
Abstract
Targeting CD40 with agonist antibodies is a promising approach to cancer immunotherapy. CD40 acts as a master regulator of immunity by mobilizing multiple arms of the immune system to initiate highly effective CD8 + T-cell-mediated responses against foreign pathogens and tumors. The clinical development of CD40 agonist antibodies requires careful optimization of the antibody to maximize therapeutic efficacy while minimizing adverse effects. Both epitope specificity and isotype are critical for CD40 agonist antibody mechanism of action and potency. We developed a novel antibody, APX005M, which binds with high affinity to the CD40 ligand-binding site on CD40 and is optimized for selective interaction with Fcγ receptors to enhance agonistic potency while limiting less desirable Fc-effector functions like antibody-dependent cellular cytotoxicity of CD40-expressing immune cells. APX005M is a highly potent inducer of innate and adaptive immune effector responses and represents a promising CD40 agonist antibody for induction of an effective anti-tumor immune response with a favorable safety profile.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Targeting of immune checkpoint inhibitor (ICI) proteins including cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) and program cell death 1 (PD-1) or its ligand, PD-L1, is able to partially reverse cancer immunosuppression of tumor-specific cytotoxic effector cells by unleashing them to kill tumor cells. However, durable responses to treatment are achieved only in a minority of cases. Most patients never respond to treatment or have an initial response followed by tumor progression which highlights the need for new immunotherapies such as immune agonists.
CD40 remains an attractive target for cancer immunotherapy, representing a key component of both the innate and adaptive immune response. CD40 is a member of the TNF receptor superfamily expressed on a variety of immune cells, including B cells, dendritic cells, macrophages and monocytes [1]. CD40 agonist antibodies can potently activate anti-tumor immunity via multiple mechanisms. First, CD40-mediated activation of antigen presenting cells (APCs) is a prerequisite for T-cell activation and thus can promote the generation of tumor-specific cytotoxic T cells even in the absence of CD4 T-cell help [2,3,4]. Second, CD40 agonists can directly activate macrophages to infiltrate and deplete tumor stroma, converting ‘cold’ tumors into ‘hot’ tumors [5]. Finally, CD40-activated APCs produce IL-12 and IL-15, cytokines that are essential for NK cell-mediated killing of tumor cells [6].
Preclinical mouse models have provided significant rationale for the clinical development of CD40 agonist antibodies as single agent therapies or in combination with standard of care in a variety of tumor types [7,8,9]. In a mouse model of bladder cancer, CD40 agonist antibodies show single-agent activity when administered either systemically or locally [10]. CD40 agonist antibodies induce tumor regression in mouse models of pancreatic cancer as a single-agent [5] and when combined with ICI [11, 12]. Numerous vaccine strategies for cancer therapy are currently being evaluated [13], and the addition of CD40 agonist antibodies enhances the activity of tumor vaccines in nearly every case studied [14,15,16] providing strong rationale for combination.
For almost a decade, CD40 agonist antibodies have been investigated clinically in a range of tumor types with compelling initial results [7, 17] [18] [19]. Clinical efficacy in melanoma was demonstrated in front line therapy after only a single injection of the CD40 agonist CP-870,893 [20, 21]. In a phase I study combining CD40 agonist and CTLA-4 blockade in patients with metastatic melanoma, long-term protection was observed with 9 of 24 patients with survival currently beyond 3 years (18.2% PR plus 9.1% CR = ORR is 27.3%; mean OS 23.6 months) [21]. While encouraging data have been observed, on-target immune-related adverse events (IRAEs) including cytokine release syndrome and liver enzyme elevation have historically hampered the enthusiasm for further clinical development. Preclinical studies have shed light on strategies that could fine-tune CD40 agonist antibodies to improve safety profiles and enhance efficacy.
Epitope specificity and isotype of CD40 agonist antibodies contribute to their function and clinical activity [14, 22,23,24]. CD40 agonist antibodies are able to induce CD40 signaling to various degrees whether or not they block natural ligand binding. Studies using anti-mouse CD40 antibodies have shown that stronger agonist activity correlated with blockade of CD40 ligand (CD40L) binding, likely because these antibodies better recapitulate the natural ligand’s ability to activate APCs [25]. Moreover, the anti-tumor efficacy of CD40 agonists in mouse models was greatly improved by engineering Fc variants with enhanced binding to FcγRIIb, including the S267E variant that confers 30-fold increased binding affinity to FcγRIIb [14, 26, 27]. Enhanced binding to FcγRIIb may contribute to enhanced efficacy by mediating crosslinking of the antibody and clustering of CD40 receptors in a way that mimics the natural CD40L interaction and by inhibiting ADCC activity on CD40-expressing APCs due to the lack of binding to FcγRIIIa. Importantly, CD40 agonists that depend on FcγRIIb crosslinking should have a more favorable safety profile because FcγRIIb-mediated crosslinking is most prevalent in tumor tissues and tumor-draining lymph nodes but limited in the circulation which could lead to less systemic APC activation and cytokine release.
We generated a panel of high affinity CD40 antibodies using the APXiMAB™ platform. APX005M was selected based on its potent agonist activity and binding to the CD40L binding site. APX005M was further engineered to include the S267E mutation which significantly enhanced its agonistic activity and minimized ADCC effector function. We show here that APX005M’s unique structural attributes give it desirable biological and pharmacological activities which are differentiated from other CD40 agonists in the clinic. Thus, APX005M represents a promising immune checkpoint activator for cancer immunotherapy. Multiple clinical trials are being conducted with APX005M alone and in combination with other therapies across several tumor types [28, 29].
Materials and methods
CD40 antibodies
APX005M is a humanized IgG1κ antibody generated by Apexigen using its APXiMAB™ technology consisting of a rabbit hybridoma platform and mutational lineage-guided (MLG) humanization method [30,31,32]. A point mutation was introduced in the Fc-domain at position 267 from serine to aspartic acid (S267E mutation). A version of APX005M lacking S267E mutation (APX005), as well as recombinant analogues of anti-CD40 agonistic antibodies CP-870,893, SGN-40 and ADC-1013 were synthesized based on published data [33,34,35]. F(ab’)2 fragments of APX005M were generated by pepsin digest (Rockland Immunochemicals).
Reagents
Human cytokines were purchased from Peprotech (Rocky Hill, NJ). CD40-his and CD40L were purchased from Cedarlane (Burlington, NC). Isotype and flow cytometry antibodies (CD4-PE, CD8-PE, CD40-APC, CD14-FITC, CD14-PECy7, HLA-DR APC-Cy7 or FITC, CD11c-Alexa Fluor 488, CD86-PE) were obtained from Biolegend or Becton Dickinson (San Jose, CA; CD80-APC, CD83-PE). Rituximab was purchased from InvivoGen (San Diego, CA).
CD40 binding assay
For B cell binding, 1 × 106 human peripheral blood mononuclear cells (PBMC) per 96-well were incubated with increasing concentrations of CD40-targeted antibodies at 4 °C for 1 h, then washed and stained at 4 °C for 30 min with anti-human CD20-FITC to gate the human B-cell population and with goat F(ab′)2 anti-human Fcγ PE to detect anti-CD40 antibodies. For DC binding, monocyte-derived DC (moDC) were generated as follows: human PBMC were isolated from buffy coats (Stanford Blood Center, Palo Alto, CA) using Ficoll® Hypaque (GE Healthcare, Pittsburgh, PA) density gradient centrifugation. After washing, CD14 + monocytes were isolated using magnetic beads (Miltenyi Biotech, Auburn, CA). Cells were cultured for 7 days in RPMI-1640 with 10% fetal bovine serum (Hyclone, GE Healthcare), penicillin–streptomycin, and 2 mM L-glutamine (Life Technologies, Grand Island, NY) with 100 ng/mL GM-CSF and 50 ng/mL IL-4. DCs were stained as described above. All samples were analyzed on a MACSQuant® flow cytometer. For each antibody titration, results are expressed as mean fluorescence intensity (MFI) for CD40 on CD40+ cells.
APC activation
Freshly isolated CD19+ human B cells were incubated at 37 °C in 96-well round-bottom plates (1 × 105 cells/100 µL) in the presence of CD40 agonist antibodies. After 48 h, cells were washed and stained with mouse anti-human CD86 PE. For HLA-DR expression on B cells, 200 µL of human whole blood was stimulated with APX005M in 96-well round bottom plates for 24 h then stained with anti-human HLA-DR FITC antibody. RBCs were then lysed with ACK lysis buffer and re-suspended in PBS + 0.5% BSA + 2 mM EDTA. All samples were analyzed on the MACSQuant® flow cytometer. For DC activation, moDC were generated as described above and 5 × 105 cells per well were cultured in 96-well plates in the presence of CD40 agonist or control antibodies for 48 h. Supernatants were harvested and assayed for IL-12p70 by ELISA (R&D Systems) and upregulation of CD86 was determined as described above.
Epitope mapping of APX005M
Chemically linked peptides on scaffolds (CLIPS) technology and peptide arrays were used for epitope mapping (Pepscan Presto, Lelystad, The Netherlands) [36]. In brief, peptide cysteine residues were coupled to CLIPS templates on the peptide array. After incubation for 30–60 min, the peptide array was washed with H2O and sonicated in the presence of 1% sodium-dodecyl sulphate-1% 2-mercaptoethanol in PBS at 70 °C for 30 min followed by sonication in H2O for 45 min. Antibody binding to the peptides was examined by ELISA. Peptide arrays were incubated with primary antibody overnight at 4 °C. After washing, peroxidase-conjugated secondary antibody was added and incubated for 30 min and, after additional washing, ABTS peroxidase substrate and 3% H2O2 was added. Color change was quantified after 1 h using a CCD camera and an image processing system.
CD40L blocking assay
ELISA plates were coated with 0.5 µg/ml CD40-his overnight. After blocking with 1% BSA in PBS, serial dilutions of APX005M or isotype control were added and plates were incubated for 1 h at room temperature. 3 µg/ml CD40L was added and the plate was incubated for 1 h. After washing, mouse anti-human CD40L (R&D Systems) was added and incubated for 1 h. Donkey anti-mouse IgG (Jackson ImmunoResearch) was added for one hour and detected using TMB substrate (Kirkegaard & Perry, Gaithersburg, MD) read at OD 450 nm.
Fc receptor binding assay
CHO cells were transfected with cDNAs encoding human Fc receptors. Expression was confirmed by flow cytometry (Eureka Therapeutics, Emeryville, CA). Cells were detached using 2.5 mM EDTA in PBS, washed and added to 96-well plates. Serial dilutions of antibodies were added and samples incubated on ice for 1 h. After washing, PE-conjugated F(ab)’2 fragment of goat anti-human IgG (Jackson ImmunoResearch) was added and cells were incubated for 30 min on ice and analyzed by FACS. Saturation binding curves were generated using nonlinear regression and one-site binding model provided in GraphPad Prism software (GraphPad Software, San Diego, CA) to derive KD values.
Antibody-dependent cell cytotoxicity assay (ADCC)
Daudi (ATCC® CCL-213™) B cells (40,000 per well) in RPMI-1640 with 10% ultra-low IgG were added to 96-well plates. Serial dilutions of indicated antibodies were added. CD56 + CD16 + NK cells were obtained from PBMC using magnetic bead separation (Miltenyi Biotech) and were added to plates (200,000 per well) resulting in an effector to target (E:T) ratio of 5:1. Plates were incubated for 4 h at 37 °C. After incubation, supernatant was transferred to 96-well plates and CytoTox® substrate mix added to each well (Promega; Madison, WI). Plates were incubated in the dark for 30 min then read at OD = 492 nm using SpectraMax® M3 spectrophotometer. Percent cytotoxicity of target cells was reported from triplicate measures and at least three donors as % cytotoxicity = (effector (spontaneous)—target (spontaneous))/(target (maximal)—target (spontaneous))*100, where medium background was subtracted from all values.
Mixed lymphocyte reaction (MLR)
Human T cells were labeled with eFluor 670 (eBioscience) and cultured (2 × 105 cells per well) with allogeneic moDC (5 × 104 cells per well) in flat-bottomed 96-well plates. moDCs were pre-activated with CD40 agonistic or control antibodies for 24 h prior to the addition of T cells. After 6 days of co-culture, supernatants were harvested and assessed for IFN-γ by ELISA (R&D Systems). To measure T-cell proliferation, cells were stained with anti-CD8 PE antibodies and T-cell divisions were evaluated using a MACS Quant® analyzer.
Viral antigen recall assay
PBMC were suspended in culture media and plated in 96-well plates at 106 per well. EBV peptides (Miltenyi Biotech; 130–099-764) were added at a final concentration of 1μg/mL. CD40 agonist antibodies were added and plates were incubated at 37˚C. After 5 days of culture, supernatants were removed and assayed for IFN-γ by ELISA.
Ex vivo tumor experiments
Tumor biopsies from consented patients undergoing surgery for human non-small cell lung cancer (NSCLC) were harvested and dispersed into single cell suspensions (Nilogen Oncosystems, Tampa, FL). APX005M (67 nM) or vehicle was added and cultures were incubated for 5 days. T cells were stained with antibodies to CD3 and Ki67 along with viability dye, and analyzed by flow cytometry. Gates were set to exclude dead cells and debris, and the number of CD3 + Ki67 + T cells was determined. Data are representative of three donors.
APX005M washout assay
Human B cells were incubated with APX005M or control antibody for 7 days. In indicated samples, antibodies were washed out after 24 h. Following 7 day incubation, B cells were harvested and expression of CD86 and HLA-DR was assessed by flow cytometry as described above.
CD40 receptor occupancy and maximal B-cell activation
A weighed least-square procedure in Phoenix WinNonlin 6.4 (Pharsight, Inc. Mountain View, CA) was used to establish a correlation between CD40 receptor occupancy on B cells and cellular activation according to the equation: E = E0 + EmaxCγ/ECγ50 + Cγ where E is the effect, E0 is the effect at baseline, Emax is the maximum effect and EC50 is the concentration at half maximum effect. The predicted CD40 receptor occupancy at maximum B-cell activation (CD86 expression) was derived from this fit.
Preclinical safety assessment of APX005M
GLP tissue cross-reactivity study on a full panel of normal human and cynomolgus monkey tissues was conducted at Charles River Laboratories, Frederick, MD. APX005M or isotype control was applied at 10 and 50 µg/mL.
Non-human primate (NHP) studies were carried out in cynomolgus monkeys by Charles River Laboratories, Reno, NV. Ten monkeys in each group (2.5–4 kg, five males and five females per group) received a once-weekly intravenous bolus injection of 0.3, 3 or 30 mg/kg body weight of APX005M for 4 weeks. Standard in-life and terminal analyses were conducted, including peripheral lymphocyte analysis by flow cytometry and serum analysis for cytokines (Luminex) and APX005M (antigen capture ELISA). PK analysis was performed by PharmaPolaris International (North Potma, MD).
Statistical analyses
Statistical analyses were performed using Graph Pad Prism® (Graph Pad Software, San Diego, CA) as indicated.
Results
Characterization of APX005M binding specificity and activity
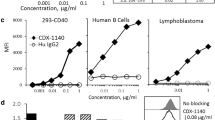
APX005M was selected from a panel of rabbit monoclonal antibodies based on binding specificity for human CD40 and potent bioactivity. APX005M was humanized as previously described [32]. APX005M binds with high affinity (KD = 1.2 × 10−10 M) to B-cell-expressed human CD40 (Fig. 1a) and cynomolgus CD40 (KD = 3.7 × 10−10 M), but does not recognize rodent CD40 (data not shown). APX005M does not bind closely related TNFR family members, including OX-40, RANK, DR5, TWEAK-R and 4-1BBL (data not shown). The primary mode of action of CD40 agonist antibodies is the activation of APCs. In vitro APX005M stimulation of human B cells up-regulated immune co-stimulatory molecules CD86/B7.2 (Fig. 1b) and MHC II (Fig. 1c).

APX005M binds to CD40 with high affinity and potently activates B cells. a Binding of APX005M on human B cells was determined by flow cytometry and expressed as mean fluorescent intensity (MFI). B-cell activation was measured by MFI of CD86 expression after 48-h incubation (b) or HLA-DR expression after 24 h (c). APX005M is labeled with green squares and isotype control by black circles. Experiments were repeated with at least two donors. d Competition ELISA showing that APX005M (green squares) but not isotype control (black circles) can compete with CD40L for binding to CD40. e ELISA results for 20‐mer overlapping peptides. X‐axis indicates the residue number of the C‐terminal position (i.e.1EPPTACREKQYLINSQCCSL20 is positioned on X‐value 20). Residues 92TSEACESCVLHRSCSP107 (P1) and 122ICEP125 (P2) are visualized on the cartoon rendering of structure 3QD6. (F) Surface rendering of 3QD6 indicating CD40L binding sites (green and light blue), APX005M binding sites (red and purple) and residues identified as overlapping (yellow)
Previous studies using anti-mouse CD40 antibodies have shown agonist activity positively correlates with their ability to inhibit CD40L binding [25]. Using a competitive ELISA, we show that APX005M blocks binding of CD40 to CD40L (Fig. 1d) via a unique epitope identified using a comprehensive peptide array consisting of 20-mer overlapping peptides derived from CD40. Based on the published structure of CD40 [37], 12 potential binding-hotspots were identified for single-position mutational analysis and were included in our peptide array. Evaluation of these peptides revealed that APX005M binds two distinct regions: 92TSEACESCVLHRSCSP107 (P1) and 125PCPVGFFSNVSSAFEKCHPW144 (P2), with the residues 92TSEACESCVLH102 anticipated to be the most relevant for binding (Fig. 1e). Visualization of these areas on the crystal structure of CD40 shows that P1 forms a looped structure facing the CD40L trimer (Fig. 1f). Of the identified binding residues, four (T92, E97 and 100VL102) are known to be contact residues between CD40 and CD40L [37]. These data indicate that APX005M binds to a unique epitope on CD40 that overlaps with the ligand binding site and effectively competes for binding with CD40L.
APX005M engages FcγRIIb and requires crosslinking for optimal activity
While APX005M-F(ab’)2 fragments bind CD40 with the same affinity as intact APX005M (data not shown), they fail to stimulate B cells (Fig. 2a) indicating the agonist activity of APX005M is critically dependent upon FcR binding. To increase Fc receptor-mediated crosslinking via FcγRIIb, we engineered the S267E mutation [27] into APX005M with the intention of enhancing agonist activity and improving its safety profile. To determine the effect of the S267E mutation on FcγR binding of APX005M, CHO cell lines expressing individual human Fc receptors were incubated with serial dilutions of APX005M or a version of APX005M with the wildtype IgG1 Fc lacking the S267E mutation (APX005). As shown in Fig. 2b, APX005M strongly bound FcγRIIb (KD = 5.8 × 10–8 M), whereas APX005 exhibited negligible binding. APX005M also lost binding to FcγRIIIa but retained binding to other FcγRs in comparison to APX005 (Supplemental Fig. 1). The increase of binding to FcγRIIb resulted in enhanced B cell activation as determined by CD86 upregulation (Fig. 2c).

APX005M agonist activity requires FcR-mediated crosslinking. a Activation of B cells after 48 h by APX005M intact (green squares) or APX005M F(ab’)2 fragments (gray circles) as measured by MFI of CD86 expression. Data are representative of two donors. b Binding of APX005M (green squares), APX005 (black circles) and isotype control (open circle) to human FcγRIIb—expressing CHO cells. c Activation of B cells by APX005M (green squares), APX005 (black circles) and isotype control (open circle) for 48 h as measured by MFI of CD86 expression. Data are representative of three donors. d NK cell-mediated ADCC activity of APX005M (green squares) and APX005 (black circles) on human Daudi B cells. Rituximab (red triangles) and human IgG1 isotype control (open circles) were used as controls. All conditions were performed in triplicate. Graph is representative of three donors
APX005M does not mediate antibody-dependent cell cytotoxicity
We assessed the ADCC activity of APX005M alongside APX005 to confirm lack of effector function due to loss of binding to FcγRIIIa. Dose-dependent killing of CD40 + Daudi B-cell targets by CD56 + CD16 + NK cells was evaluated after APX005M or APX005 treatment by monitoring release of lactate dehydrogenase into the culture media. As expected, APX005M was unable to mediate ADCC of CD40-expressing cells (Fig. 2d) as compared to APX005.
APX005M enhances T-cell responses in vitro
Since the primary mode of action (MOA) of APX005M is to activate APCs and prime tumor-specific T-cell responses, we performed a viral antigen recall assay using primary human PBMC from donors previously exposed to EBV challenged with EBV antigens. APX005M potently enhanced IFN-γ secretion in a dose-dependent manner in cultures stimulated with EBV antigens (Fig. 3a) supporting this MOA.

APX005M enhances T-cell responses in vitro. a Enhancement of antigen-specific T-cell response measured by IFN-γ production by human PBMCs stimulated with EBV peptides in the presence of indicated concentrations of APX005M for 5 days. Culture supernatants were assayed for IFN-γ in triplicate and data are shown as mean ± SEM. b Ex vivo cultures of NSCLC biopsies were treated with APX005M and T-cell proliferation was assessed by Ki67 expression on CD3 + T cells. Representative FACS data are shown in (c). Graph depicts MFI of Ki67 on CD3 + cells and represents data from three different NSCLC patients. *p value < 0.01
To assess the therapeutic potential of APX005M to generate anti-tumor T-cell responses, ex vivo assays from tumor biopsies were performed. Single-cell suspensions containing both tumor cells and tumor-infiltrating immune cells derived from freshly isolated human non-small cell lung cancer (NSCLC) biopsies from consented patients were stimulated with APX005M (67 nM) for 5 days and then T-cell proliferation was determined. As shown in Fig. 3b, c, APX005M significantly enhanced tumor-infiltrating T-cell proliferation. These data suggest APX005M may promote antigen presentation by APCs, leading to the activation of tumor-infiltrating T-cell responses.
APX005M is functionally distinct from other CD40 agonist antibodies
APX005M has unique structural characteristics compared to other reported CD40 agonist antibodies in clinical development. To determine whether these differences impart distinct functionality of APX005M, we compared the biological activity of analogs of CP-870,893, SGN-40 and ADC-1013. Though all antibodies tested showed binding to CD40 on DC (albeit with different affinities) (Fig. 4a), only CP-870,893 analog and APX005M were able to stimulate APCs (Fig. 4b). Interestingly, only APX005M was able to induce substantial secretion of IL-12p70 from human DCs, which is a key functional difference as IL-12 is critical for the activation of naïve T cells (Fig. 4c).

APX005M induces potent activation of dendritic cells. a Binding of CD40 agonist antibodies to monocyte-derived DC (moDCs). Activation of moDCs by CD40 agonist antibodies for 48 h was measured by b MFI of CD86 expression and c IL-12p70 production in culture supernatants. All data are represented as mean ± SEM of triplicate wells and assays were performed in at least two donors. d The effect of CD40 agonist antibodies on T cells was assessed in MLR. moDCs were stimulated with serial dilutions of CD40 agonist antibodies for 24 h, and allogeneic eFluor 670-labeled CD8 T cells were added. Cell proliferation indicated by eFluor 670 dye dilution was assessed on day 6 using a MACSQuant Analyzer. IFN-γ secretion was measured from cell supernatants of MLR cultures (e). All data are represented as mean ± SEM of triplicate wells and assays were performed in at least two donors
To evaluate whether the observed differences in DC activation for these agonists translate to similar differences in downstream T-cell activation, we performed mixed lymphocyte reactions (MLR) using DC pre-activated with varying concentrations of antibody then co-cultured with allogeneic human CD8 T cells to examine degree of proliferation and IFN-γ induction. APX005M-activated APCs induced proliferation of CD8 T cells (Fig. 4d). CP-870,893 also induced proliferation of T cells, though to a lesser extent than APX005M. In contrast, SGN-40 and ADC-1013 were weak inducers of T-cell proliferation. Interestingly, only APX005M was able to induce significant secretion of IFN-γ (Fig. 4e), correlating with IL-12 secretion from DC (Fig. 4c). Similar results were obtained with CD4 T cells (data not shown). These data suggest that the epitope and Fc mutation of APX005M drive more potent activation of both the innate and adaptive immune responses as compared the other CD40 agonists tested.
APX005M induces long-lasting APC activation and achieves maximum potency at 10% receptor occupancy
As shown above, APX005M possesses unique attributes that may translate into better safety and efficacy in patients. To support clinical development for first-in-human studies, we explored the pharmacodynamics of APX005M. We first examined the relationship between receptor occupancy and bioactivity. The dose–response relationship between CD40 receptor occupancy (Fig. 5a) and B-cell activation (Fig. 5b) revealed that only 10% receptor occupancy (1 nM) is sufficient to produce maximum activity (CD86 expression on B cells) (Fig. 5c), suggesting that saturation of CD40 receptors is not required for APX005M to achieve optimal agonistic activity and a low clinical dose may be sufficient to achieve maximal pharmacological effect.

APX005M induces long-lasting APC activation and achieves maximum potency at 10% receptor occupancy. A weighed least-square procedure in Phoenix WinNonlin 6.4 was used to establish a correlation between CD40 receptor occupancy on B cells (a) and B-cell activation (b). The predicted CD40 receptor occupancy at the predicted maximum B-cell activation as expressed of CD86 expression was derived from this fit (c). CD19 + B cells were cultured with indicated concentrations of APX005M or isotype control antibody for 1 week. In washed conditions, antibodies were washed out of the cultures after 24-h incubation. In control conditions, antibodies were maintained throughout culture. At the end of the week, B cells were harvested and expression of HLA-DR (d) and CD86 (e) was assessed by flow cytometry. Data are presented as mean + SEM and represents at least three different experiments
Next, we performed assays to determine the duration of B-cell activation following short-term exposure to APX005M. B cells were stimulated with APX005M or isotype control antibody for 24 h and then antibodies were washed out. After washout, B cells were continuously cultured for 1, 2 or 3 weeks and HLA-DR (Fig. 5d) and CD86 (Fig. 5e) were measured. As a control, B cells were cultured with APX005M or isotype control antibody without washout for 1 week. B cell activation was similar in B cell cultures that had been exposed to APX005M throughout the culture for 1 week to those that had been exposed to APX005M for only 24 h (Fig. 5d, e). B-cell activation was detected up to 3 weeks following 24-h exposure, though not to the same levels as B cells exposed to APX005M throughout the culture (data not shown). These results indicate that short-term exposure to APX005M can induce long-lasting activation of APC which should allow for reduced dosing frequency.
APX005M pharmacology in non-human primates
A tissue-cross-reactivity study of APX005M was performed on human and cynomolgus macaque tissue panels. Positive staining by APX005M was considered to be generally consistent with known sites of CD40 antigen expression and positive specific staining was seen in multiple cell types including follicular lymphocytes, bone marrow cells, macrophages, endothelial cells and reticular cells in the thymic medulla. Staining was similar between human and cynomolgus tissues which supported use of the cynomolgus monkey as an appropriate species for toxicologic evaluation.
The safety profile of APX005M was thus assessed in a repeat-dose study in cynomolgus monkeys. APX005M was administered IV at 0.3, 3 and 30 mg/kg weekly for 4 weeks (a total of 5 doses). The observed PK properties of APX005M (Table 1) appeared typical of other humanized monoclonal antibodies in monkeys. Increases in the dose of APX005M (0.3 mg/kg to 30 mg/kg) led to dose-proportional increases in Cmax and more than dose-proportional increases in AUC(0–168). The no observable adverse effect level (NOAEL) was determined to be 30 mg/kg.
The major pharmacodynamic effect was a reduction in peripheral B lymphocyte numbers observed within 24 h of APX005M administration in most animals dosed at 3 and 30 mg APX005M/kg (data not shown). This effect was observed over the course of the 28-day observation period following the final administration of APX005M. Since APX005M does not mediate ADCC and CDC on CD40 + cells but can activate B cells, it is possible that APX005M activated circulating B cells leading to extravasation of B cells to peripheral lymphoid tissues such as lymph nodes as has been hypothesized for other CD40 agonists [20]. There were no findings suggestive of adverse effects on organ system function including central nervous, respiratory, and cardiovascular systems, indicating that APX005M was well-tolerated in monkeys. While increases in several cytokines were observed in human PBMC cultures stimulated with APX005M, including TNF-α, IL-6, IL-12 and IFN-γ (Supplemental Fig. 2), there were no drug-associated changes in IL-1β, IL-2, IL-4, IL-5, IL 6, IL-12/23 (p40), IL-13, IL-17, G-CSF, GM-CSF, IFN-γ, and TNF-α in cyno monkey PBMC culture. Since the binding of APX005M to cyno CD40 is similar to human, these differences may be attributed to differences in Fc receptor expression between the species.
Discussion
The treatment of cancer by manipulation of the immune system is a therapeutic modality with potentially curative effect. Antibodies targeting ICIs have shown significant and long-lasting anti-tumor activity in some patients, but many fail to respond to treatment or become refractory suggesting that simply blocking T-cell checkpoints is not sufficient to restore anti-tumor immunity in every case. One promising alternative to ICIs is the use of CD40 agonist antibodies to prime APCs and invigorate anti-tumor T cells that have been inhibited by the suppressive TME. In addition, CD40 agonism can potentially prime immune responses to novel T-cell epitopes further facilitating cancer eradication.
Agonistic antibodies tend to have different functionality and potency depending upon the affinity, epitope and FcR binding profile emphasizing the importance of careful selection of the appropriate binding epitope, affinity, and Fc engineering to achieve the desired profile of agonist antibodies for cancer immunotherapy. We demonstrate that APX005M is a potent CD40 agonist antibody that binds the CD40L binding site and depends on Fc receptor-mediated crosslinking for bioactivity. These properties make APX005M unique among the CD40 agonists currently in clinical development. APX005M potently activates APCs leading to expression of costimulatory molecules and inflammatory cytokine secretion followed by robust T-cell proliferation in response to antigens, including tumor-specific antigen. APX005M was the only agonist tested in our studies that induced potent secretion of IL-12 p70, which is essential for activation of naïve T cells and NK cells, indicating that APX005M may play an important role in initiating anti-tumor immune responses by multiple immune cell types. We hypothesize that the unique features of APX005M are necessary to enable in vivo priming of effective anti-tumor immune responses with less systemic toxicities.
Conflicting data have been published regarding the role of epitope specificity in conferring agonistic activity to CD40 antibodies. For example, it was recently suggested that CD40 agonist antibodies that bind membrane-distal epitopes of CD40 without blocking ligand binding are more potent agonists [22]. One hypothesis was that membrane-distal epitopes allow better access to the Fc portion of the antibody for more efficient cross-linking. Here, we show that APX005M, which binds to the CD40L binding site and effectively competes with CD40L, elicits more potent agonistic activity than antibodies that do not block ligand binding, suggesting Fc-crosslinking can still occur when the epitope on CD40 is proximal to the cell membrane. To the best of our knowledge, aside from APX005M, all other CD40 agonist antibodies in clinical development bind epitopes that do not compete with CD40L for binding [20, 38,39,40]. Our data argue that the specific binding epitope of APX005M may have biological and therapeutic significance when coupled with a requirement for FcR-dependent crosslinking. APX005M likely activates CD40 in a manner similar to endogenous CD40L because both bind overlapping sites on CD40 and likely have similar modes of action.
In addition to the unique epitope of APX005M, the Fc region of APX005M plays a critical role in mediating its functions, as F(ab’)2 fragments of APX005M lack biological activity. This contrasts with the Fc-independent activity seen following stimulation with CP-870,893 [22, 41] suggesting that the combination of binding epitope and interaction with FcRs is essential for optimal agonistic activity of CD40 agonist antibodies. Fitting with this hypothesis, the two commonly used anti-mouse CD40 agonist antibody clones, FGK45 and IC10, bind the CD40L binding site of CD40 and depend on FcR crosslinking and both show potent anti-tumor efficacy in preclinical mouse models [24]. These antibodies therefore represent the best mouse-specific surrogates for APX005M.
The requirement for Fc-crosslinking varies among CD40 agonist antibodies currently in clinical development. ADC-1013, SGN-40, and ChiLob 7/4 all require crosslinking for agonist activity whereas CP-870,893 agonist activity is independent of crosslinking [39, 41,42,43]. In mouse models, enhanced engagement of CD40 agonist antibodies by FcγRIIb results in optimal CD40 agonist activity [14, 23, 26]. As such, APX005M was optimized for engagement to FcγRIIb. Our data are consistent with studies that showing CD40 agonist activity is significantly enhanced by binding to FcγRIIb, which mediates crosslinking of the antibody leading to efficient clustering of CD40 receptors facilitating downstream signaling [23, 26]. In addition to enhancing the potency of APX005M, the S267E mutation reduces its binding to FcγRIIIa resulting in abrogation of ADCC activity. This feature of APX005M further enhances its therapeutic value by preventing the depletion of CD40-expressing APCs, which are key mediators of the anti-tumor response. The requirement for FcγR binding may translate into a better safety profile for APX005M in the clinic.
Because cells expressing FcγRIIb are most prevalent in tumor tissues and tumor draining lymph nodes (TDLNs), we hypothesize that APX005M may not be as active in the circulation due to limited FcR crosslinking. This should reduce on-target toxicities observed with other CD40 agonists, such as cytokine release syndrome. In contrast, TDLNs and tumor environment provides more cell–cell interaction promoting effective CD40 crosslinking through FcγRIIb [44] and allowing for maximum potency of APX005M in the tumor itself.
To better understand the pharmacology of APX005M and inform dosing regimen for first-in-human studies, we performed a correlation between receptor occupancy and activity. We found that maximal APX005M activity could be achieved at only 10% receptor occupancy, suggesting efficacy may be seen in humans at relatively low doses. In addition, we found that short-term exposure to APX005M resulted in long-term APC activation, suggesting that APX005M can be administered less frequently. Our initial results from a phase 1 dose-escalation study with APX005M (Study NCT02482168) revealed potent and dose-dependent on-target bioactivity demonstrated by the induction of peripheral immune activation based on cell phenotyping and serum cytokines at relatively low doses and with every 3 week dosing [28]. These data further validate the concept that APX005M stimulates immune activation in patients with cancer with a well tolerated safety profile
Abbreviations
- APC:
-
Antigen presenting cell
- ICA:
-
Immune checkpoint activator
- ICI:
-
Immune checkpoint inhibitor
- IRAE:
-
Immune-related adverse events
- PBMC:
-
Peripheral blood mononuclear cells
References
Grewal IS, Flavell RA (1998) CD40 and CD154 in cell-mediated immunity. Annu Rev Immunol 16:111–135. https://doi.org/10.1146/annurev.immunol.16.1.111
Diehl L, den Boer AT, Schoenberger SP, van der Voort EI, Schumacher TN, Melief CJ, Offringa R, Toes RE (1999) CD40 activation in vivo overcomes peptide-induced peripheral cytotoxic T-lymphocyte tolerance and augments anti-tumor vaccine efficacy. Nat Med 5(7):774–779. https://doi.org/10.1038/10495
French RR, Chan HT, Tutt AL, Glennie MJ (1999) CD40 antibody evokes a cytotoxic T-cell response that eradicates lymphoma and bypasses T-cell help. Nat Med 5(5):548–553. https://doi.org/10.1038/8426
Sotomayor EM, Borrello I, Tubb E, Rattis FM, Bien H, Lu Z, Fein S, Schoenberger S, Levitsky HI (1999) Conversion of tumor-specific CD4+ T-cell tolerance to T-cell priming through in vivo ligation of CD40. Nat Med 5(7):780–787. https://doi.org/10.1038/10503
Beatty GL, Chiorean EG, Fishman MP, Saboury B, Teitelbaum UR, Sun W, Huhn RD, Song W, Li D, Sharp LL, Torigian DA, O’Dwyer PJ, Vonderheide RH (2011) CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science 331(6024):1612–1616. https://doi.org/10.1126/science.1198443
Sartorius R, D’Apice L, Barba P, Guardiola J, Santoni A, Velotti F, De Berardinis P (2003) Induction of human NK cell-mediated cytotoxicity by CD40 triggering on antigen presenting cells. Cell Immunol 221(2):81–88
Khong A, Nelson DJ, Nowak AK, Lake RA, Robinson BW (2012) The use of agonistic anti-CD40 therapy in treatments for cancer. Int Rev Immunol 31(4):246–266. https://doi.org/10.3109/08830185.2012.698338
Khalil M, Vonderheide RH (2007) Anti-CD40 agonist antibodies: preclinical and clinical experience. Update Cancer Ther 2(2):61–65. https://doi.org/10.1016/j.uct.2007.06.001
Rech AJ, Dada H, Kotzin JJ, Henao-Mejia J, Minn AJ, Twyman-Saint Victor C, Vonderheide RH (2018) Radiotherapy and CD40 activation separately augment immunity to checkpoint blockade in cancer. Cancer Res. https://doi.org/10.1158/0008-5472.CAN-17-3821
Sandin LC, Orlova A, Gustafsson E, Ellmark P, Tolmachev V, Totterman TH, Mangsbo SM (2014) Locally delivered CD40 agonist antibody accumulates in secondary lymphoid organs and eradicates experimental disseminated bladder cancer. Cancer Immunol Res 2(1):80–90. https://doi.org/10.1158/2326-6066.CIR-13-0067
Winograd R, Byrne KT, Evans RA, Odorizzi PM, Meyer AR, Bajor DL, Clendenin C, Stanger BZ, Furth EE, Wherry EJ, Vonderheide RH (2015) Induction of T-cell immunity overcomes complete resistance to PD-1 and CTLA-4 blockade and improves survival in pancreatic carcinoma. Cancer Immunol Res 3(4):399–411. https://doi.org/10.1158/2326-6066.CIR-14-0215
Luheshi NM, Coates-Ulrichsen J, Harper J, Mullins S, Sulikowski MG, Martin P, Brown L, Lewis A, Davies G, Morrow M, Wilkinson RW (2016) Transformation of the tumour microenvironment by a CD40 agonist antibody correlates with improved responses to PD-L1 blockade in a mouse orthotopic pancreatic tumour model. Oncotarget 7(14):18508–18520. https://doi.org/10.18632/oncotarget.7610
Guo C, Manjili MH, Subjeck JR, Sarkar D, Fisher PB, Wang XY (2013) Therapeutic cancer vaccines: past, present, and future. Adv Cancer Res 119:421–475. https://doi.org/10.1016/B978-0-12-407190-2.00007-1
Li F, Ravetch JV (2011) Inhibitory Fcgamma receptor engagement drives adjuvant and anti-tumor activities of agonistic CD40 antibodies. Science 333(6045):1030–1034. https://doi.org/10.1126/science.1206954
Rolph MS, Kaufmann SH (2001) CD40 signaling converts a minimally immunogenic antigen into a potent vaccine against the intracellular pathogen Listeria monocytogenes. J Immunol 166(8):5115–5121
Nimanong S, Ostroumov D, Wingerath J, Knocke S, Woller N, Gurlevik E, Falk CS, Manns MP, Kuhnel F, Wirth TC (2017) CD40 signaling drives potent cellular immune responses in heterologous cancer vaccinations. Cancer Res 77(8):1918–1926. https://doi.org/10.1158/0008-5472.CAN-16-2089
Vonderheide RH (2018) The immune revolution: a case for priming, Not Checkpoint. ancer Cell 33(4):563–569. https://doi.org/10.1016/j.ccell.2018.03.008
Vonderheide RH, Dutcher JP, Anderson JE, Eckhardt SG, Stephans KF, Razvillas B, Garl S, Butine MD, Perry VP, Armitage RJ, Ghalie R, Caron DA, Gribben JG (2001) Phase I study of recombinant human CD40 ligand in cancer patients. J Clin Oncol 19(13):3280–3287. https://doi.org/10.1200/JCO.2001.19.13.3280
Beatty GL, Li Y, Long KB (2017) Cancer immunotherapy: activating innate and adaptive immunity through CD40 agonists. Expert Rev Anticancer Ther 17(2):175–186. https://doi.org/10.1080/14737140.2017.1270208
Vonderheide RH, Flaherty KT, Khalil M, Stumacher MS, Bajor DL, Hutnick NA, Sullivan P, Mahany JJ, Gallagher M, Kramer A, Green SJ, O’Dwyer PJ, Running KL, Huhn RD, Antonia SJ (2007) Clinical activity and immune modulation in cancer patients treated with CP-870,893, a novel CD40 agonist monoclonal antibody. J Clin Oncol 25(7):876–883. https://doi.org/10.1200/JCO.2006.08.3311
Bajor DL, Xu X, Torigian DA, Mick R, Garcia LR, Richman LP, Desmarais C, Nathanson KL, Schuchter LM, Kalos M, Vonderheide RH (2014) Immune activation and a 9-year ongoing complete remission following CD40 antibody therapy and metastasectomy in a patient with metastatic melanoma. Cancer Immunol Res 2(11):1051–1058. https://doi.org/10.1158/2326-6066.CIR-14-0154
Yu X, Chan HTC, Orr CM, Dadas O, Booth SG, Dahal LN, Penfold CA, O’Brien L, Mockridge CI, French RR, Duriez P, Douglas LR, Pearson AR, Cragg MS, Tews I, Glennie MJ, White AL (2018) Complex interplay between epitope specificity and isotype dictates the biological activity of anti-human CD40 antibodies. Cancer Cell 33(4):664-675 e664. https://doi.org/10.1016/j.ccell.2018.02.009
White AL, Chan HT, Roghanian A, French RR, Mockridge CI, Tutt AL, Dixon SV, Ajona D, Verbeek JS, Al-Shamkhani A, Cragg MS, Beers SA, Glennie MJ (2011) Interaction with FcgammaRIIB is critical for the agonistic activity of anti-CD40 monoclonal antibody. J Immunol 187(4):1754–1763. https://doi.org/10.4049/jimmunol.1101135
Richman LP, Vonderheide RH (2014) Role of crosslinking for agonistic CD40 monoclonal antibodies as immune therapy of cancer. Cancer Immunol Res 2(1):19–26. https://doi.org/10.1158/2326-6066.CIR-13-0152
Barr TA, Heath AW (2001) Functional activity of CD40 antibodies correlates to the position of binding relative to CD154. Immunology 102(1):39–43
Dahan R, Barnhart BC, Li F, Yamniuk AP, Korman AJ, Ravetch JV (2016) Therapeutic activity of agonistic, human anti-CD40 monoclonal antibodies requires selective FcgammaR engagement. Cancer Cell 29(6):820–831. https://doi.org/10.1016/j.ccell.2016.05.001
Chu SY, Vostiar I, Karki S, Moore GL, Lazar GA, Pong E, Joyce PF, Szymkowski DE, Desjarlais JR (2008) Inhibition of B cell receptor-mediated activation of primary human B cells by coengagement of CD19 and FcgammaRIIb with Fc-engineered antibodies. Mol Immunol 45(15):3926–3933. https://doi.org/10.1016/j.molimm.2008.06.027
Bendell JC, Fakih M, Infante JR, Bajor DL, Cristea MC, Tremblay T, Trifan OC, Vonderheide RH (2016) Phase 1 study to evaluate the safety and tolerability of the CD40 agonistic monoclonal antibody APX005M in subjects with solid tumors. J Clin Oncol 34(15_suppl):TPS3110–TPS3110. https://doi.org/10.1200/JCO.2016.34.15_suppl.TPS3110
Johnson DH, Bentebibel SE, Lecagoonporn S, Bernatchez C, Haymaker CL, Murthy R, Tam A, Yee C, Amaria RN, Patel SP, Tawbi HA-H, Glitza IC, Davies MA, Hwu W-J, Hwu P, Overwijk WW, Diab A (2018) Phase I/II dose escalation and expansion cohort safety and efficacy study of image guided intratumoral CD40 agonistic monoclonal antibody APX005M in combination with systemic pembrolizumab for treatment naive metastatic melanoma. J Clin Oncol 36(15_suppl):TPS3133–TPS3133. https://doi.org/10.1200/JCO.2018.36.15_suppl.TPS3133
Spieker-Polet H, Sethupathi P, Yam PC, Knight KL (1995) Rabbit monoclonal antibodies: generating a fusion partner to produce rabbit-rabbit hybridomas. Proc Natl Acad Sci USA 92(20):9348–9352
Zhu W (2009) Rabbit Hybridoma. In: therapeutic monoclonal antibodies: from bench to clinic, pp 151–168. doi:https://doi.org/10.1002/9780470485408.ch6
Yu Y, Lee P, Ke Y, Zhang Y, Yu Q, Lee J, Li M, Song J, Chen J, Dai J, Do Couto FJ, An Z, Zhu W, Yu GL (2010) A humanized anti-VEGF rabbit monoclonal antibody inhibits angiogenesis and blocks tumor growth in xenograft models. PLoS ONE 5(2):e9072. https://doi.org/10.1371/journal.pone.0009072
Ellmark PBJ (2014) Anti-CD40 antibodies, uses and methods. United States Patent, 20140348836
Presta LG (2012) Humanized anti-CD40 antibodies and thier methods of use. United States Patent, 8303955
Beidan V (2008) Methods of treating cancer and enhancing immune responses with antibodies that bind CD40. United States Patent, 7338660
Timmerman P, Puijk WC, Meloen RH (2007) Functional reconstruction and synthetic mimicry of a conformational epitope using CLIPS technology. J Mol Recognit 20(5):283–299. https://doi.org/10.1002/jmr.846
An HJ, Kim YJ, Song DH, Park BS, Kim HM, Lee JD, Paik SG, Lee JO, Lee H (2011) Crystallographic and mutational analysis of the CD40-CD154 complex and its implications for receptor activation. J Biol Chem 286(13):11226–11235. https://doi.org/10.1074/jbc.M110.208215
Johnson P, Challis R, Chowdhury F, Gao Y, Harvey M, Geldart T, Kerr P, Chan C, Smith A, Steven N, Edwards C, Ashton-Key M, Hodges E, Tutt A, Ottensmeier C, Glennie M, Williams A (2015) Clinical and biological effects of an agonist anti-CD40 antibody: a Cancer Research UK phase I study. Clin Cancer Res 21(6):1321–1328. https://doi.org/10.1158/1078-0432.CCR-14-2355
Mangsbo SM, Broos S, Fletcher E, Veitonmaki N, Furebring C, Dahlen E, Norlen P, Lindstedt M, Totterman TH, Ellmark P (2015) The human agonistic CD40 antibody ADC-1013 eradicates bladder tumors and generates T-cell-dependent tumor immunity. Clin Cancer Res 21(5):1115–1126. https://doi.org/10.1158/1078-0432.CCR-14-0913
Vitale LA, O’Neill T, Widger J, Crocker A, He L-Z, Weidlick J, Sundarapandiyan K, Storey J, Thomas L, Goldstein J, Marsh HC, Keler T (2016) Abstract 4866: Development and characterization of novel CD40 antibody agonists for cancer immunotherapy. Can Res 76(14 Supplement):4866–4866. https://doi.org/10.1158/1538-7445.Am2016-4866
White AL, Chan HT, French RR, Willoughby J, Mockridge CI, Roghanian A, Penfold CA, Booth SG, Dodhy A, Polak ME, Potter EA, Ardern-Jones MR, Verbeek JS, Johnson PW, Al-Shamkhani A, Cragg MS, Beers SA, Glennie MJ (2015) Conformation of the human immunoglobulin G2 hinge imparts superagonistic properties to immunostimulatory anticancer antibodies. Cancer Cell 27(1):138–148. https://doi.org/10.1016/j.ccell.2014.11.001
Lapalombella R, Gowda A, Joshi T, Mehter N, Cheney C, Lehman A, Chen C-S, Johnson AJ, Caligiuri MA, Tridandapani S, Muthusamy N, Byrd JC (2009) The humanized CD40 antibody SGN-40 demonstrates pre-clinical activity that is enhanced by lenalidomide in chronic lymphocytic leukaemia. Br J Haematol 144(6):848–855. https://doi.org/10.1111/j.1365-2141.2008.07548.x
Chowdhury F, Johnson PW, Glennie MJ, Williams AP (2014) Ex vivo assays of dendritic cell activation and cytokine profiles as predictors of in vivo effects in an anti-human CD40 monoclonal antibody ChiLob 7/4 phase I trial. Cancer Immunol Res 2(3):229–240. https://doi.org/10.1158/2326-6066.CIR-13-0070
Cassard L, Cohen-Solal JF, Fournier EM, Camilleri-Broet S, Spatz A, Chouaib S, Badoual C, Varin A, Fisson S, Duvillard P, Boix C, Loncar SM, Sastre-Garau X, Houghton AN, Avril MF, Gresser I, Fridman WH, Sautes-Fridman C (2008) Selective expression of inhibitory Fcgamma receptor by metastatic melanoma impairs tumor susceptibility to IgG-dependent cellular response. Int J Cancer 123(12):2832–2839. https://doi.org/10.1002/ijc.23870
Acknowledgements
The authors would like to thank Ovid Trifan for his input on the study and manuscript. We would also like to thank Ryan Alvarado for formatting the manuscript. We are grateful to Emil Samara for help with NHP PK analysis.
Funding
This work was funded by Apexigen, Inc.
Author information
Authors and Affiliations
Contributions
Erin L. Filbert: study design, methodology, data analysis, and manuscript preparation. Pia K Bjorck: study design, methodology, and data analysis. Minu K. Srivastava: manuscript preparation. Frances R. Bahjat: manuscript preparation. Xiaodong Yang: study design, methodology, data analysis, and manuscript preparation.
Corresponding author
Ethics declarations
Conflict of interest
All authors are current or former employees and stockholders of Apexigen, Inc.
Ethics approval and consent
Informed consent for human blood samples was obtained by Stanford Blood Center. Nilogen, Inc. obtained informed consent for tumor biopsies. Non-human primate studies were approved by the Charles River IACUC and were handled according to AAALAC rules.
Cell line authentication
Cell lines were sourced directly from vendors that provide authentication.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Filbert, E.L., Björck, P.K., Srivastava, M.K. et al. APX005M, a CD40 agonist antibody with unique epitope specificity and Fc receptor binding profile for optimal therapeutic application. Cancer Immunol Immunother 70, 1853–1865 (2021). https://doi.org/10.1007/s00262-020-02814-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00262-020-02814-2