
Abstract
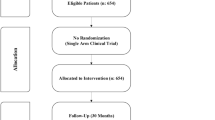
Over the past 20 years, hydroxyurea (HU) has emerged as an effective therapeutic agent in thalassemic patients to improve anemia and decrease the transfusion dependency. We evaluated long-term safety and clinical response to HU in patients with non-transfusion-dependent β-thalassemia (NTDT). In this retrospective study, medical records of 181 patients with NTDT were evaluated during October to December 2020 in Southern Iran. No requirement to blood transfusion was considered as sustained transfusion independence response. All patients were regularly examined and monitored for the occurrence of any adverse event (AE) of HU. The mean duration of HU consumption ± SD was 18.2 ± 4.0 (8–22) years. Overall, 149 patients (82.3%) had sustained transfusion independence response. β-globin gene mutations and XmnI polymorphisms were not significantly associated with clinical response (P > 0.05). Mild and transient AEs were reported in 60 patients (33%) with no requirement to drug interruption. Hydroxyurea with the dose of 8–15 mg/kg can be used as a safe and effective treatment in NTDT patients. It was well tolerated in long term without any serious complication or secondary malignancy. No relationship between XmnI or β-globin gene mutations with HU response was observed in this geographic area of the world.
Similar content being viewed by others


Data availability statement
The data presented in this study are available on request from the corresponding author.
References
Cao A, Galanello R (2010) Beta-thalassemia. Genet Med 12(2):61–76
Taher AT, Musallam KM, Cappellini MD (2021) β-Thalassemias. N Engl J Med 384(8):727–743
Karimi M, Cohan N, De Sanctis V, Mallat NS, Taher A (2014) Guidelines for diagnosis and management of Beta-thalassemia intermedia. Pediatr Hematol Oncol 31(7):583–596. https://doi.org/10.3109/08880018.2014.937884
Musallam KM, Cappellini MD, Daar S, Taher AT (2021) Morbidity-free survival and hemoglobin level in non-transfusion-dependent β-thalassemia: a 10-year cohort study. Ann Hematol. https://doi.org/10.1007/s00277-020-04370-2
Musallam KM, Cappellini MD, Taher AT (2021) Variations in hemoglobin level and morbidity burden in non-transfusion-dependent β-thalassemia. Ann Hematol 100(7):1903–1905
Algiraigri AH, Wright NAM, Paolucci EO, Kassam A (2017) Hydroxyurea for nontransfusion-dependent β-thalassemia: a systematic review and meta-analysis. Hematol Oncol Stem Cell Ther 10(3):116–125. https://doi.org/10.1016/j.hemonc.2017.02.002
Pourfarzad F, von Lindern M, Azarkeivan A, Hou J, Kia SK, Esteghamat F, van IJcken W, Philipsen S, Najmabadi H, Grosveld F (2013) Hydroxyurea responsiveness in β-thalassemic patients is determined by the stress response adaptation of erythroid progenitors and their differentiation propensity. Haematologica 98(5):696–704
Banan M (2013) Hydroxyurea treatment in β-thalassemia patients: to respond or not to respond? Ann Hematol 92(3):289–299
Ronchi A, Ottolenghi S (2013) To respond or not to respond to hydroxyurea in thalassemia: a matter of stress adaptation? Haematologica 98(5):657
Kosaryan M, Zafari M, Alipur A, Hedayatizadeh-Omran A (2014) The effect and side effect of hydroxyurea therapy on patients with β-thalassemia: a systematic review to December 2012. Hemoglobin 38(4):262–271. https://doi.org/10.3109/03630269.2014.927770
Keikhaei B, Yousefi H, Bahadoram M (2015) Clinical and haematological effects of hydroxyurea in β-thalassemia intermedia patients. J Clin Diagn Res 9(10):Om01-03. https://doi.org/10.7860/jcdr/2015/14807.6660
Algiraigri AH, Kassam A (2017) Hydroxyurea for hemoglobin E/β-thalassemia: a systematic review and meta-analysis. Int J Hematol 106(6):748–756. https://doi.org/10.1007/s12185-017-2307-0
Ehsani MA, Hedayati-Asl AA, Bagheri A, Zeinali S, Rashidi A (2009) Hydroxyurea-induced hematological response in transfusion-independent beta-thalassemia intermedia: case series and review of literature. Pediatr Hematol Oncol 26(8):560–565. https://doi.org/10.3109/08880010903271671
Musallam KM, Taher AT, Cappellini MD, Sankaran VG (2013) Clinical experience with fetal hemoglobin induction therapy in patients with β-thalassemia. Blood 121(12):2199–2212. https://doi.org/10.1182/blood-2012-10-408021 (quiz 2372)
Menzel S, Thein SL (2009) Genetic architecture of hemoglobin F control. Curr Opin Hematol 16(3):179–186. https://doi.org/10.1097/moh.0b013e328329d07a
Lettre G, Sankaran VG, Bezerra MA, Araújo AS, Uda M, Sanna S, Cao A, Schlessinger D, Costa FF, Hirschhorn JN, Orkin SH (2008) DNA polymorphisms at the BCL11A, HBS1L-MYB, and beta-globin loci associate with fetal hemoglobin levels and pain crises in sickle cell disease. Proc Natl Acad Sci U S A 105(33):11869–11874. https://doi.org/10.1073/pnas.0804799105
Sedgewick AE, Timofeev N, Sebastiani P, So JCC, Ma ESK, Chan LC, Fucharoen G, Fucharoen S, Barbosa CG, Vardarajan BN, Farrer LA, Baldwin CT, Steinberg MH, Chui DHK (2008) BCL11A is a major HbF quantitative trait locus in three different populations with beta-hemoglobinopathies. Blood Cells Mol Dis 41(3):255–258. https://doi.org/10.1016/j.bcmd.2008.06.007
Banan M, Bayat H, Azarkeivan A, Mohammadparast S, Kamali K, Farashi S, Bayat N, Khani MH, Neishabury M, Najmabadi H (2012) The XmnI and BCL11A single nucleotide polymorphisms may help predict hydroxyurea response in Iranian β-thalassemia patients. Hemoglobin 36(4):371–380. https://doi.org/10.3109/03630269.2012.691147
Karimi M, Haghpanah S, Farhadi A, Yavarian M (2012) Genotype-phenotype relationship of patients with β-thalassemia taking hydroxyurea: a 13-year experience in Iran. Int J Hematol 95(1):51–56. https://doi.org/10.1007/s12185-011-0985-6
Chalikiopoulou C, Tavianatou AG, Sgourou A, Kourakli A, Kelepouri D, Chrysanthakopoulou M, Kanelaki VK, Mourdoukoutas E, Siamoglou S, John A, Symeonidis A, Ali BR, Katsila T, Papachatzopoulou A, Patrinos GP (2016) Genomic variants in the ASS1 gene, involved in the nitric oxide biosynthesis and signaling pathway, predict hydroxyurea treatment efficacy in compound sickle cell disease/β-thalassemia patients. Pharmacogenomics 17(4):393–403. https://doi.org/10.2217/pgs.16.1
Kazazian HH Jr, Boehm CD (1988) Molecular basis and prenatal diagnosis of beta-thalassemia. Blood 72(4):1107–1116
Najmabad H, Teymourian S, Jalilnezhad S, Azad M, Khatibi T, Neyshabouri M, Pourfarzad F, Oberkanins C, Krugluger W (2001) Amplification Refractory Mutation System (ARMS) and reverse hybridization in the detection of beta-thalassemia mutations. Arch Iran Med 4(4):165–170
Hosseinpour Feizi MA, Hosseinpour Feizi AA, Pouladi N, Haghi M, Azarfam P (2008) Molecular spectrum of beta-thalassemia mutations in Northwestern Iran. Hemoglobin 32(3):255–261. https://doi.org/10.1080/03630260802004145
Motovali-Bashi M, Ghasemi T (2015) Role of XmnIgG polymorphism in hydroxyurea treatment and fetal hemoglobin level at Isfahanian intermediate β-thalassemia patients. Iran Biomed J 19(3):177–182. https://doi.org/10.7508/ibj.2015.03.008
Betts M, Flight PA, Paramore LC, Tian L, Milenković D, Sheth S (2020) Systematic literature review of the burden of disease and treatment for transfusion-dependent β-thalassemia. Clin Ther 42(2):322-337.e322. https://doi.org/10.1016/j.clinthera.2019.12.003
Asadov C, Alimirzoeva Z, Mammadova T, Aliyeva G, Gafarova S, Mammadov J (2018) β-Thalassemia intermedia: a comprehensive overview and novel approaches. Int J Hematol 108(1):5–21. https://doi.org/10.1007/s12185-018-2411-9
Zohaib M, Ansari SH, Shamsi TS, Zubarev RA, Zarina S (2019) Pharmacoproteomics profiling of plasma from β-thalassemia patients in response to hydroxyurea treatment. J Clin Pharmacol 59(1):98–106. https://doi.org/10.1002/jcph.1297
Iqbal A, Ansari SH, Parveen S, Khan IA, Siddiqui AJ, Musharraf SG (2018) Hydroxyurea treated β-thalassemia children demonstrate a shift in metabolism towards healthy pattern. Sci Rep 8(1):15152. https://doi.org/10.1038/s41598-018-33540-6
Karimi M, Darzi H, Yavarian M (2005) Hematologic and clinical responses of thalassemia intermedia patients to hydroxyurea during 6 years of therapy in Iran. J Pediatr Hematol Oncol 27(7):380–385. https://doi.org/10.1097/01.mph.0000174386.13109.28
Bohara VV, Ray S, Chakrabarti P, Ray SS, Nath UK, Chaudhuri U (2014) Optimizing the dose of hydroxyurea therapy for patients with β-thalassemia intermedia (Hb E-β-thalassemia): a single center study from Eastern India. Hemoglobin 38(1):44–48. https://doi.org/10.3109/03630269.2013.845844
Karimi M, Cohan N, Mousavizadeh K, Falahi MJ, Haghpanah S (2010) Adverse effects of hydroxyurea in beta-thalassemia intermedia patients: 10 years′ experience. Pediatr Hematol Oncol 27(3):205–211. https://doi.org/10.3109/08880011003639952
Bayanzay K, Khan R (2015) Meta-analysis on effectiveness of hydroxyurea to treat transfusion-dependent beta-thalassemia. Hematology 20(8):469–476. https://doi.org/10.1179/1607845414Y.0000000222
Alebouyeh M, Moussavi F, Haddad-Deylami H, Vossough P (2004) Hydroxyurea in the treatment of major beta-thalassemia and importance of genetic screening. Ann Hematol 83(7):430–433. https://doi.org/10.1007/s00277-003-0836-5
Italia KY, Jijina FJ, Merchant R, Panjwani S, Nadkarni AH, Sawant PM, Nair SB, Ghosh K, Colah RB (2009) Response to hydroxyurea in beta thalassemia major and intermedia: experience in western India. Clin Chim Acta 407(1–2):10–15. https://doi.org/10.1016/j.cca.2009.06.019
Tafrali C, Paizi A, Borg J, Radmilovic M, Bartsakoulia M, Giannopoulou E, Giannakopoulou O, Stojiljkovic-Petrovic M, Zukic B, Poulas K, Stavrou EF, Lambropoulou P, Kourakli A, Felice AE, Papachatzopoulou A, Philipsen S, Pavlovic S, Georgitsi M, Patrinos GP (2013) Genomic variation in the MAP3K5 gene is associated with β-thalassemia disease severity and hydroxyurea treatment efficacy. Pharmacogenomics 14(5):469–483. https://doi.org/10.2217/pgs.13.31
Gravia A, Chondrou V, Kolliopoulou A, Kourakli A, John A, Symeonidis A, Ali BR, Sgourou A, Papachatzopoulou A, Katsila T, Patrinos GP (2016) Correlation of SIN3A genomic variants with β-hemoglobinopathies disease severity and hydroxyurea treatment efficacy. Pharmacogenomics 17(16):1785–1793. https://doi.org/10.2217/pgs-2016-0076
Borg J, Phylactides M, Bartsakoulia M, Tafrali C, Lederer C, Felice AE, Papachatzopoulou A, Kourakli A, Stavrou EF, Christou S, Hou J, Karkabouna S, Lappa-Manakou C, Ozgur Z, van Ijcken W, von Lindern M, Grosveld FG, Georgitsi M, Kleanthous M, Philipsen S, Patrinos GP (2012) KLF10 gene expression is associated with high fetal hemoglobin levels and with response to hydroxyurea treatment in β-hemoglobinopathy patients. Pharmacogenomics 13(13):1487–1500. https://doi.org/10.2217/pgs.12.125
Kolliopoulou A, Siamoglou S, John A, Sgourou A, Kourakli A, Symeonidis A, Vlachaki E, Chalkia P, Theodoridou S, Ali BR, Katsila T, Patrinos GP, Papachatzopoulou A (2019) Role of genomic biomarkers in increasing fetal hemoglobin levels upon hydroxyurea therapy and in β-thalassemia intermedia: a validation cohort study. Hemoglobin 43(1):27–33. https://doi.org/10.1080/03630269.2019.1597732
Karimi M, Zarei T, Haghpanah S, Moghadam M, Ebrahimi A, Rezaei N, Heidari G, Vazin A, Khavari M, Miri HR (2017) Relationship between some single-nucleotide polymorphism and response to hydroxyurea therapy in iranian patients with β-thalassemia intermedia. J Pediatr Hematol Oncol 39(4):e171–e176. https://doi.org/10.1097/mph.0000000000000779
Acknowledgements
We would like to thank Shiraz University of Medical Sciences for their approval support.
Funding
This research was funded by the Research Vice-Chancellor of Shiraz University of Medical Sciences, grant number (21488).
Author information
Authors and Affiliations
Contributions
Conceptualization, M.K.; methodology, S.H., T.Z.; data curation: T. Z., A.A; software, S.H. A.B; validation, S.H.; formal analysis, S.H., A.B., A.A., and S.D.; writing original draft preparation, S.H. and T.Z.; writing review and editing, M.K. and S.H; supervision, M.K. and S.H.; project administration, M.K. and S.H.; funding acquisition, S.H. All authors have read and agreed to the published version of the manuscript. All authors have contributed substantially to the work reported.
Corresponding author
Ethics declarations
Institutional review board statement
The study was conducted according to the guidelines of the Declaration of Helsinki, and approved by the Ethics Committee of Shiraz University of Medical Sciences (protocol code IR.SUMS.REC.1399.805 on September 23th, 2020).
Informed consent statement
Written informed consent has been obtained from the patients to publish this paper.
Conflict of interest
The authors declare no competing interests.
Disclaimer
The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Karimi, M., Zarei, T., Bahmanimehr, A. et al. Long-term safety and efficacy of hydroxyurea in patients with non-transfusion-dependent β-thalassemia: a comprehensive single-center experience. Ann Hematol 100, 2901–2907 (2021). https://doi.org/10.1007/s00277-021-04627-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00277-021-04627-4