
Abstract
Despite advances in the understanding of the pathophysiology of cytomegalovirus (CMV) infection, it remains as one of the most common infectious complications after allogeneic hematopoietic stem cell transplantation (allo-HSCT). The aim of this study was to determine the genotype of cytokines and chemokines in donor and recipient and their association with CMV reactivation. Eighty-five patients receiving an allo-HSCT from an HLA-identical sibling donor were included in the study. Fifty genes were selected for their potential role in the pathogenesis of CMV infection. CMV DNAemia was evaluated until day 180 after allo-HSCT. CMV reactivation was observed in 51/85 (60%) patients. Of the 213 genetic variants selected, 11 polymorphisms in 7 different genes (CXCL12, IL12A, KIR3DL1, TGFB2, TNF, IL1RN, and CD48) were associated with development or protection from CMV reactivation. A predictive model using five of such polymorphisms (CXCL12 rs2839695, IL12A rs7615589, KIR3DL1 rs4554639, TGFB2 rs5781034 for the recipient and CD48 rs2295615 for the donor) together with the development of acute GVHD grade III/IV improved risk stratification of CMV reactivation. In conclusion, the data presented suggest that the screening of five polymorphisms in recipient and donor pre-transplantation could help to predict the individual risk of CMV infection development after HLA-identical allo-HSCT.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cytomegalovirus (CMV) infection is one of the most common viral infections after allogeneic hematopoietic stem cell transplantation (allo-HSCT). In patients without CMV prophylaxis and depending on the transplant setting, incidences of CMV reactivation after allo-HSCT among CMV-seropositive patients are as high as 80%. In addition to the direct effects of CMV reactivation, tissue-invasive CMV disease may be associated with increased risk of graft-versus-host-disease (GVHD) and infections [1]. T-cell mediated cellular immunity is the most important factor in controlling CMV replication. CMV induces a strong CD8 + cytotoxic T-lymphocyte (CTL) response; therefore, immunosuppression significantly contributes to the loss of CMV specific adaptive immune control [2, 3]. However, the observation that only a fraction of patients with similar degrees of immunosuppression develops CMV infection suggests that other factors not yet defined contribute to susceptibility to reactivations.
Cytokines and chemokines are the first line of defense against viral infections [4]. High post-transplant proinflammatory cytokine levels have been associated with the risk for developing CMV infection [5]. Recent studies demonstrated that cytokine gene polymorphisms result in inter-individual differences in cytokine production [6]. To date, several groups have demonstrated that genetic differences, particularly single-nucleotide polymorphisms (SNPs), in non-HLA genes between recipients and donors influence transplant outcome and can be used as biomarkers to anticipate post-transplantation complications such as GVHD [7]. With the development of next-generation sequencing methods, an extensive genetic characterization of donor and recipient is possible in order to determine the contributions of a large number of different polymorphisms in immune-related genes to the allo-HSCT outcomes.
Numerous reports have also demonstrated that polymorphisms in different genes influence the outcome and course of infections, particularly CMV infection [8, 9]. SNPs in genes coding for cytokines or chemokines have been reported to be associated with increased risks for CMV infections [10,11,12,13,14]. Such polymorphisms may influence the rate and regulatory dynamics of gene transcription, the stability of the mRNA, and the production and biological activity of the resulting protein. Corrales et al. reported that patients carrying the CCR5 A/A genotype displayed episodes of active CMV infection with higher CMV viral load [12]. Other authors have found that heterozygosity for the toll-like receptor (TLR) 2 and TLR4 SNPs was associated with lower risk of CMV infection and lower level of viremia, respectively and, therefore, these polymorphisms appear to be protective factors in CMV replication [13]. In addition, allo-HSCT recipients that experienced active CMV infection usually present higher frequencies of the genotype T/T of the CD28 gene. Recently, the genotype of the donor-activating killer immunoglobulin-like receptor (KIR), which regulates NK cell function, has been demonstrated to influence the development of CMV infection after allo-HSCT [15]. Two studies showed that allo-HSCT recipients of donor haplotypes containing the activating KIR2DS2 and KIR2DS4 genes had a reduced risk of CMV infection compared to those who received grafts with other haplotypes [16, 17].
Therefore, there are increasing evidences indicating that polymorphisms in genes coding for cytokines or chemokines and their receptors may modulate the susceptibility to, as well as the dynamics and outcomes, of CMV infections. The present study aims to determine whether the genotype of the donor and recipient for 50 immune-system related genes influences CMV reactivation in patients receiving an allo-HSCT from an HLA-identical sibling donor.
Methods
Study design
Ninety consecutive patients who received allo-HSCT from an HLA-identical sibling donor from 2000 to 2015 at Gregorio Marañón General University Hospital (HGUGM) and with available sample for analysis were included in the study. Simultaneously, their sibling donors (n = 90) were also analyzed. The study period comprised the first 180 days following allo-HSCT. However, five patients (5.4%) who died before day 180 without CMV reactivation were excluded from the analyses. The ethics committee of HGUGM approved the study. All recipients and donors provided written informed consent according to the Declaration of Helsinki.
All patients and donors were classified according to epidemiological risk factors of clinical interest (Table 1). Graft source was unmanipulated mobilized peripheral blood stem cells (PBSCs) in most patients (80 patients, 94.1%). The conditioning regimen for allo-HSCT was myeloablative for 48 patients and reduced intensity conditioning for 37 patients, according to standard clinical practices. GVHD prophylaxis included conventional prophylaxis with Cyclosporine A (CsA) 5 mg/kg per day from day − 1 and Methotrexate (MTX) 15 mg/m2 on day + 1 and 10 mg/m2 on days + 3, + 6, and + 11.
Virological monitoring
Antiviral prophylaxis with 800 mg acyclovir twice daily since admission to 1 year after allo-HSCT was administered to every patient. After allo-HSCT and until day 180 after the infusion, all patients were monitored for CMV reactivation/infection. CMV DNAemia was evaluated twice a week during the first month after allo-HSCT and in a weekly basis thereafter until day 100. CMV assessments were performed on plasma specimen obtained from peripheral blood (PB) using a quantitative real-time PCR assay. CMV reactivation or infection was defined as the detection of CMV DNAemia (threshold level of 100 copies/mL or 155 UI/mL) in one plasma specimen. A new CMV episode was defined as CMV DNAemia detection (> 100 copies/mL) after 2 weeks PCR negativity off antiviral therapy.
Gene selection
From a predesigned 235-gene panel, we conducted a PubMed search to identify published studies reporting significant associations between one or more human genetic variants and a CMV-related phenotype. A total of 50 genes were selected for their potential role in the pathogenesis of CMV or in any viral infection (Supplementary Table 1). The selected genes correspond to immune-related genes, most of them being cytokines, chemokines and their receptors and some of them have showed association with CVM infection in previous studies.
Next generation sequencing
Genomic DNA was purified using Maxwell® RSC Blood DNA Kit (Promega, USA) from 170 PB samples obtained from 85 patients and 85 donors at the pre-transplant evaluation. DNA libraries were performed using a custom enrichment-capture gene panel according to the manufacturer’s protocol (SureSelect, Agilent). Paired-end 2 × 101 bp sequencing was performed using the HiSeq platform (Illumina, USA). FASTQ files were aligned against the human reference genome version GRCh37/hg19 using the Burrows Wheeler Alignment tool v0.7.15-r1140. Variant calling was performed using GATK version 2.8–1, VarScan algorithms and in-house scripts to combine and filter variants. Integrative Genomics Viewer (Broad Institute, USA) was used to visualize variants aligned against the reference genome to confirm the accuracy of the variant calls by checking for possible strand biases and sequencing errors.
Variant annotation and filtration
The Genesystems software (Sistemas Genómicos, Spain) was used for variant annotation, providing the infrastructure and interface for bioinformatic analysis. Transcript annotation of variants was based on all human transcripts obtained from Ensembl Released v81. We selected both SNPs and small insertions and deletions (INDELs). Algorithm for variant filtration is described in Supplementary Fig. 1. Variants located in coding regions, in splicing sites of canonical isoforms, and intronic variants were analyzed. Attending to its consequence, synonymous variants were excluded. We selected polymorphisms for which variants had a depth ≥ 30 × and a variant allele frequency (VAF) ≥ 0.4.
In addition, population databases (GenomAD and 1000 genomes) were used to the consult minor allele frequency (MAF) of each variant, in order to identify polymorphisms. We selected variants with MAF greater ≥ 10% in the European population to select polymorphisms that can be applied to routine clinical practice. Finally, Genecards and Uniprot were used to obtain gene information about the function of the encoded protein, critical domains, etc.
Statistical analysis
Patient’s characteristics were summarized by means of frequency (n) and percentage (%) for categorical variables and by means of median and range for continuous variables. Differences among groups were evaluated in univariable analysis by the Fisher Exact Test. The Statistical Package for the Social Sciences (SPSS, Chicago, USA) was used for all statistical tests except for cumulative incidence (CI) rates that were calculated using the R Statistical Software (version 3.3.2). Probability values < 0.05 were considered statistically significant.
Multiple logistic regression models were performed with the selected genetic variants that could be applied to clinical practice to anticipate CMV infection and frequency of CMV episodes. Only polymorphisms and clinical variables with p < 0.05 in the analysis were included as predictors. The predictive capacity of CMV infection of each model was evaluated using the area under the curve (AUC). Subsequently, the models with the highest AUC value and the lowest number of genetic variants used were selected. To build the risk score, the coefficients of each genetic variant obtained by the logistic model were used. Sensitivity, specificity, positive, and negative predictive values (PPV and NPV) for the different cutoff values were studied. Finally, the score obtained from the chosen predictive model was used to classify patients as low- or high-risk according to the selected cutoff point.
Results
Incidence of CMV infection and clinical risk factors
CMV reactivation was observed in 51/85 (60%) patients. In the CMV infection cohort (n = 51), 24 patients (47.1%) had only one episode of CMV reactivation, 12 (23.5%) had two different episodes, and 15 patients (29.4%) had more than two episodes within 180 days following allo-HSCT. Initial episodes of CMV reactivation (DNAemia over 100 copies/ml) occurred at a median of 48 days (2–151) after transplant. None of these clinical variants (age, gender, stem cells, hematological disease, conditioning regimen, TBI, prior autologous transplant, and CMV serostatus) were associated with CMV reactivation; only aGVHD was correlated with higher CMV reactivation (Table 1).
Additionally, the relationship between clinical variables with the occurrence of more than 2 CMV reactivations after allo-HSCT was also tested (n = 15) Only aGVHD and D/R serology were correlated. Twelve out of fifteen patients with > 2 episodes of CMV reactivation had aGVHD grades II–IV after allo-HSCT, p = 0.045; OR = 4.0 (1.0–15.4). In addition, 13 cases had D-/R + CMV serology. Six out of thirteen of these patients suffered more than 2 CMV reactivations after allo-HSCT, p = 0.009; OR = 6.0 (1.6–21.9).
Variant analysis
Using previously defined bioinformatic filters in our material and methods, 213 SNPs and INDELs were detected in 85 donor-recipient pairs (n = 170; Supplementary Table 2, Supplementary Fig. 1). Three variants were specifically detected in recipients (TGFB1 rs1989457) or donors (LTA rs1041981 and rs2229094) and 210 variants were common for both.
Association between variants in recipient and/or donor and CMV reactivation
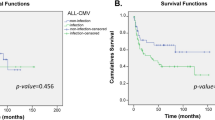
Genetic analyses for 213 selected SNPs and INDELs were correlated with the development of CMV reactivation in the first 180 days after allo-HSCT. Although 202 variants studied had no apparent impact on CMV reactivation, we found that eleven variants in seven different genes (CXCL12, IL12A, KIR3DL1, TGFB2, TNF, IL1RN, and CD48) were significantly associated with the risk or protection against CMV reactivation (Table 2, Fig. 1). In the table, we represented variants with p < 0.05 (11 variants) and p

Influence of the genotype of the patient and donor for the polymorphisms selected on the CMV reactivation after allo-HSCT
Development of a genetic risk score for CMV reactivation
In order to anticipate CMV reactivation, different predictive models were built using combinations of the 11 polymorphisms selected in the analysis. The parameters used to select the best model were the number of genetic variables and the AUC value (Supplementary Table 3). A model with five genetic polymorphisms (CXCL12 rs2839695, IL12A rs7615589, KIR3DL1 rs4554639, TGFB2 rs5781034 for the recipient and CD48 rs2295615 for the donor; Supplementary Table 4) was selected (AUC: 0.81401, 95% CI: 0.71493–0.89020, Supplementary Fig. 2). Score values, odds ratios, and coefficients assigned to each genotype in the prediction model selected are shown in Table 3. The optimal cutoff value to predict the risk of CMV infection, as derived from the analysis of the ROC curves, was defined as 0.49 (Supplementary Table 4). This cutoff value allowed to classify patients as low-risk (< 0.49) or high-risk (≥ 0.49) of suffering CMV infection at pre-transplantation. The sensitivity and specificity of the predictive model were 84.3% (95% CI: 72.0–91.8) and 67.6% (95% CI
Considering that no clinical variables were included in the pre-transplant, polymorphisms based, predictive model constructed, aGVHD grades III–IV were included to re-stratify patients classified as low-risk since this variable was an important risk factor for CMV reactivation in our cohort. Specifically, 4 patients stratified as low-risk of CMV infection (< 0.49) with the pre-transplantation predictive model who suffered grades III–IV aGVHD (Supplementary Table 5) were re-stratified as high-risk. Then, we calculated the CMV risk score for each patient (defined by score value ≥ 0.49 or grade III/IV GVHD) to test the usefulness of the model to identify patients at high-risk of experiencing a CMV reactivation after allo-HSCT. Out of the 85 participants, 58 (68.2%) were classified as high-risk and 27 (31.8%) as low-risk. At 100 days after allo-HSCT, 70.7% of patients with a high-risk score experienced CMV reactivation compared to 14.8% of those with a low-risk score (Fig. 2). Therefore, the CMV predictive score was able to correctly stratify patients according to their risk of developing CMV reactivation (p

Stratification of the whole cohort of patients according to the risk of CMV reactivation. Risk was calculated using the proposed predictive model which includes five genetic polymorphisms and the cutoff value to predict the risk of CMV infection used was 0.49
We also calculated the CMV risk score for only R + patient. Out of the 73 patients, 46 were classified as high risk and 27 as low risk. At 180 days after allo-HSCT, 78.2% of patients with a high-risk score experienced CMV reactivation compared to 29.6% of those with a low-risk score. Therefore, the CMV predictive score was able to correctly stratify patients according to their risk of developing CMV reactivation in R + cohort.
Discussion
CMV remains as one of the most common clinically significant viral infections after allo-HSCT. Over the past decade, most centers have adopted a preemptive strategy in which CMV surveillance and detection in blood by different methods trigger antiviral treatment to prevent clinical CMV disease and minimize the toxic effects of these antiviral agents [18]. In recent years, gene-polymorphism studies have shown clinical utility in different settings. Polymorphisms in certain cytokine and chemokine genes showed significant association with the presentation of viral infections in allo-HSCT recipients, essentially with CMV infection [19,20,21,22,23]. Recently, Casto A et al. suggested that the G allele for rs1045642 in ABCB1 in donors reduces the risk of CMV reactivation by approximately 20%, because these are related with lower intracellular calcineurin inhibitor concentrations [24].
Our data suggest a significantly lower risk of CMV reactivation in recipients and donors with particular cytokine loci variants. In particular, our data showed that the TNF-α SNP rs3093662 (GA/GG) genotype in donors and recipients was associated with protection from CMV infection after allo-HSCT. The presence of allele G in this position has been associated with an increase in transcriptional activity and high in vitro production of TNF-α [25]. CMV is a potent inducer of TNF-α production but this cytokine has direct antiviral effects and, together with IL1, potentiates the lytic activity of NK cells [26]. On other hand, IL12 plays a crucial role in anti-infectious immune responses, especially by stimulating IFNγ production [27]. Hoffmann TW et al. investigated the 3´UTR polymorphism (rs3212227) of the IL12p40 gene and shown that the C allele is associated with lower production of the p40 polypeptide, which might explain the pathogenic link between the IL12B genotype and CMV replication [28]. In contrast to Hoffmann et al., we noted an association between the IL12A polymorphisms rs7615589 and rs2243123 in the recipient and protection against CMV infection. Although TGFB1 alleles were found to be significantly associated with CMV reactivation, our data also suggest a significantly lower risk of CMV reactivation in patients with TGFB2 rs5781034 SNP. This gene encodes a secreted ligand of the TGF-beta superfamily of proteins. TGFβ-2 protein helps control the growth, differentiation and proliferation of cells.
On the other hand, we demonstrated that CD48 rs2295615 (GC/GG) and IL1RN rs439154 (AG/AA) in the donor was associated with protection for CMV reactivation. However, the exact mechanism by which these SNPs exert their activity is not well established. These could culminate in trans-activation of a repertoire of pro-inflammatory cytokines that would favor the elimination of the infectious agents. Individuals homozygous for specific alleles for the IL1RN gene present a high prolonged and severe proinflammatory immune response [29]. In our cohort, IL1RN rs439154 (AG/AA) in the donor was also associated with protection from CMV reactivation. Hurme M et al. have shown that the number of 86-bp tandem repeats in intron 2 of the IL1RN gene are associated with an increased inflammatory response provoked by an infectious disease [30]. In conclusion, our data suggested that particular polymorphisms in cytokine genes such as TNF-α, LTA, IL12, TGBF2, and IL1RN could play an important role in increasing pro-inflammatory cytokine expression in T cells and NK cells, which might be beneficial in protection against CMV infection. On the other hand, our data showed that the CXCL12 rs2839695 (GG/AG) SNP in the recipient was associated with an increased risk of CMV infection. An increased expression of CXCL12 would promote local inflammatory responses that trigger CMV reactivation and induce CMV replication. CXCL12 signaling was amplified in CMV-infected cells [31].
Most of the KIR genes exhibit allelic polymorphism and this serves to diversify KIR haplotypes, which define different levels of cell surface expression. Several authors reported associations between the expression of KIR genes in allo-HSCT donors and patients and the risk of CMV infection [32, 33]. De Rham et al. explored the expression of KIR3DL1 on NK cells during acute CMV infection and showed high levels of expression, which implies that this receptor could be involved in clearing CMV infection [34]. Our analysis showed a significantly higher risk of CMV reactivation in recipients with KIR3DL1 rs45542639 (AG/AA), rs149123986 (GA/GG), rs144994606 (AG/AA), and rs143159382 (TC/TT). However, the exact mechanism by which these SNPs influence KIR3DL1 expression is not well established, so an extensive study would better elucidate whether these KIR3DL1 genetic variants may modify the risk of CMV infections after allo-HSCT.
Finally, with the objective of increasing the clinical utility of our results, we built the best-possible predictive model with our data to improve the prediction of CMV reactivation after allo-HSCT. Our results suggest that particular SNPs in recipient and donors (pre-transplantation model), in combination with the development of grades III-IV aGVHD, could predict CMV reactivation/infection. The model proposed can be readily applied by other centers using the predictive score built in this study.
Regarding clinical variables, important risk factors for CMV infections are the serological status of donor and recipient, aGVHD, and T-cell depletion. It has been established that CMV seropositive patients show a significantly higher incidence of CMV infection than CMV-seronegative recipients [35]. Our study is unique in that CMV serologic status did not influence the rate of CMV infection. The presented cohort has 85% of R + patients, so only a minority of patients was CMV-seronegative recipients. It is possible that the impact of CMV serostatus will vary by increasing the number of R- and, consequently, CMV serostatus may include in the CMV predictive score. In our cohort, CMV D-/R + serologic status only influenced the occurrence of more than two CMV reactivation episodes. According to other authors, we also confirmed a strong association between aGVHD (grades II/IV and III/IV) and CMV infections [36].
To our knowledge, this is the first preliminary report contributing a pre-transplant genetic risk score useful to identify patients at high-risk of experiencing a CMV reactivation after allo-HSCT. This approach could facilitate personalized risk-adapted clinical management of patients undergoing allo-HSCT. Nevertheless, the study reported in this manuscript is exploratory and cannot be considered complete until further work is done to verify the performance of the model in an independent cohort. As it stands, the predictive model was optimized to fit the observed data in a relatively small cohort. Therefore, the results should be considered preliminary and an external validation is needed. In addition, these polymorphisms should be validated in a larger cohort and in other hematopoietic stem cell transplant settings such as the currently widely used haploidentical stem cell transplantation with post-transplant cyclophosphamide (PT/Cy) based GVHD prophylaxis. Retrospective studies of allo-HSCT with PT/Cy have shown a high incidence of CMV viremia and large collaborative studies in PT/Cy patients would elucidate whether genetic variants impact viral infections in this setting [37].
In summary, the data presented here suggest that screening of patients and donors’ pre-transplantation helps to predict the individual risk of the development of CMV infection and disease after an HLA-identical allo-HSCT. These results might also allow the identification of patients at high-risk of CMV reactivation after transplantation, enabling pre-emptive therapy or attempts to cure the infection by administering antiviral therapy or CMV-specific T lymphocytes.
References
Yong MK, Ananda-Rajah M, Cameron PU et al (2017) Cytomegalovirus reactivation is associated with increased risk of late-onset invasive fungal disease after allogeneic hematopoietic stem cell transplantation: a multicenter study in the current era of viral load monitoring. Biol Blood Marrow Transplant 23:1961–1967. https://doi.org/10.1016/j.bbmt.2017.07.025
Ciáurriz M, Beloki L, Zabalza A et al (2017) Functional specific-T-cell expansion after first cytomegalovirus reactivation predicts viremia control in allogeneic hematopoietic stem cell transplant recipients. Transplant Infect Dis 19. https://doi.org/10.1111/tid.12778
Widmann T, Sester U, Gärtner BC et al (2008) Levels of CMV specific CD4 T cells are dynamic and correlate with CMV viremia after allogeneic stem cell transplantation. PLoS ONE 3:e3634. https://doi.org/10.1371/journal.pone.0003634
McSharry B, Avdic S, Slobedman B (2012) Human Cytomegalovirus encoded homologs of cytokines, chemokines and their receptors: roles in immunomodulation. Viruses 4:2448–2470. https://doi.org/10.3390/v4112448
Kato T, Nishida T, Ito Y et al (2014) Correlations of programmed death 1 expression and serum IL-6 level with exhaustion of cytomegalovirus-specific T cells after allogeneic hematopoietic stem cell transplantation. Cell Immunol 288:53–59. https://doi.org/10.1016/j.cellimm.2014.02.007
Mitsani D, Nguyen MH, Girnita DM et al (2011) A polymorphism linked to elevated levels of interferon-γ is associated with an increased risk of cytomegalovirus disease among Caucasian lung transplant recipients at a single center. J Heart Lung Transplant 30:523–529. https://doi.org/10.1016/j.healun.2010.11.008
Martínez-Laperche C, Buces E, Aguilera-Morillo MC et al (2018) A novel predictive approach for GVHD after allogeneic SCT based on clinical variables and cytokine gene polymorphisms. Blood Adv 2:1719–1737. https://doi.org/10.1182/bloodadvances.2017011502
Cano P, Han FS, Wang H-L et al (2012) Cytokine gene polymorphisms affect reactivation of cytomegalovirus in patients with cancer. Cytokine 60:417–422. https://doi.org/10.1016/j.cyto.2012.07.018
Kielsen K, Enevold C, Heilmann C et al (2018) Donor genotype in the interleukin-7 receptor α-chain predicts risk of graft-versus-host disease and cytomegalovirus infection after allogeneic hematopoietic stem cell transplantation. Front Immunol 9:109. https://doi.org/10.3389/fimmu.2018.00109
Annibali O, Piccioni L, Tomarchio V et al (2018) Impact of IFN lambda 3/4 single nucleotide polymorphisms on the cytomegalovirus reactivation in autologous stem cell transplant patients. PLoS ONE 13:e0200221. https://doi.org/10.1371/journal.pone.0200221
Mezger M, Steffens M, Semmler C et al (2008) Investigation of promoter variations in dendritic cell-specific ICAM3-grabbing non-integrin (DC-SIGN) (CD209) and their relevance for human cytomegalovirus reactivation and disease after allogeneic stem-cell transplantation. Clin Microbiol Infect 14:228–234. https://doi.org/10.1111/j.1469-0691.2007.01902.x
Corrales I, Giménez E, Solano C et al (2015) Incidence and dynamics of active cytomegalovirus infection in allogeneic stem cell transplant patients according to single nucleotide polymorphisms in donor and recipient CCR5, MCP-1, IL-10, and TLR9 genes. J Med Virol 87:248–255. https://doi.org/10.1002/jmv.24050
Carvalho A, Cunha C, Carotti A et al (2009) Polymorphisms in toll-like receptor genes and susceptibility to infections in allogeneic stem cell transplantation. Exp Hematol 37:1022–1029. https://doi.org/10.1016/j.exphem.2009.06.004
Sellathamby S, Lakshmi KM, Busson M et al (2012) Polymorphisms in the immunoregulatory genes are associated with hematopoietic recovery and increased susceptibility to bacterial infections in patients with thalassaemia major undergoing matched related hematopoietic stem cell transplantation. Biol Blood Marrow Transplant 18:1219–1226. https://doi.org/10.1016/j.bbmt.2012.01.011
Sobecks RM, Askar M, Thomas D et al (2011) Cytomegalovirus reactivation after matched sibling donor reduced-intensity conditioning allogeneic hematopoietic stem cell transplant correlates with donor killer immunoglobulin-like receptor genotype. Exp Clin Transplant 9:7–13
Zaia JA, Sun JY, Gallez-Hawkins GM et al (2009) The effect of single and combined activating killer immunoglobulin-like receptor genotypes on cytomegalovirus infection and immunity after hematopoietic cell transplantation. Biol Blood Marrow Transplant 15:315–325. https://doi.org/10.1016/j.bbmt.2008.11.030
Gallez-Hawkins GM, Franck AE, Li X et al (2011) Expression of activating KIR2DS2 and KIR2DS4 genes after hematopoietic cell transplantation: relevance to cytomegalovirus infection. Biol Blood Marrow Transplant 17:1662–1672. https://doi.org/10.1016/j.bbmt.2011.04.008
Ljungman P, Boeckh M, Hirsch HH et al (2017) Definitions of cytomegalovirus infection and disease in transplant patients for use in clinical trials: table 1. Clin Infect Dis 64:87–91. https://doi.org/10.1093/cid/ciw668
Boeckh M, Ljungman P (2009) How we treat cytomegalovirus in hematopoietic cell transplant recipients. Blood 113:5711–5719. https://doi.org/10.1182/blood-2008-10-143560
Bogunia-Kubik K, Jaskula E, Lange A (2007) The presence of functional CCR5 and EBV reactivation after allogeneic haematopoietic stem cell transplantation. Bone Marrow Transplant 40:145–150. https://doi.org/10.1038/sj.bmt.1705703
Karimi MH, Motazedian M, Geramizadeh B et al (2011) Association of the co-stimulatory molecules polymorphisms with CMV infection in liver transplant recipients. Int J Organ Transplant Med 2:171–177
Jaskula E, Dlubek D, Duda D et al (2009) Interferon gamma 13-CA-repeat homozygous genotype and a low proportion of CD4+ lymphocytes are independent risk factors for cytomegalovirus reactivation with a high number of copies in hematopoietic stem cell transplantation recipients. Biol Blood Marrow Transplant 15:1296–1305. https://doi.org/10.1016/j.bbmt.2009.06.008
Loeffler J, Steffens M, Arlt E-M et al (2006) Polymorphisms in the genes encoding chemokine receptor 5, interleukin-10, and monocyte chemoattractant protein 1 contribute to cytomegalovirus reactivation and disease after allogeneic stem cell transplantation. J Clin Microbiol 44:1847–1850. https://doi.org/10.1128/JCM.44.5.1847-1850.2006
A C, S S, D L, et al (2021) Genetic variants associated with cytomegalovirus infection after allogeneic hematopoietic cell transplantation. Blood. https://doi.org/10.1182/BLOOD.2021012153
Kroeger KM, Carville KS, Abraham LJ (1997) The -308 tumor necrosis factor-alpha promoter polymorphism effects transcription. Mol Immunol 34:391–399
Zheng Q, Tao R, Gao H et al (2012) HCMV-encoded UL128 enhances TNF-α and IL-6 expression and promotes PBMC proliferation through the MAPK/ERK pathway in vitro. Viral Immunol 25:98–105. https://doi.org/10.1089/vim.2011.0064
Popescu I, Pipeling MR, Mannem H et al (2016) IL-12–Dependent cytomegalovirus-specific CD4 + T cell proliferation, T-bet induction, and effector multifunction during primary infection are key determinants for early immune control. J Immunol 196:877–890. https://doi.org/10.4049/jimmunol.1501589
Hoffmann TW, Halimi J-M, Büchler M et al (2008) Association between a polymorphism in the IL-12p40 gene and cytomegalovirus reactivation after kidney transplantation. Transplantation 85:1406–1411. https://doi.org/10.1097/TP.0b013e31816c7dc7
Witkin SS, Gerber S, Ledger WJ (2002) Influence of interleukin-1 receptor antagonist gene polymorphism on disease
Hurme M, Helminen M (1998) Resistance to human cytomegalovirus infection may be influenced by genetic polymorphisms of the tumour necrosis factor-alpha and interleukin-1 receptor antagonist genes. Scand J Infect Dis 30:447–449
Tu CC, Arnolds KL, O’Connor CM, Spencer JV (2017) Human cytomegalovirus UL111A and US27 gene products enhance the CXCL12/CXCR4 signaling axis via distinct mechanisms. J Virol 92. https://doi.org/10.1128/JVI.01981-17
Foley BA, Santis DD, Van Beelen E et al (2008) The reactivity of Bw4+ HLA-B and HLA-A alleles with KIR3DL1: implications for patient and donor suitability for haploidentical stem cell transplantations. Blood 112:435–443. https://doi.org/10.1182/blood-2008-01-132902
Beziat V, Liu LL, Malmberg J-A et al (2013) NK cell responses to cytomegalovirus infection lead to stable imprints in the human KIR repertoire and involve activating KIRs. Blood 121:2678–2688. https://doi.org/10.1182/blood-2012-10-459545
de Rham C, Hadaya K, Bandelier C et al (2014) Expression of killer cell immunoglobulin-like receptors (KIRs) by natural killer cells during acute CMV infection after kidney transplantation. Transpl Immunol 31:157–164. https://doi.org/10.1016/j.trim.2014.08.002
Boeckh M, Nichols WG (2004) The impact of cytomegalovirus serostatus of donor and recipient before hematopoietic stem cell transplantation in the era of antiviral prophylaxis and preemptive therapy. Blood 103:2003–2008
Özdemir E, Saliba RM, Champlin RE et al (2007) Risk factors associated with late cytomegalovirus reactivation after allogeneic stem cell transplantation for hematological malignancies. Bone Marrow Transplant 40:125–136. https://doi.org/10.1038/sj.bmt.1705699
Slade M, Goldsmith S, Romee R et al (2017) Epidemiology of infections following haploidentical peripheral blood hematopoietic cell transplantation. Transpl Infect Dis 19:e12629. https://doi.org/10.1111/tid.12629
Acknowledgements
The authors would also like to acknowledge the patients who participated in this study, as well as the staff of the Hematology Department, Hospital General Universitario Gregorio Marañón (Madrid, Spain), who made the study possible.
Funding
This study was partially supported by the Ministry of Economy and Competitiveness ISCIII-FIS Grants PI14/01731 and PI17/01880 and co-financed by the European Regional Development Fund from the European Commission, the “A way of making Europe” initiative, as well as grants from Fundación LAIR and Asociación Madrileña de Hematología y Hemoterapia.
Author information
Authors and Affiliations
Contributions
M.V., P.M., D.C., M.C., C.M.-L., and I.B were responsible for conception and design; M.K., L.S., R.B., N.D., J.L.D.-M. provided patients and samples; M.V., P.M., D.C., M.C., J.S-G., P.C., N.R., C.M.-L., and I.B. collected and assembled data; M.V., J.S-G., J.M.B., J.C.T., D.G., C.M.-L., and I.B. were responsible for data analysis and interpretation; M.V., C.M.-L., and I.B. wrote the manuscript; and all authors gave final approval of the manuscript and are accountable for all aspects of the work.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Vallejo, M., Muñiz, P., Kwon, M. et al. Risk prediction of CMV reactivation after allogeneic stem cell transplantation using five non-HLA immunogenetic polymorphisms. Ann Hematol 101, 1567–1576 (2022). https://doi.org/10.1007/s00277-022-04841-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00277-022-04841-8