
Abstract
Recombinant human granulocyte colony-stimulating factor (filgrastim) has multiple hematologic and oncologic indications as Neupogen® (Amgen filgrastim). Hospira has developed a biosimilar filgrastim (Nivestim™). Here, results are reported from a phase I trial, primarily designed to compare the pharmacokinetic profiles of Hospira filgrastim and Amgen filgrastim. A phase I, single-center, open-label, randomized trial was undertaken to demonstrate equivalence of the pharmacokinetic characteristics of Hospira filgrastim and Amgen filgrastim. Forty-eight healthy volunteers were randomized to receive intravenous (i.v.) or subcutaneous (s.c.) dosing and then further randomized to order of treatment. Volunteers in each of the two dosing groups received a single 10µg/kg dose of Hospira filgrastim or Amgen filgrastim, with subsequent crossover. Bioequivalence was evaluated by analysis of variance; if the estimated 90% confidence intervals (CIs) for the ratio of ‘test’ to ‘reference’ treatment means were within the conventional equivalence limits of 0.80–1.25, then bioequivalence was concluded. Forty-six volunteers completed the study. Geometric mean area under the curve from time 0 to the last time point (primary endpoint) was similar in volunteers given Hospira filgrastim or Amgen filgrastim following i.v. (ratio of means: 0.96; 90% CI: 0.90–1.02) or s.c. (ratio of means: 1.02; 90% CI: 0.95–1.09) dosing; 90% CIs were within the predefined range necessary to demonstrate bioequivalence. Hospira filgrastim was well tolerated with no additional safety concerns over Amgen filgrastim. Hospira filgrastim is bioequivalent with Amgen filgrastim in terms of its pharmacokinetic properties and may provide a clinically effective alternative.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Granulocyte colony-stimulating factor (G-CSF) is a naturally occurring cytokine that stimulates the proliferation and differentiation of hematopoietic stem and progenitor cells committed to the neutrophil and granulocyte lineages. Fully differentiated neutrophilic granulocytes are functionally activated by G-CSF [1–4]. Due to its hematopoietic activity, recombinant G-CSF (filgrastim) is used as an integral part of supportive therapy across multiple indications under the trade name Neupogen® (Amgen filgrastim; Amgen Inc., Thousand Oaks, CA, USA) [5–7]. In the oncology setting, Amgen filgrastim is licensed for use in patients with non-myeloid malignancies who are receiving myelosuppressive cytotoxic chemotherapy [8]. In these patients, Amgen filgrastim has been shown to significantly reduce the incidence, severity and duration of severe neutropenia and febrile neutropenia, and decrease the rate of infections compared with chemotherapy alone [5, 6, 9–14]. Other indications of Amgen filgrastim include the mobilization of peripheral blood progenitor cells, and the treatment of severe congenital, cyclic or idiopathic neutropenia [8].
Following the patent expiry of Amgen filgrastim in 2006, Hospira has developed a biosimilar version of filgrastim (Hospira filgrastim, Nivestim™), which could potentially provide a clinically effective alternative to Amgen filgrastim. Owing to the complex nature of the manufacturing process of biopharmaceuticals, and the potential for heterogeneity between reference products and biosimilar medicines, guidelines are in place for the development of biosimilars in terms of quality, safety, and efficacy [15]. In accordance with these guidelines, the similarity of Hospira filgrastim and Amgen filgrastim with respect to their physicochemical profile was demonstrated using a wide range of rigorous analytical techniques [16]. Further to this, we report results from a randomized phase I trial primarily designed to compare the pharmacokinetic (PK) profile of Hospira filgrastim with that of Amgen filgrastim following administration of a single intravenous (i.v.) or subcutaneous (s.c.) dose in healthy volunteers. Pharmacodynamic (PD) and safety assessments were secondary objectives. This is the first of two studies: a second phase I study primarily designed to compare the PD profile of Hospira filgrastim with that of Amgen filgrastim will also be reported in this journal (Annals of Hematology in press).
Design and methods
Eligibility
The study protocol and all amendments were approved by a local research ethics committee and the UK Medicines and Healthcare products Regulatory Agency. All volunteers gave written informed consent before any study-specific procedures were undertaken. Volunteers were free to withdraw from the study at any time.
Healthy, non-smoking, male or female volunteers aged 18–45 were recruited. All volunteers were required to have a body mass index of 19–30 kg/m2 and a body weight of 50–100 kg. Volunteers with clinically significant symptoms, pathologic laboratory findings, vital signs or 12-lead electrocardiogram (ECG) results were excluded from the study. Additional exclusion criteria included: a positive test for human immunodeficiency virus or hepatitis; a history of pulmonary infiltrates or pneumonia in the previous 6 months; a history or evidence of drug or alcohol abuse; hereditary fructose intolerance; a history of hypotensive episodes associated with i.v. drug administration; blood donation ≥500 ml in the previous 12 weeks; and pregnancy or lactation. Concurrent use of prescription or over-the-counter medicines (excluding hormonal contraceptives and occasional use of paracetamol) was not permitted. Similarly, caffeine- and alcohol-containing beverages were not allowed during the assessment periods or in the 24 h prior to first dose.
Study design and outcomes
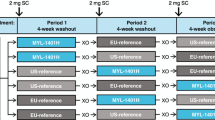
Volunteers were treated at Charles River Clinical Services, Edinburgh, UK, between October 18 and December 28, 2006, as part of a randomized, comparator-controlled, two-way crossover trial (Fig. 1). Volunteers were randomized to one of two parallel groups (i.v. or s.c. routes), then further randomized to order of agent administration. Study drugs were administered on an open-label basis. Volunteers in each of the two parallel groups received a single 10-µg/kg dose of Hospira filgrastim and Amgen filgrastim in random order. Randomization was performed according to a computer-generated randomization list, produced by Constella Group Ltd, Abingdon, UK. Conditions relating to provision of meals, fasting periods, overnight stays, and levels of permitted physical activity were standardized throughout.

Study design and volunteer disposition. (a) All volunteers who received ≥1 dose of study medication; (b) all volunteers who completed the study with a sufficient number of quantifiable concentrations to warrant parameter estimation in both assessment periods; (c) all volunteers for whom an evaluable PD parameter was obtained in both assessment periods
An initial screening period of up to 21 days was followed by two 6-day assessment periods, separated by a washout period of at least 13 days. Hospira filgrastim and Amgen filgrastim were administered via s.c. injection or via i.v. infusion over 30 min.
The primary endpoint was the area under the curve from time 0 to the last time point (AUC0–tlast) for plasma G-CSF concentration. Secondary PK endpoints were: maximum observed plasma concentration of G-CSF (C max); time at which C max occurred (T max); elimination half life (T 1/2); AUC from time 0 extrapolated to infinity (AUC0–infinity); terminal elimination rate constant (λ z ); and clearance. Secondary PD endpoints included ANC AUC0–tlast, ANC T max, ANCmax, and ANCmin. Safety was also assessed as a secondary endpoint.
Blood samples were collected for measurement of plasma G-CSF at −1, 0.25, 0.5, 0.75, 1, 2, 3, 4, 6, 8, 10, 12, 16, 24, and 48 h post dose. Samples were also collected at −1, 0.5, 1, 2, 4, 6, 8, 24, 48, 72, 96, and 120 h following dose administration for PD (ANC) analysis. G-CSF concentrations were assessed by the Charles River Laboratories Central Laboratory using a validated commercial assay [17–21].
Safety was assessed based on observed adverse events (AEs), clinical laboratory tests (hematology, biochemistry and urinalysis), physical examination, results of 12-lead and 30-min continuous ECG and vital sign assessments.
Statistical analysis
The PK population consisted of all volunteers who completed the study with a sufficient number of quantifiable concentrations to warrant parameter estimation in both assessment periods. The PD population consisted of all volunteers for whom an evaluable PD parameter was obtained in both assessment periods. The safety population comprised all volunteers who received at least one dose of study medication.
Tmaxλ z , clearance for plasma concentration of G-CSF and ANC Tmax were summarized descriptively only. All other PK and PD parameters were loge-transformed prior to statistical analysis and presented as geometric means, along with other summary statistics. Missing concentrations were deleted, resulting in an interpolation between the nearest two values. Outliers were identified using an outlier check (Dixon test or T procedure). PK data were analyzed by non-compartmental methods using WinNonlin® (Pharsight® Corporation, Mountain View, CA, USA).
Bioequivalence was assessed for the primary and secondary endpoints using a mixed effects analysis of variance, with terms including subject within sequence as a random effect, and study drug and period as fixed effects. A 90% confidence interval (CI) for the ratio of ‘test’ to ‘reference’ means was calculated using least squares estimates of the means and residual variance from the model. If the 90% CI was completely within the conventional bioequivalence limits of 0.80–1.25, then bioequivalence could be concluded.
A total of 44 evaluable volunteers were required for 80% power (α = 0.05) to demonstrate bioequivalence between Hospira filgrastim and Amgen filgrastim, assuming a within-subject coefficient of variation of 21% (i.v. volunteers) or 23% (s.c. volunteers) and a bioequivalence range of 0.80–1.25 in the ‘test’ to ’reference’ mean ratio for AUC0–tlast.
Results
Subject disposition
Forty-eight volunteers were enrolled: 22 into the i.v. group and 26 into the s.c. group (Fig. 1). Two volunteers in the i.v. group withdrew from the study owing to AEs (moderate nausea, dizziness, agitation, pharynx dysesthesia, dyspnea, and a respiratory disorder) and withdrawal of consent, respectively, after receiving a single dose of Amgen filgrastim. These volunteers were excluded from the PK and PD analyses. Three additional volunteers in the i.v. group were excluded from the PD analyses due to insufficient data (one subject had three missing values in a row due to discontinuation, two volunteers did not complete both assessment periods). During review of the PD data, it was noted that there were two consecutive missing ANC results for five volunteers (two in the i.v. group and three in the s.c. group). Therefore, for these patients, ANC estimates were interpolated from the nearest two known ANC values. All volunteers were evaluable for safety.
Investigators permitted several volunteers to take paracetamol for pain relief during the study; however, this was not considered to affect the study outcomes and therefore volunteers were not withdrawn. Baseline demographics were generally well matched between groups (Table 1).
Pharmacokinetics
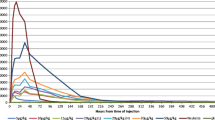
The primary PK endpoint (geometric mean AUC0–tlast) was similar in volunteers given Hospira filgrastim or Amgen filgrastim following either i.v. (ratio of means, 0.96; 90% CI, 0.90–1.02) or s.c. (ratio of means, 1.02; 90% CI, 0.95–1.09) administration (Table 2). For both routes of administration, 90% CIs were within the predefined range of 0.80–1.25 necessary to demonstrate bioequivalence of the two agents. In volunteers who received Hospira filgrastim or Amgen filgrastim via the s.c. route, the geometric mean AUC0–tlast was slightly lower than in those who received study medication via the i.v. route (ratio of means: 0.75; 90% CI: 0.67–0.84). Analysis of secondary PK parameters showed that geometric means for AUC0–infinity, C max, T 1/2, T max, λ z, and clearance were similar in volunteers who received Hospira filgrastim or Amgen filgrastim following either i.v. or s.c. administration (Table 2). As with the primary endpoint, 90% CIs for AUC0–tlast, C max, and T 1/2 were within the predefined range required to demonstrate bioequivalence of the two agents, irrespective of administration route.
Pharmacodynamics
The PD parameters analyzed were generally similar in volunteers given Hospira filgrastim or Amgen filgrastim, in both the s.c. and i.v. groups. In the i.v. group, the ratios of geometric means for ANC AUC0–tlast, ANCmax and ANCmin were 1.03 (90% CI, 0.99–1.08), 1.07 (90% CI, 1.00–1.14), and 0.83 (90% CI, 0.68–1.02), respectively. The 90% CIs for AUC0–tlast and ANCmax were within the range required to show bioequivalence of Hospira filgrastim and Amgen filgrastim, while those for ANCmin were outside the equivalence range. Mean values for ANC Tmax were similar in volunteers who received i.v. Hospira filgrastim or i.v. Amgen filgrastim (17.99 vs. 19.05 h, respectively).
Similar results were obtained in the s.c. group, with geometric mean ratios of 1.03 (90% CI: 0.99–1.06), 1.04 (90% CI: 0.98–1.11), and 1.13 (90% CI: 0.83–1.54) reported for ANC AUC0–tlast, ANCmax, and ANCmin, respectively. As in the i.v. group, s.c. Hospira filgrastim and s.c. Amgen filgrastim were shown to be bioequivalent for ANC AUC0–tlast and ANCmax, but not for ANCmin. Mean ANC T max was 19.44 h in volunteers who received s.c. Hospira filgrastim and 21.49 h in those who received s.c. Amgen filgrastim.
Safety
The overall incidence of AEs was lower in volunteers who received i.v. Hospira filgrastim compared with those who received i.v. Amgen filgrastim (60 vs. 82%, respectively), whereas it was similar in volunteers who received study drugs via the s.c. route (77% vs. 73%, respectively; Table 3). This pattern was also reflected in the incidence of AEs related to the study drug (50% vs. 68%, respectively, for the i.v. route; 58% vs. 50%, respectively, for the s.c. route).
The most common AEs were headache, back pain and nausea (Table 3), which were generally related to the study drug. Only minor differences in AE profiles were observed between Hospira filgrastim and Amgen filgrastim (Table 3). All AEs were mild or moderate in intensity and no serious AEs were reported.
No clinically significant changes in heart rate, 12-lead ECG, hematology, biochemistry, urinalysis, and physical examination were reported.
Discussion
Guidance issued by the European Medicines Agency (EMEA) states that new biosimilar medicinal products containing filgrastim should demonstrate comparability with the reference product, Amgen filgrastim [15]. The EMEA recommends that extensive preclinical and clinical studies are conducted, including PK, PD and safety investigations. PK investigations should include single-dose, crossover trials of s.c. and i.v. administration, with AUC as the primary endpoint, and C max and T 1/2 as secondary parameters. The phase I PK study reported here was designed in accordance with these guidelines and forms part of the regulatory assessment for Hospira filgrastim.
All PK parameters examined were found to be similar following a single dose of Hospira filgrastim or Amgen filgrastim, irrespective of dosing route (i.v. or s.c.). Importantly, therapeutic bioequivalence was demonstrated for AUC0–tlast (primary endpoint), AUC0–infinity, C max, and T 1/2 with i.v. or s.c. dosing. As reported previously for G-CSF [22], the bioavailability of Hospira filgrastim and Amgen filgrastim was higher when administered via the i.v. versus the s.c. route.
The two drugs were also shown to be bioequivalent for all PD parameters except ANCmin, regardless of the administration route. There was some slight variation in T max between the two drugs, but these minor differences are unlikely to be of clinical importance and, overall, the data overwhelmingly support the bioequivalence of Hospira filgrastim and Amgen filgrastim. This observed bioequivalence is likely to be attributed to the almost identical physicochemical (including formulation, strength, composition, purity, and presentation) and molecular characteristics of the two drugs [16].
Hospira filgrastim was well tolerated and there were no unexpected safety concerns associated with its use. The AE profiles of Hospira filgrastim and Amgen filgrastim, in terms of their nature and intensity, were comparable, and similar to those reported previously for recombinant G-CSF in healthy volunteers [23].
This study, together with a second phase I study (also reported in this journal) (Annals of Hematology in press), provide a comprehensive analysis of the PK and PD characteristics of Hospira filgrastim. Similar analyses have been described in the literature for the biosimilar filgrastim XM02, although fewer endpoints were reported [24]. Taken together, the two phase I characterization studies provide a strong rationale for the further clinical evaluation of Hospira filgrastim. In this regard, a large, randomized, double-blind, phase III trial of Hospira filgrastim versus Amgen filgrastim (5 µg/kg s.c.) in patients with breast cancer receiving myelosuppressive chemotherapy has been conducted [25]. Overall, data suggest that Hospira filgrastim is effective and well tolerated, and may provide an alternative to Amgen filgrastim for the treatment of chemotherapy-induced neutropenia and other indications for which G-CSF is routinely used.
References
Gomez RC, Pinto MA, Gonzalez BM (2006) Colony-stimulating factors: clinical evidence for treatment and prophylaxis of chemotherapy-induced febrile neutropenia. Clin Transl Oncol 8:729–734
Bronchud MH, Scarffe JH, Thatcher N, Crowther D, Souza LM, Alton NK, Testa NG, Dexter TM (1987) Phase I/II study of recombinant human granulocyte colony-stimulating factor in patients receiving intensive chemotherapy for small cell lung cancer. Br J Cancer 56:809–813
Bronchud MH, Potter MR, Morgenstern G, Blasco MJ, Scarffe JH, Thatcher N, Crowther D, Souza LM, Alton NK, Testa NG (1988) In vitro and in vivo analysis of the effects of recombinant human granulocyte colony-stimulating factor in patients. Br J Cancer 58:64–69
Roberts AW (2005) G-CSF: a key regulator of neutrophil production, but that's not all! Growth Factors 23:33–41
Aapro MS, Cameron DA, Pettengell R, Bohlius J, Crawford J, Ellis M, Kearney N, Lyman GH, Tjan-Heijnen VC, Walewski J, Weber DC, Zielinski C (2006) EORTC guidelines for the use of granulocyte-colony stimulating factor to reduce the incidence of chemotherapy-induced febrile neutropenia in adult patients with lymphomas and solid tumours. Eur J Cancer 42:2433–2453
Dale DC (2002) Colony-stimulating factors for the management of neutropenia in cancer patients. Drugs 62(Suppl 1):1–15
Welte K, Gabrilove J, Bronchud MH, Platzer E, Morstyn G (1996) Filgrastim (r-metHuG-CSF): the first 10 years. Blood 88:1907–1929
Neupogen 30MU and 48MU solution for injection. Summary of product characteristics. Available at http://emc.medicines.org.uk/document.aspx?documentId=7907 Accessed 2 November 2009
Bohlius J, Herbst C, Reiser M, Schwarzer G, Engert A (2008) Granulopoiesis-stimulating factors to prevent adverse effects in the treatment of malignant lymphoma. Cochrane Database Syst Rev CD003189
Clark OA, Lyman GH, Castro AA, Clark LG, Djulbegovic B (2005) Colony-stimulating factors for chemotherapy-induced febrile neutropenia: a meta-analysis of randomized controlled trials. J Clin Oncol 23:4198–4214
Garcia-Carbonero R, Mayordomo JI, Tornamira MV, Lopez-Brea M, Rueda A, Guillem V, Arcediano A, Yubero A, Ribera F, Gomez C, Tres A, Perez-Gracia JL, Lumbreras C, Hornedo J, Cortes-Funes H, Paz-Ares L (2001) Granulocyte colony-stimulating factor in the treatment of high-risk febrile neutropenia: a multicenter randomized trial. J Natl Cancer Inst 93:31–38
Kuderer NM, Dale DC, Crawford J, Lyman GH (2007) Impact of primary prophylaxis with granulocyte colony-stimulating factor on febrile neutropenia and mortality in adult cancer patients receiving chemotherapy: a systematic review. J Clin Oncol 25:3158–3167
Lyman GH, Kuderer NM, Djulbegovic B (2002) Prophylactic granulocyte colony-stimulating factor in patients receiving dose-intensive cancer chemotherapy: a meta-analysis. Am J Med 112:406–411
Sung L, Nathan PC, Alibhai SM, Tomlinson GA, Beyene J (2007) Meta-analysis: effect of prophylactic hematopoietic colony-stimulating factors on mortality and outcomes of infection. Ann Intern Med 147:400–411
Committee for Medicinal Products for Human Use (CHMP). Annex to guideline on similar biological medicinal products containing biotechnology-derived proteins as active substance: non-clinical and clinical issues. Guidance on similar medicinal products containing recombinant granulocyte-colony stimulating factor. Available at www.emea.europa.eu/pdfs/human/biosimilar/3132905en_pdf. Accessed 28 January 2009
Skrlin A, Radic I, Vuletic M, Schwinke D, Runac D (2009) The physicochemical properties of a biosimilar filgrastim are comparable to those of Neupogen®. Poster presentation at the 14th Congress of the European Hematology Association, Berlin, Germany;Abstract 0568
Duhrsen U, Villeval JL, Boyd J, Kannourakis G, Morstyn G, Metcalf D (1988) Effects of recombinant human granulocyte colony-stimulating factor on hematopoietic progenitor cells in cancer patients. Blood 72:2074–2081
Martinez C, Urbano-Ispizua A, Marin P, Merino A, Rovira M, Carreras E, Montserrat E (1999) Efficacy and toxicity of a high-dose G-CSF schedule for peripheral blood progenitor cell mobilization in healthy donors. Bone Marrow Transplant 24:1273–1278
Souza LM, Boone TC, Gabrilove J, Lai PH, Zsebo KM, Murdock DC, Chazin VR, Bruszewski J, Lu H, Chen KK (1986) Recombinant human granulocyte colony-stimulating factor: effects on normal and leukemic myeloid cells. Science 232:61–65
Stroncek DF, Clay ME, Petzoldt ML, Smith J, Jaszcz W, Oldham FB, McCullough J (1996) Treatment of normal individuals with granulocyte-colony-stimulating factor: donor experiences and the effects on peripheral blood CD34+ cell counts and on the collection of peripheral blood stem cells. Transfusion 36:601–610
Welte K, Bonilla MA, Gillio AP, Boone TC, Potter GK, Gabrilove J, Moore MAS, O'Reilly J, Souza LM (1987) Recombinant human granulocyte colony-stimulating factor. J Exp Med 165:941–948
Sugiura M, Yamamoto K, Sawada Y, Iga T (1997) Pharmacokinetic/pharmacodynamic analysis of neutrophil proliferation induced by recombinant granulocyte colony-stimulating factor (rhG-CSF): comparison between intravenous and subcutaneous administration. Biol Pharm Bull 20:684–689
Anderlini P (2009) Effects and safety of granulocyte colony-stimulating factor in healthy volunteers. Curr Opin Hematol 16:35–40
Lubenau H, Bias P, Maly AK, Siegler KE, Mehltretter K (2009) Pharmacokinetic and pharmacodynamic profile of new biosimilar filgrastim XM02 equivalent to marketed filgrastim Neupogen®: single-blind, randomized, crossover trial. BioDrugs 23:43–51
Waller CF, Semiglazov VF, Chan S, Challand R (2009) Biosimilar filgrastim is an effective primary prophylactic therapy for neutropenia in patients (pts) receiving doxorubicin and docetaxel (AT) for breast cancer (BC). Poster presentation at the joint ECCO 15 and 34th ESMO Multidisciplinary Congress; Abstract E15-1238
Acknowledgments
The authors kindly acknowledge the contribution of Hannah FitzGibbon and Nigel C Eastmond from GeoMed for medical writing assistance in the preparation of this manuscript, with financial support from Hospira.
Authorship and disclosures
CW contributed to the study design, the interpretation of the data and the consideration of its relevance to clinical practice. MB contributed to the interpretation of the data and the consideration of its relevance to clinical practice. SM was the Principal Investigator and had overall responsibility for the conduct of the study. RC is the Clinical Program Director at Hospira UK Ltd. All authors critically reviewed each draft of the manuscript prior to submission. CW and MB have consultancy agreements with Hospira UK Ltd, and SM is the Medical Director of Operations at Quotient Clinical. No other conflicts of interest were reported.
Funding
This work was supported by Hospira.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Waller, C.F., Bronchud, M., Mair, S. et al. Pharmacokinetic profiles of a biosimilar filgrastim and Amgen filgrastim: results from a randomized, phase I trial. Ann Hematol 89, 927–933 (2010). https://doi.org/10.1007/s00277-010-0961-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00277-010-0961-x