardavas CI, Nikitara K (2020) COVID-19 and smoking: a systematic review of the evidence. Tob Induc Dis 18:20. https://doi.org/10.18332/tid/119324
Butenas S, Orfeo T, Mann KG (2009) Tissue factor in coagulation: which? where? when? Arterioscler Thromb Vasc Biol 29(12):1989–1996
McGonagle D, O'Donnell JS, Sharif K, Emery P, Bridgewood C (2020) Immune mechanisms of pulmonary intravascular coagulopathy in COVID-19 pneumonia. Lancet Rheumatol 2(7):e437–e445. https://doi.org/10.1016/S2665-9913(20)30121-1
Gorog DA, Storey RF, Gurbel PA et al (2022) Current and novel biomarkers of thrombotic risk in COVID-19: a Consensus Statement from the International COVID-19 Thrombosis Biomarkers Colloquium. Nat Rev Cardiol 19(7):475–495. https://doi.org/10.1038/s41569-021-00665-7
Hottz ED, Martins-Gonçalves R, Palhinha L et al (2022) Platelet-monocyte interaction amplifies thromboinflammation through tissue factor signaling in COVID-19. Blood Adv. https://doi.org/10.1182/bloodadvances.2021006680
Skendros P, Mitsios A, Chrysanthopoulou A et al (2020) Complement and tissue factor–enriched neutrophil extracellular traps are key drivers in COVID-19 immunothrombosis. J Clin Investig 130(11):6151–6157. https://doi.org/10.1172/JCI141374
Semeraro N, Colucci M (2021) The prothrombotic state associated with SARS-CoV-2 infection: pathophysiological aspects. Mediterr J Hematol Infect Dis 13(1):e2021045. https://doi.org/10.4084/mjhid.2021.045
Rosell A, Havervall S, von Meijenfeldt F et al (2021) Patients with COVID-19 have elevated levels of circulating extracellular vesicle tissue factor activity that is associated with severity and mortality-brief report. Arterioscler Thromb Vasc Biol 41(2):878–882. https://doi.org/10.1161/atvbaha.120.315547
Mast AE, Wolberg AS, Gailani D et al (2021) SARS-CoV-2 suppresses anticoagulant and fibrinolytic gene expression in the lung. Elife 10. https://doi.org/10.7554/eLife.64330
FitzGerald ES, Jamieson AM (2022) Comment on SARS-CoV-2 suppresses anticoagulant and fibrinolytic gene expression in the lung. Elife 11. https://doi.org/10.7554/eLife.74268
Lippi G, Sanchis-Gomar F, Favaloro EJ, Lavie CJ, Henry BM (2021) Coronavirus Disease 2019-Associated Coagulopathy. Mayo Clin Proc 96(1):203–217. https://doi.org/10.1016/j.mayocp.2020.10.031
Pelle MC, Zaffina I, Lucà S et al (2022) Endothelial dysfunction in COVID-19: potential mechanisms and possible therapeutic options. Life 12(10):1605
Tabibian S, Shiravand Y, Shams M et al (2019) A comprehensive overview of coagulation factor V and congenital factor V deficiency. Semin Thromb Hemost 45(5):523–543. https://doi.org/10.1055/s-0039-1687906
Shams M, Hassani S, Dorgalaleh A, Zamani F, Ahmadi A (2023) Factor VII Padua in Iran: clinical and laboratory findings of three unrelated patients. Blood Coagul Fibrinolysis. https://doi.org/10.1097/mbc.0000000000001195
Stefely JA, Christensen BB, Gogakos T, Cone Sullivan JK, Montgomery GG, Barranco JP, Van Cott EM (2020) Marked factor V activity elevation in severe COVID-19 is associated with venous thromboembolism. Am J Hematol 95(12):1522–1530. https://doi.org/10.1002/ajh.25979
Wang J, Kotagiri P, Lyons PA et al (2022) Coagulation factor V is a T-cell inhibitor expressed by leukocytes in COVID-19. iScience 25(3):103971. https://doi.org/10.1016/j.isci.2022.103971
von Meijenfeldt FA, Havervall S, Adelmeijer J et al (2021) Elevated factor V activity and antigen levels in patients with Covid-19 are related to disease severity and 30-day mortality. Am J Hematol 96(4):E98–E100. https://doi.org/10.1002/ajh.26085
Zhang Y, Cao W, Jiang W et al (2020) Profile of natural anticoagulant, coagulant factor and anti-phospholipid antibody in critically ill COVID-19 patients. J Thromb Thrombolysis 50(3):580–586. https://doi.org/10.1007/s11239-020-02182-9
Begbie M, Notley C, Tinlin S, Sawyer L, Lillicrap D (2000) The factor VIII acute phase response requires the participation of NFkappaB and C/EBP. Thromb Haemost 84(2):216–222
Wang IE, Cooper G, Mousa SA (2021) Diagnostic approaches for COVID-19 and its associated complications. Diagnostics (Basel) 11(11). https://doi.org/10.3390/diagnostics11112071
Ceballos FC, Ryan P, Blancas R et al (2021) Are reduced levels of coagulation proteins upon admission linked to COVID-19 severity and mortality? Front Med (Lausanne) 8:718053. https://doi.org/10.3389/fmed.2021.718053
Purroy F, Arqué G (2021) Influence of thromboembolic events in the prognosis of COVID-19 hospitalized patients. Results from a cross sectional study. PLoS One 16(6):e0252351. https://doi.org/10.1371/journal.pone.0252351
Al-Tamimi AO, Yusuf AM, Jayakumar MN et al (2022) SARS-CoV-2 infection induces soluble platelet activation markers and PAI-1 in the early moderate stage of COVID-19. Int J Lab Hematol 44(4):712–721. https://doi.org/10.1111/ijlh.13829
Leentjens J, van Haaps TF, Wessels PF, Schutgens REG, Middeldorp S (2021) COVID-19-associated coagulopathy and antithrombotic agents-lessons after 1 year. Lancet Haematol 8(7):e524–e533. https://doi.org/10.1016/s2352-3026(21)00105-8
White D, MacDonald S, Edwards T et al (2021) Evaluation of COVID-19 coagulopathy; laboratory characterization using thrombin generation and nonconventional haemostasis assays. Int J Lab Hematol 43(1):123–130. https://doi.org/10.1111/ijlh.13329
Fish RJ, Neerman-Arbez M (2012) Fibrinogen gene regulation. Thromb Haemost 108(3):419–426. https://doi.org/10.1160/th12-04-0273
Ranucci M, Ballotta A, Di Dedda U et al (2020) The procoagulant pattern of patients with COVID-19 acute respiratory distress syndrome. J Thromb Haemost 18(7):1747–1751. https://doi.org/10.1111/jth.14854
Polimeni A, Leo I, Spaccarotella C et al (2021) Differences in coagulopathy indices in patients with severe versus non-severe COVID-19: a meta-analysis of 35 studies and 6427 patients. Sci Rep, 11 (1):10464. https://doi.org/https://doi.org/10.1038/s41598-021-89967-x
Renné T, Schmaier AH, Nickel KF, Blombäck M, Maas C (2012) In vivo roles of factor XII. Blood 120(22):4296–4303. https://doi.org/10.1182/blood-2012-07-292094
van der Meijden PE, Munnix IC, Auger JM et al (2009) Dual role of collagen in factor XII-dependent thrombus formation. Blood 114(4):881–890. https://doi.org/10.1182/blood-2008-07-171066
Gould TJ, Vu TT, Swystun LL, Dwivedi DJ, Mai SH, Weitz JI, Liaw PC (2014) Neutrophil extracellular traps promote thrombin generation through platelet-dependent and platelet-independent mechanisms. Arterioscler Thromb Vasc Biol 34(9):1977–1984. https://doi.org/10.1161/atvbaha.114.304114
Rahi MS, Jindal V, Reyes S-P, Gunasekaran K, Gupta R, Jaiyesimi I (2021) Hematologic disorders associated with COVID-19: a review. Ann Hematol 100(2):309–320. https://doi.org/10.1007/s00277-020-04366-y
Englert H, Rangaswamy C, Deppermann C et al (2021) Defective NET clearance contributes to sustained FXII activation in COVID-19-associated pulmonary thrombo-inflammation. EBioMedicine 67:103382. https://doi.org/10.1016/j.ebiom.2021.103382
Dorgalaleh A, Rashidpanah J (2016) Blood coagulation factor XIII and factor XIII deficiency. Blood Rev 30(6):461–475. https://doi.org/10.1016/j.blre.2016.06.002
von Meijenfeldt FA, Havervall S, Adelmeijer J et al (2021) COVID-19 is associated with an acquired factor XIII deficiency. Thromb Haemost 121(12):1668–1669
Gupta A, Qaisar R, Halwani R, Kannan M, Ahmad F (2022) TFPI and FXIII negatively and S100A8/A9 and cystatin C positively correlate with D-dimer in COVID-19. Exp Biol Med (Maywood) 247(17):1570–1576. https://doi.org/10.1177/15353702221102117
Wright FL, Vogler TO, Moore EE et al (2020) Fibrinolysis shutdown correlation with thromboembolic events in severe COVID-19 infection. J Am Coll Surg 231(2):193–203.e191. https://doi.org/10.1016/j.jamcollsurg.2020.05.007
Heinz C, Miesbach W, Herrmann E et al (2021) Greater fibrinolysis resistance but no greater platelet aggregation in critically ill COVID-19 patients. Anesthesiology 134(3):457–467. https://doi.org/10.1097/aln.0000000000003685
Juneja GK, Castelo M, Yeh CH et al (2021) Biomarkers of coagulation, endothelial function, and fibrinolysis in critically ill patients with COVID-19: a single-center prospective longitudinal study. J Thromb Haemost 19(6):1546–1557. https://doi.org/10.1111/jth.15327
Kwaan HC (2020) Coronavirus disease 2019: the role of the fibrinolytic system from transmission to organ injury and sequelae. Semin Thromb Hemost 46(7):841–844. https://doi.org/10.1055/s-0040-1709996
Henry BM, Benoit SW, Hoehn J, Lippi G, Favaloro EJ, Benoit JL (2020) Circulating plasminogen concentration at admission in patients with coronavirus disease 2019 (COVID-19). Semin Thromb Hemost 46(07):859–862
Hammer S, Häberle H, Schlensak C et al (2021) Severe SARS-CoV-2 infection inhibits fibrinolysis leading to changes in viscoelastic properties of blood clot: a descriptive study of fibrinolysis in COVID-19. Thromb Haemost 121(11):1417–1426. https://doi.org/10.1055/a-1400-6034
Nougier C, Benoit R, Simon M et al (2020) Hypofibrinolytic state and high thrombin generation may play a major role in SARS-COV2 associated thrombosis. J Thromb Haemost 18(9):2215–2219. https://doi.org/10.1111/jth.15016
Francischetti IMB, Toomer K, Zhang Y et al (2021) Upregulation of pulmonary tissue factor, loss of thrombomodulin and immunothrombosis in SARS-CoV-2 infection. EClinicalMedicine 39:101069. https://doi.org/10.1016/j.eclinm.2021.101069
Tabatabai A, Rabin J, Menaker J et al (2020) Factor VIII and functional protein C activity in critically ill patients with coronavirus disease 2019: a case series. A A Pract 14(7):e01236. https://doi.org/10.1213/xaa.0000000000001236
Corrêa TD, Cordioli RL, Campos Guerra JC et al (2020) Coagulation profile of COVID-19 patients admitted to the ICU: An exploratory study. PLoS One 15(12):e0243604. https://doi.org/10.1371/journal.pone.0243604
Sehgal T, Gupta N, Kohli S et al (2021) A prospective study of specialized coagulation parameters in admitted COVID-19 patients and their correlation with acute respiratory distress syndrome and outcome. Cureus 13(8):e17463. https://doi.org/10.7759/cureus.17463
Gerotziafas GT, Sergentanis TN, Voiriot G et al (2020) Derivation and validation of a predictive score for disease worsening in patients with COVID-19. Thromb Haemost 120(12):1680–1690. https://doi.org/10.1055/s-0040-1716544
Stanne TM, Pedersen A, Gisslén M, Jern C (2021) Low admission protein C levels are a risk factor for disease worsening and mortality in hospitalized patients with COVID-19. Thromb Res 204:13–15. https://doi.org/10.1016/j.thromres.2021.05.016
Won T, Wood MK, Hughes DM et al (2022) Endothelial thrombomodulin downregulation caused by hypoxia contributes to severe infiltration and coagulopathy in COVID-19 patient lungs. EBioMedicine 75:103812. https://doi.org/10.1016/j.ebiom.2022.103812
Cani E, Dwivedi DJ, Liaw KL et al (2021) Immunothrombosis biomarkers for distinguishing coronavirus disease 2019 patients from noncoronavirus disease septic patients with pneumonia and for predicting ICU Mortality. Crit Care Explor 3(12):e0588. https://doi.org/10.1097/cce.0000000000000588
López-Aguirre Y, Páramo JA (1999) Endothelial cell and hemostatic activation in relation to cytokines in patients with sepsis. Thromb Res 94(2):95–101. https://doi.org/10.1016/s0049-3848(98)00200-x
Vassiliou AG, Keskinidou C, Jahaj E et al (2021) Could soluble endothelial protein C receptor levels recognize SARS-CoV2-positive patients requiring hospitalization? Shock 56(5):733–736. https://doi.org/10.1097/shk.0000000000001780
Liaw PC, Neuenschwander PF, Smirnov MD, Esmon CT (2000) Mechanisms by which soluble endothelial cell protein C receptor modulates protein C and activated protein C function. J Biol Chem 275(8):5447–5452. https://doi.org/10.1074/jbc.275.8.5447
Bayrakci N, Ozkan G, Mutlu LC, Erdem L, Yildirim I, Gulen D, Celikkol A (2021) Relationship between serum soluble endothelial protein C receptor level and COVID-19 findings. Blood Coagul Fibrinolysis 32(8):550–555. https://doi.org/10.1097/mbc.0000000000001070
Han H, Yang L, Liu R, Liu F, Wu KL, Li J, Liu XH, Zhu CL (2020) Prominent changes in blood coagulation of patients with SARS-CoV-2 infection. Clin Chem Lab Med 58(7):1116–1120. https://doi.org/10.1515/cclm-2020-0188
Gross O, Moerer O, Weber M, Huber TB, Scheithauer S (2020) COVID-19-associated nephritis: early warning for disease severity and complications? Lancet 395(10236):e87–e88. https://doi.org/10.1016/s0140-6736(20)31041-2
Buijsers B, Yanginlar C, de Nooijer A et al (2020) Increased plasma heparanase activity in COVID-19 patients. Front Immunol 11:575047. https://doi.org/10.3389/fimmu.2020.575047
Nimah M, Brilli RJ (2003) Coagulation dysfunction in sepsis and multiple organ system failure. Crit Care Clin 19(3):441–458. https://doi.org/10.1016/s0749-0704(03)00008-3
Gazzaruso C, Paolozzi E, Valenti C et al (2020) Association between antithrombin and mortality in patients with COVID-19. A possible link with obesity. Nutr Metab Cardiovasc Dis 30(11):1914–1919. https://doi.org/10.1016/j.numecd.2020.07.040
Anaklı İ, Ergin Özcan P, Polat Ö et al (2021) Prognostic value of antithrombin levels in COVID-19 patients and impact of fresh frozen plasma treatment: a retrospective study. Turk J Haematol 38(1):15–21. https://doi.org/10.4274/tjh.galenos.2021.2020.0695
Boknäs N, Laine C, Hillarp A, Macwan AS, Gustafsson KM, Lindahl TL, Holmström M (2022) Associations between hemostatic markers and mortality in COVID-19 - compounding effects of D-dimer, antithrombin and PAP complex. Thromb Res 213:97–104. https://doi.org/10.1016/j.thromres.2022.03.013
Lippi G, Henry BM, Sanchis-Gomar F (2020) Plasma antithrombin values are significantly decreased in coronavirus disease 2019 (COVID-19) patients with severe illness. Semin Thromb Hemost 47(04):460–462
Rostami M, Mansouritorghabeh H (2022) Trend of fluctuations of antithrombin in plasma of patients with COVID-19: a meta-analysis and systematic review. Expert Rev Hematol 15(8):747–755. https://doi.org/10.1080/17474086.2022.2104708
Tang H, Ivanciu L, Popescu N et al (2007) Sepsis-induced coagulation in the baboon lung is associated with decreased tissue factor pathway inhibitor. Am J Pathol 171(3):1066–1077. https://doi.org/10.2353/ajpath.2007.070104
Al Otair HA, Abdel Gader AG, Khurshid SM et al (2016) The levels of tissue factor pathway inhibitor in sepsis patients receiving prophylactic enoxaparin. Turk J Haematol 33(2):112–118. https://doi.org/10.4274/tjh.2014.0312
Cacciola R, Gentilini Cacciola E, Vecchio V, Cacciola E (2022) Cellular and molecular mechanisms in COVID-19 coagulopathy: role of inflammation and endotheliopathy. J Thromb Thromb 53(2):282–290. https://doi.org/10.1007/s11239-021-02583-4
Kelliher S, Weiss L, Cullivan S et al (2022) Non-severe COVID-19 is associated with endothelial damage and hypercoagulability despite pharmacological thromboprophylaxis. J Thromb Haemost 20(4):1008–1014. https://doi.org/10.1111/jth.15660
Dupont A, Rauch A, Staessens S et al (2021) Vascular endothelial damage in the pathogenesis of organ injury in severe COVID-19. Arterioscler Thromb Vasc Biol 41(5):1760–1773. https://doi.org/10.1161/ATVBAHA.120.315595
Yau JW, Teoh H, Verma S (2015) Endothelial cell control of thrombosis. BMC Cardiovasc Disord 15:130. https://doi.org/10.1186/s12872-015-0124-z
McPherson RA, Pincus MR (2021) Henry's clinical diagnosis and management by laboratory methods, 4th edn. Elsevier, Philadelphia
Gralinski LE, Bankhead A 3rd, Jeng S et al (2013) Mechanisms of severe acute respiratory syndrome coronavirus-induced acute lung injury. mBio 4(4). https://doi.org/10.1128/mBio.00271-13
Varga Z, Flammer AJ, Steiger P et al (2020) Endothelial cell infection and endotheliitis in COVID-19. Lancet 395(10234):1417–1418. https://doi.org/10.1016/s0140-6736(20)30937-5
Price LC, McCabe C, Garfield B, Wort SJ (2020) Thrombosis and COVID-19 pneumonia: the clot thickens, vol 56. Eur Respiratory Soc
Guervilly C, Burtey S, Sabatier F et al (2020) Circulating endothelial cells as a marker of endothelial injury in severe COVID -19. J Infect Dis 222(11):1789–1793. https://doi.org/10.1093/infdis/jiaa528
Hadid T, Kafri Z, Al-Katib A (2021) Coagulation and anticoagulation in COVID-19. Blood Rev 47:100761. https://doi.org/10.1016/j.blre.2020.100761
Conway EM, Mackman N, Warren RQ et al (2022) Understanding COVID-19-associated coagulopathy. Nat Rev Immunol 22(10):639–649. https://doi.org/10.1038/s41577-022-00762-9
Luo W-R, Yu H, Gou J-Z et al (2020) Histopathologic findings in the explant lungs of a patient with COVID-19 treated with bilateral orthotopic lung transplant. Transplantation 104(11):329–331. https://doi.org/10.1097/tp.0000000000003412
Johnson JE, McGuone D, Xu ML, Jane-Wit D, Mitchell RN, Libby P, Pober JS (2022) Coronavirus disease 2019 (COVID-19) coronary vascular thrombosis: correlation with neutrophil but not endothelial activation. Am J Pathol 192(1):112–120
Copin M-C, Parmentier E, Duburcq T, Poissy J, Mathieu D (2020) Time to consider histologic pattern of lung injury to treat critically ill patients with COVID-19 infection. Intensive Care Med 46(6):1124–1126
Su H, Yang M, Wan C et al (2020) Renal histopathological analysis of 26 postmortem findings of patients with COVID-19 in China. Kidney Int 98(1):219–227
Chioh FW, Fong S-W, Young BE et al (2021) Convalescent COVID-19 patients are susceptible to endothelial dysfunction due to persistent immune activation. Elife 10:e64909
Keller TT, Mairuhu AT, de Kruif MD et al (2003) Infections and endothelial cells. Cardiovasc Res 60(1):40–48
Otifi HM, Adiga BK (2022) Endothelial dysfunction in Covid-19 infection. Am J Med Sci 363 (4):281–287. https://doi.org/10.1016/j.amjms.2021.12.010
Lim EHT, van Amstel RBE, de Boer VV et al (2023) Complement activation in COVID-19 and targeted therapeutic options: a scoping review. Blood Rev 57:100995. https://doi.org/10.1016/j.blre.2022.100995
Oncul S, Afshar-Kharghan V (2020) The interaction between the complement system and hemostatic factors. Curr Opin Hematol 27(5):341–352. https://doi.org/10.1097/moh.0000000000000605
Yan B, Freiwald T, Chauss D et al (2021) SARS-CoV-2 drives JAK1/2-dependent local complement hyperactivation. Sci Immunol 6(58). https://doi.org/10.1126/sciimmunol.abg0833
Holter JC, Pischke SE, de Boer E et al (2020) Systemic complement activation is associated with respiratory failure in COVID-19 hospitalized patients. Proc Natl Acad Sci U S A 117(40):25018–25025. https://doi.org/10.1073/pnas.2010540117
Carvelli J, Demaria O, Vely F et al (2020) Association of COVID-19 inflammation with activation of the C5a-C5aR1 axis. Nature 588(7836):146–150. https://doi.org/10.1038/s41586-020-2600-6
Sinkovits G, Mezo B, Reti M et al (2021) Complement overactivation and consumption predicts in-hospital mortality in SARS-CoV-2 infection. Front Immunol 12:663187. https://doi.org/10.3389/fimmu.2021.663187
Magro C, Mulvey JJ, Berlin D et al (2020) Complement associated microvascular injury and thrombosis in the pathogenesis of severe COVID-19 infection: a report of five cases. Trans Res 220:1–13. https://doi.org/10.1016/j.trsl.2020.04.007
Pellegrini D, Kawakami R, Guagliumi G et al (2021) Microthrombi as a major cause of cardiac injury in COVID-19: a pathologic study. Circulation 143(10):1031–1042. https://doi.org/10.1161/CIRCULATIONAHA.120.051828
Perico L, Morigi M, Galbusera M et al (2022) SARS-CoV-2 spike protein 1 activates microvascular endothelial cells and complement system leading to platelet aggregation. Front Immunol 13:827146. https://doi.org/10.3389/fimmu.2022.827146
Marlar RA, Potts RM, Welsh CH (2007) A systems biology approach to the diagnosis of venous thrombosis risk. Blood Coagul Fibrinolysis 18(3):215–217. https://doi.org/10.1097/MBC.0b013e3280116c84
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethical approval
The study was approved by the Ethics Committee at the Arak University of Medical Sciences (No. IR.ARAKMU.REC.1401.223). This article does not contain any studies with human participants or animals performed by any of the authors.
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Sayyadi, M., Hassani, S., Shams, M. et al. Status of major hemostatic components in the setting of COVID-19: the effect on endothelium, platelets, coagulation factors, fibrinolytic system, and complement. Ann Hematol 102, 1307–1322 (2023). https://doi.org/10.1007/s00277-023-05234-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00277-023-05234-1