
Abstract
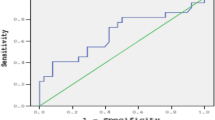
Since iron overload is the commonest cause of morbidity and mortality in β thalassemia major (β-TM), it represents one major target in therapeutic management of the disease. The recently discovered erythroid regulator, erythroferrone (ERFE), governed by high levels of erythropoietin, was found to suppress hepcidin expression, thus increasing iron availability for developing erythroid progenitors. We aimed to investigate ERFE levels in Egyptian β-TM patients as an attempt to understand its role in the prediction of iron overload states. Our study included 70 β-TM patients, divided into two subgroups according to the degree of iron overload, and 30 sex and age-matched healthy subjects. ERFE gene expression was analyzed by quantitative real-time polymerase chain reaction (qRT-PCR), and serum hepcidin was measured using enzyme-linked immunosorbent assay (ELISA) technique. Both ERFE gene expression levels and transferrin saturation (TS%) values were able to discriminate among cases with different degrees of iron overload, in contrast to hepcidin. TS% was acknowledged as the best predictor of iron overload (AUC 0.893) in comparison with serum hepcidin and ERFE gene levels (AUC 0.807 and 0.677, respectively), and ERFE gene expression was an independent predictor for the estimated TS%. In conclusion, we suggest that using the ERFE gene expression, combined with serum hepcidin estimation, can substantiate the role of estimated TS% as a promising tool in screening for iron overload in β-TM patients.


Similar content being viewed by others


References
Origa R (2017) Beta-thalassemia. Genetics in Medicine 19:609–619
Adly A, Ebeid F (2015) Cultural preferences and limited public resources influence the spectrum of thalassemia in Egypt. J Pediatr Hematol Oncol 37(4):281–284
Sherief L, Abd El-Salam S, Kamal N et al (2014) Nutritional biomarkers in children and adolescents with beta-thalassemia-major: an Egyptian center experience. Biomed Res Int 2014:261761
El-Shanshory M, Hagag A, Shebl S et al (2014) Spectrum of beta globin gene mutations in Egyptian children with β-thalassemia. Mediterr J Hematol Infect Dis 6(1):e2014071
Elmezayen A, Kotb S, Sadek N et al (2015) β-Globin mutations in Egyptian patients with β-thalassemia. Lab Med 46(1):8–13
Hoffbrand A, Taher A, Cappellini M (2012) How I treat transfusional iron overload. Blood. 120(18):3657–3669
Olivieri N, Nathan D, MacMillan J, Wayne AS, Liu PP, McGee A, Martin M, Koren G, Cohen AR (1994) Survival in medically treated patients with homozygous beta-thalassemia. N Engl J Med 331:574–578
Telfer PT, Prestcott E, Holden S, Walker M, Hoffbrand AV, Wonke B (2000) Hepatic iron concentration combined with long-term monitoring of serum ferritin to predict complications of iron overload in thalassaemia major. Br J Haematol 110:971–977
Puliyel M, Sposto R, Berdoukas V, Hofstra TC, Nord A, Carson S, Wood J, Coates TD (2014) Ferritin trends do not predict changes in total body iron in patients with transfusional iron overload. Am J Hematol 89(4):391–394
Musallam K, Cappellini M, Wood J, Motta I, Graziadei G, Tamim H, Taher AT (2011) Elevated liver iron concentration is a marker of increased morbidity in patients with beta thalassemia intermedia. Haematologica 96(11):1605–1612
Wood J (2014) Guidelines for quantifying iron overload. ASH Education Book 1:210–215
Jacobs E, Hendriks J, van Tits B, Evans PJ, Breuer W, Liu DY, Jansen EH, Jauhiainen K, Sturm B, Porter JB, Scheiber-Mojdehkar B, von Bonsdorff L, Cabantchik ZI, Hider RC, Swinkels DW (2005) Results of an international round robin for the quantification of serum non-transferrin-bound iron: need for defining standardization and a clinically relevant isoform. Anal Biochem 341(2):241–250
Wood J (2007) Diagnosis and management of transfusion iron overload: the role of imaging. Am J Hematol 82(12 Suppl):1132–1135
Kautz L, Jung G, Valore E, Rivella S, Nemeth E, Ganz T (2014) Identification of erythroferrone as an erythroid regulator of iron metabolism. Nat Genet 46(7):678–684
Pasricha S, McHugh K, Drakesmith H (2016) Regulation of hepcidin by erythropoiesis: the story so far. Annu Rev Nutr 36:417–434
Moura I, Hermine O (2015) Erythroferrone: the missing link in β-thalassemia? Blood 126(17):1974–1975
Koury M. Erythroferrone: a missing link in iron regulation. The Hematologist. 2015;12(1):10
Musallam K, Taher A, Rachmilewitz E (2013) β-Thalassemia intermedia: a clinical perspective. Cold Spring Harbor perspectives in medicine 2(7):a013482
Shander A, Cappellini M, Goodnough L (2009) Iron overload and toxicity: the hidden risk of multiple blood transfusions. Vox Sang 97(3):185–197
Wang W, Knovich M, Coffman L et al (2010) Serum ferritin: past, present and future. Biochim Biophys Acta 1800(8):760–769
Shander A, Sazama K (2010) Clinical consequences of iron overload from chronic red blood cell transfusions, its diagnosis, and its management by chelation therapy. Transfusion 50(5):1144–1155
Mishra A, Tiwari A (2013) Iron overload in beta thalassaemia major and intermedia patients. Maedica 8(4):328
Chauhan R, Sharma S, Chandra J (2014) What regulates hepcidin in poly-transfused β-thalassemia major: erythroid drive or store drive? Indian J Pathol Microbiol 57(1):39
Kaddah A, Abdel-Salam A, Farhan M, Ragab R (2017) Serum hepcidin as a diagnostic marker of severe iron overload in beta-thalassemia major. The Indian Journal of Pediatrics 84(10):745–750
Eissa D, El-Gamal R (2014) Iron overload in transfusion-dependent β-thalassemia patients: defining parameters of comorbidities. The Egyptian Journal of Haematology 39(3):164–170
Kautz L, Jung G, Du X et al (2015) Erythroferrone contributes to hepcidin suppression and iron overload in a mouse model of β-thalassemia. Blood 126:2031–2037
Makis A, Hatzimichael E, Papassotiriou I, Voskaridou E (2016) Clinical trials update in new treatments of β-thalassemia. Am J Hematol 91(11):1135–1145
Jiang X, Gao M, Chen Y et al (2016) EPO-dependent induction of erythroferrone drives hepcidin suppression and systematic iron absorption under phenylhydrazine-induced hemolytic anemia. Blood Cell Mol Dis 58:45–51
Ravasi G, Pelucchi S, Trombini P, Mariani R, Tomosugi N, Modignani GL, Pozzi M, Nemeth E, Ganz T, Hayashi H, Barisani D, Piperno A (2012) Hepcidin expression in iron overload diseases is variably modulated by circulating factors. PLoS One 7(5):e36425
Aboul-Enein A, El-Beshlawy A, Hamdy M et al (2015) Peripheral expression of hepcidin gene in Egyptian β-thalassemia major. Gene. 564(2):206–209
Galesloot T, Vermeulen S, Geurts-Moespot A et al (2011) Serum hepcidin: reference ranges and biochemical correlates in the general population. Blood 117(25):218–225
Haghpanah S, Esmaeilzadeh M, Honar N, Hassani F, Dehbozorgian J, Rezaei N, Abdollahi M, Bardestani M, Safaei S, Karimi M (2015) Relationship between serum hepcidin and ferritin levels in patients with thalassemia major and intermedia in Southern Iran. Iran Red Crescent Med J 17(7):e28343
Cheng P, Jiao X, Wang X, Lin JH, Cai YM (2011) Hepcidin expression in anemia of chronic disease and concomitant iron-deficiency anemia. Clin Exp Med 11(1):33–42
D'Angelo G (2013) Role of hepcidin in the pathophysiology and diagnosis of anemia. Blood research 48(1):10–15
Pratummo K, Jetsrisuparb A, Fucharoen S, Tripatara A (2014) Hepcidin expression from monocyte of splenectomized and non-splenectomized patients with HbE-β-thalassemia. Hematology 19(3):175–180
Kaddah N, El-Gindi H, Mostafa N et al (2011) Role of hepcidin in the pathogenesis of iron overload in children with β-thalassemia. International Journal of Academic Research 3(4):62–69
Hendy O, Allam M, Allam A et al (2010) Hepcidin levels and iron status in beta-thalassemia major patients with hepatitis c virus infection. The Egyptian Journal of Immunology 17(2):33–44
Honda H, Kobayashi Y, Onuma S, Shibagaki K, Yuza T, Hirao K, Yamamoto T, Tomosugi N, Shibata T (2016) Associations among erythroferrone and biomarkers of erythropoiesis and Iron metabolism, and treatment with long-term erythropoiesis-stimulating agents in patients on hemodialysis. PLoS One 11(3):e0151601. https://doi.org/10.1371/journal.pone.0151601
Schrier S, Bacon B. Clinical manifestations and diagnosis of hereditary hemochromatosis. UpToDate. 2016. https://www.uptodate.com/contents/clinical-manifestations-and-diagnosis-of-hereditary-hemochromatosis. last updated Jan 25, 2018
Porto G, Brissot P, Swinkels D, Zoller H, Kamarainen O, Patton S, Alonso I, Morris M, Keeney S (2016) EMQN best practice guidelines for the molecular genetic diagnosis of hereditary hemochromatosis (HH). Eur J Hum Genet 24(4):479–495
Danjou F, Cabantchik Z, Origa R, Moi P, Marcias M, Barella S, Defraia E, Dessì C, Foschini ML, Giagu N, Leoni GB, Morittu M, Galanello R (2014) A decisional algorithm to start iron chelation in patients with beta thalassemia. Haematologica. 99(3):e38–e40
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Verbal consent was taken from all participants or their guardians sharing in this study according to the ethical committee regulations of Ain Shams University.
Conflict of interest
The author declares that they have no conflict of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
El-Gamal, R.A.ER., Abdel-Messih, I.Y., Habashy, D.M. et al. Erythroferrone, the new iron regulator: evaluation of its levels in Egyptian patients with beta thalassemia. Ann Hematol 99, 31–39 (2020). https://doi.org/10.1007/s00277-019-03882-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00277-019-03882-w