
Abstract
Porcine circovirus type 2 (PCV2) is the primary causative agent of porcine circovirus-associated diseases in pigs. The sole structural capsid protein of PCV2, Cap, consists of major antigenic domains, but little is known about the assembly of capsid particles. The purpose of this study is to produce a large amount of Cap protein using Escherichia coli expression system for further studying the essential sequences contributing to formation of particles. By using codon optimization of rare arginine codons near the 5′-end of the cap gene for E. coli, a full-length Cap without any fusion tag recombinant protein (Cap1-233) was expressed and proceeded to form virus-like particles (VLPs) in normal Cap appearance that resembled the authentic PCV2 capsid. The N-terminal deletion mutant (Cap51-233) deleted the nuclear localization signal (NLS) domain, while the internal deletion mutant (CapΔ51-103) deleted a likely dimerization domain that failed to form VLPs. The unique Cys108 substitution mutant (CapC/S) exhibited most irregular aggregates, and only few VLPs were formed. These results suggest that the N-terminal region within the residues 1 to 103 possessing the NLS and dimerization domains are essential for self-assembly of stable Cap VLPs, and the unique Cys108 plays an important role in the integrity of VLPs. The immunogenicity of PCV2 VLPs was further evaluated by immunization of pigs followed by challenge infection. The Cap1-233-immunized pigs demonstrated specific antibody immune responses and are prevented from PCV2 challenge, thus implying its potential use for a VLP-based PCV2 vaccine.
Similar content being viewed by others


Introduction
Porcine circovirus (PCV) is a member of the family Circoviridae consisting of a circular single-stranded DNA genome. The virions are icosahedral, nonenveloped, and 20.5 nm in diameter (Crowther et al. 2003). PCV type 1 is nonpathogenic in swine and was first described in 1974 as a persistent contaminant of a porcine kidney PK-15 cell line (ATCC-CCL33, Tischer et al. 1986; Allen et al. 1995). In contrast, type 2 PCV (PCV2) is closely associated with a newly emerged disease called postweaning multisystemic wasting syndrome (PMWS) in growing pigs now known as porcine circovirus-associated diseases (Allan and Ellis 2000). The characteristic symptoms of the syndrome include severe progressive weight loss, dyspnea, tachypnea, anemia, diarrhea, and lymphadenopathy in pigs of 5–15 weeks of age (Allan and Ellis 2000; Ellis et al. 2004; Chae 2005). PMWS is endemic in the majority of the swine-producing countries, causing a severe economic impact on the swine industry worldwide.
The PCV2 genome has two major open reading frames (ORFs). They are ORF1, the rep gene, which encodes the Rep proteins responsible for virus replication and ORF2, the cap gene, which encodes the immunogenic capsid (Cap) protein (Mankertz et al. 1998; Truong et al. 2001). A recent report showed that a nonstructural protein encoded by ORF3 could induce apoptosis in the PCV2-infected cultured cells (Liu et al. 2005). The only structural protein of viral capsid, Cap, has become the major target for developing PCV2 vaccines and serological diagnostic reagents. Several recombinant Cap proteins expressed by the recombinant baculovirus expression system have been used as ELISA antigens and further as a subunit vaccine (Nawagitgul et al. 2000; Beach and Meng 2011). However, the production of recombinant proteins in the eukaryotic expression systems is more laborious and expensive. Since the N-terminal 41 amino acid (a.a.) residues of the PCV2 Cap protein possess a nuclear localization signal (NLS) containing the high incidence of arginine residues and rare codons for E. coli that was disadvantageous for full-length Cap expression (Liu et al. 2001; Zhou et al. 2005). Deletion of NLS (Trundova and Celer 2007) or fusion to a tag such as glutathione-S-transferase (Zhou et al. 2005) has been employed to improve the expression efficiency and stability of expressed protein in E. coli. The crystal structure of an N-terminally truncated PCV2 virus-like particle (VLP) at 2.3-Å resolution has been determined (Khayat et al. 2011). A full-length Cap has recently been demonstrated to express in E. coli by adapting codon usage and supplementing the host with rare tRNAs but has failed to self-assemble into VLPs (Marcekova et al. 2009).
The N-terminus of the PCV2 Cap contains residues rich in basic amino acids. Thus, it may be involved in the formation of the interior surface of the virion and interact with the negative charges of genomic DNA during virus assembly (Lekcharoensuk et al. 2004; Meehan et al. 1998). Studies on the hepatitis B virus (HBV) capsid particle assembly reveal that capsid proteins assemble into dimmers which provide the precursor, or assembly units for capsid formation (Zhou and Standring 1992a), and the Cys residues of capsid protein are not essential for particle assembly but can influence their stability (Zhou and Standring 1992b). Very little is known about the assembly of the PCV2 capsid particles, since insufficient quantities of purified virus are available for detailed studies. The purpose of this study is to produce a large amount of full-length Cap recombinant protein resembling the authentic native viral Cap using E. coli expression system, and further to study the essential sequences for particle assembly and immunogenicity of the PCV2 VLPs.
Materials and methods
Cloning of PCV2 cap gene by PCR and construction of recombinant expression plasmids
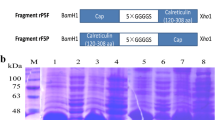
PCV2 genomic DNA was extracted with a commercial DNA extraction kit (QIAamp Tissue kit, QIAGEN) from 20 mg of frozen lymph nodes that was collected from a severe PMWS-affected piglet in a commercial farm in central Taiwan. The PCR primer pairs were designed according to the PCV2 Taiwan strain sequence (GenBank accession no. AY885225) and used for amplification of the defined coding region of PCV2 Cap (Table 1). The PCR reaction was carried out as described previously (Wu et al. 2008). The full length of the cap gene which was amplified with the primer pair mCapF1/2 and CapR233 was digested with restriction enzymes BglII/XhoI followed by cloning into the expression vector ΔpFlag which is derived from pFlag-1 (IBI) by deleting the franking region of FLAG tag (Fig. 1b) to generate the recombinant plasmid ΔpFlagCap1-233. The cap gene lacking the NLS region, which was amplified with the primer pair Cap51/BglII and CapR233, was digested with restriction enzymes BglII/XhoI followed by cloning into the expression vector ΔpFlag to generate the recombinant plasmid ΔpFlagCap51-233. The DNA fragment encoding the a.a. 1–50 of Cap was amplified with the primer pair mCapF1/2 and CapR50-2, followed by cloning to the ΔpFlagCap1-233 and replacing the fragment encoding the a.a. 1–103 of Cap with BglII/EcoRI digestion to generate the recombinant plasmid ΔpFlagCapΔ51-103. The DNA fragment encoding the a.a. 102–233 of Cap with the substitution from cysteine to serine at a.a. 108 was amplified with the primer pair CapC/S and CapR233, followed by cloning to the ΔpFlagCap1-233 and replacing the fragment encoding the a.a. 102–233 of Cap with EcoRI/XhoI digestion to generate the recombinant plasmid ΔpFlagCapC/S (Fig. 1a). All the recombinant plasmids were sequenced to confirm the accuracy of the open reading frame of cap coding sequences.

Construction of recombinant expression plasmids expressing different Cap proteins. a Schematic diagram of the expressed coding regions of PCV2 Cap recombinant subunits. Bars represent expressed coding sequences, and the amino acid residue numbers at both termini are indicated. b The cloning plasmid ΔpFlag with the flanking region of FLAG tag deleted
Expression of Cap protein in E. coli and purification of VLPs
Recombinant expression plasmids were transformed respectively into E. coli BL21-codon Plus (DE3)-RIL competent cells (Stratagene) according to the manufacturer’s manual. A single colony of transformant was grown in Luria–Bertani medium containing 50 μg/ml ampicillin at 37 °C until the OD600 reached 0.6. Then isopropyl β-d-thiogalactopyranoside was added to a final concentration of 1 mM. The culture was incubated for an additional 4 h at 30 °C. The cells were harvested by centrifugation at 6,000×g for 10 min at 4 °C and resuspended in Tris buffer [50 mM Tris (pH 7.2), 150 mM NaCl, 0.2 % Triton-X 100, 1 mM protease inhibitor, 5 % glycerol]. Cells were broken by sonication followed by centrifugation at 15,000×g for 10 min at 4 °C, and proteins were analyzed by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and Western blotting analysis. The supernatant was further transferred to a centrifuge tube containing equal volume of 40 % sucrose cushion and centrifuged at 210,000×g for 6 h at 4 °C for purification of VLPs. The pellet was resuspended with 0.5 ml of Tris buffer and examined by transmission electron microscope (TEM).
Western blotting analysis
Expressed proteins were resuspended in equal volumes of 2× SDS-PAGE sample buffer [125 mM Tris–Cl (pH 6.8), 20 % glycerol, 4 % SDS, 0.25 % bromophenol blue] in the presence or absence of 10 % β-mercaptoethanol. Proteins were separated by 12 % SDS-PAGE and then transferred by electroblotting onto PolyScreen PVDF transfer membrane (NEN) using semidry transfer cell (Bio-Rad) according to the manufacturer’s manual. The membrane was then treated sequentially with blocking solution [phosphate-buffered saline (PBS) containing 5 % skim milk], with appropriate dilution of mouse antiserum specific to Cap, and with anti-mouse IgG goat antibody conjugated to peroxidase (Zymed). Finally, the membrane was soaked in a chromogen/substrate solution (TMB single solution; Zymed) for color development.
Transmission electron microscope
Purified VLPs were adsorbed onto a copper grid for 3 min at room temperature. The grids were dried gently using filter paper and stained with saturated aqueous uranyl acetate for 40 s. The excess liquid was removed with filter, and the samples were examined under a TEM (JEOL JEM1400).
Immunization of pigs and challenge infection
Five 6-week-old SPF piglets were randomly allotted to the Cap (n = 3) and control (n = 2) groups. Each piglet was immunized intramuscularly into the neck region with 200 μg Cap VLP (Cap group) or normal saline (control group) in a 1:1 water-in-oil-in-water emulsion with the adjuvant ISA201 (SEPPIC) twice at a 2-week interval. The pigs were challenged intranasally and intramuscularly via the neck with 1 ml of 1 × 105 TCID50 PCV2 at 2 weeks after booster immunization. Pigs were checked daily for clinical signs, and the body weight was measured weekly. At 4 weeks post-challenge (p.c.) infection, the surviving animals were euthanized according to the guidelines of the Institutional Animal Care and Use Committee of National Chung Hsing University, and a necropsy was performed.
Indirect enzyme-linked immunosorbent assay
Serum blood samples were tested for PCV2 Cap-specific antibodies with an indirect enzyme-linked immunosorbent assay (ELISA). ELISA plates (Nunc) were coated at 4 °C overnight with 50 μl volume of 10 μg/ml purified Cap1-233 in coating buffer (carbonate buffer, pH 9.6). Each well was then thoroughly washed with PBS containing 0.05 % Tween-20 (PBST) and was added 50 μl volume of tested swine (1:100 diluted) serum in blocking buffer (PBS containing 1 % bovine serum albumin) containing 5 % E. coli lysate and incubated at 37 °C for 1 h. Subsequently, the plate was washed with PBST thoroughly, and each well was added 50 μl volume of 2,000-fold dilution of goat anti-swine IgG conjugated to peroxidase (Zymed) in blocking buffer at 37 °C for 45 min. The plate was washed with PBST three times followed by washing with PBS twice. Fifty microliters of freshly prepared chromogen/substrate solution (ABTS single solution, Zymed) was added into each well, and the plate was incubated at room temperature for 15 min in the dark. Finally, the reaction was stopped by adding an equal volume of stop solution (2 % SDS). Optical density (OD) of each well was read at 405 nm using a microplate reader (MRXII, Dynex). Each sample was analyzed in duplicate, and the OD405 of duplicates was averaged. The mean OD405 of SPF swine sera and an anti-PCV2 polyclone serum (VMRD) were used as negative and positive controls, respectively, to optimize the assays. Mean sample/positive (S/P) ratio of duplicate of each test was calculated.
Results
Expression of Cap protein variants in E. coli
In order to study the capsid assembly, variant Cap recombinant proteins without any fusion tag were expressed in E. coli. The defined coding regions of cap gene were amplified by PCR using carefully designed primer pairs (Table 1) and cloned into the expression vector ΔpFlag (Fig. 1b) to construct the recombinant plasmids including ΔpFlagCap1-233, ΔpFlagCap51-233, ΔpFlagCapΔ51-103, and ΔpFlagCapC/S (Fig. 1a). Four out of seven arginine codons near the utmost 5′ end of cap gene were optimized for efficient codon usage in E. coli. Those recombinant plasmids transformed respectively into E. coli BL21-codon plus (DE3)-RIL competent cells which contain extra copies of argU, ileY, and leuW genes for rare tRNAs, and the expressed proteins were characterized by Western blotting analysis. The full-length Cap protein which consisted 233 a.a. residues with the molecular weights of 25.7 kDa in size was expressed and designated Cap1-233. Two deletion mutants including an N-terminal deletion mutant (Cap51-233) lacking the NLS domain located at a.a. 1–50, and an internal deletion mutant (CapΔ51-103) lacking the portion of a.a. 51–103 were also successfully expressed with the estimated molecular weights of 20.1 and 19.9 kDa, respectively. Another full-length mutant designated CapC/S was constructed by substituting the unique cysteine (a.a. 108) of Cap to serine. All of the Cap recombinant proteins could be specifically recognized by the antiserum against PCV2 Cap, while except for CapC/S, all other three including Cap1-233, Cap51-233, and CapΔ51-103 were able to form homodimer in the absence of β-mercaptoethanol (Fig. 2).

a, b Characterization of recombinant Cap proteins by Western blotting analysis. Purified different Cap proteins were separated by 12 % SDS-PAGE in the presence (plus sign) or absence (minus sign) of β-mercaptoethanol followed by Western blotting analysis with mouse immune serum specific to PCV2 Cap. Lanes 1–5 are purified Cap1-233, Cap51-233, CapΔ51-103, CapC/S, and E. coli competent cell lysate, respectively. The expected Cap protein is indicated by an arrow and the dimmer form of Cap is indicated by an asterisk
Particles formed by the full length of Cap protein
The soluble expressed proteins were further purified by ultracentrifugation to detect whether the Cap protein is able to self-assemble into VLPs. TEM images of each purified protein revealed that only the full-length Cap protein (Cap1-233) could assemble into VLPs with diameter ranging from 15 to 20 nm (Fig. 3a). The full-length cysteine substitution mutant (CapC/S) exhibited most irregular aggregates, and only few VLPs were formed (Fig. 3d).

TEM images of Cap protein VLPs of PCV2. Purified Cap1-233 (a), Cap51-233 (b), CapΔ51-103 (c), CapC/S (d), and E. coli competent cell lysate (e) were negatively stained and observed by TEM. Scale bar 50 nm
Immunization of pigs and challenge infection
To evaluate the immunogenicity of Cap VLPs, three 6-week-old SPF piglets (nos. 1–3) were immunized intramuscularly with 200 μg Cap1-233 twice at a 2-week interval, while two control piglets (nos. 4–5) were immunized with normal saline. All Cap-immunized pigs seroconverted to Cap-specific antibody after booster immunization as determined by an indirect ELISA, while no antibody detected in the control pigs (Fig. 4a). The specificity of immune serum was further confirmed by indirect immunofluorescence with the Porcine Circovirus FA substrate slides (VMRD, USA) according to the manufacturer’s manual. After challenge infection, the control pigs exhibited growth stagnation and diarrhea, and had to be euthanized at 14 days due to severe clinical symptoms. In contrast, the Cap-immunized pigs grew healthily (Fig. 4b) without any clinical symptom, and postmortem analysis at 4 weeks p.c. revealed no evidence of PCV2 infection.

Time course of ELISA antibody development (a) and body weight (b) of the VLP-immunized pigs after vaccination and challenge
Discussion
PCV2 is a recognized essential infectious agent behind the development of PMWS, which is currently regarded as an important infectious swine viral disease, that results in a serious economic impact on the pig industry worldwide. The PCV2 Cap protein consists of major antigenic domains, suggesting its diagnostic and subunit vaccine potentials (Nawagitgul et al. 2002; Liu et al. 2004). Currently, commercially available PCV2 subunit vaccines have been developed using the baculovirus expression system. The recombinant Cap expressed in insect cells could assemble to form VLPs (Nawagitgul et al. 2002). However, the expression level is limited, and the cost of cell culture and time consumption in purification of expressed products are major concerns. In contrast, the E. coli expression systems have been utilized successfully for high-level expression of many heterologous proteins because of its relative simplicity, low cost, and fast high-density cultivation (Yin et al. 2010). The N-terminal NLS domain of the Cap protein is rich in arginine residues and contains several rare codons for E. coli that hampers a foreign gene expression (Liu et al. 2001; Zhou et al. 2005). Thus, using codon optimization and an alternative E. coli host strain which contains extra copies of genes for rare tRNAs can circumvent the difficulty of high-level expression of full-length Cap. The full-length Cap1-233 without any fusion tag was successfully expressed and proceeded to form VLPs. The small ubiquitin-like modifiers (SUMO) fusion protein expression system has recently been employed successfully to improve highly soluble full-length Cap-tag fusion protein expressed in E. coli, and the expressed Cap has the ability to self-assemble into VLPs after the protease cleavage of the SUMO fusion motif (Yin et al. 2010). Nevertheless, the tag motif may interfere with the protein conformation and antigenicity, and the cleavage and purification processes may be cumbersome and costly.
The PCV2 Cap contains sole cysteine residue at a.a. 108 (Cys108). In order to examine the role played by the Cys108 in the assembly of either dimmers or capsids, a Cys-minus mutant (CapC/S) was constructed. Those expressed proteins retaining the Cys108 such as Cap1-233, Cap51-233, and CapΔ51-103 could form homodimer in the absence of β-mercaptoethanol, while the full-length Cys108 substitution mutant CapC/S abolished this activity. This result suggests that the unique cysteine is responsible for dimmer formation of Cap via formation of a disulfide bond. In addition, in our previous study on the development of Cap subunit-based indirect ELISAs (Wu et al. 2008), the Cap subunit B representing the a.a. 51–100 exhibited both monomer and dimmer forms in the reducing condition (data not shown), leading to our speculation that this region might be related to a dimerization domain. The internal deletion mutant CapΔ51-103 did not show that the formation of VLPs seems to confirm our hypothesis. Only the full-length Cap1-233 demonstrated self-assembly into VLPs, suggesting that both regions in the N-terminal NLS domain and internal a.a. 51–103, a likely dimerization domain, are essential for capsid assembly. Indispensable sequences or motifs conferring VLPs formation are currently under study. Further, the Cys108 substitution mutant CapC/S exhibited most irregular aggregates but only few VLPs were formed, indicating that the Cys108 is dispensable for capsid assembly but formation of the disulfide bond is important for the integrity of the Cap VLPs.
VLPs mimic the structure of authentic virus particles and they are safe and very efficient at stimulating both cellular and humoral immune responses (Noad and Roy 2003; Antonis et al. 2006). Several VLPs from many viruses have been developed as vaccine candidates including two commercialized VLP-based vaccines that succeed in protecting humans from HBV and human papillomavirus infection (Grgacic and Anderson 2006; Ludwig and Wagner 2007). The immunogenicity of Cap VLPs was confirmed by the Cap1-233-immunized pigs that elicited specific antibody responses against Cap and offer prevention from PCV2 infection. A large-scale field trial is undertaken by comparing the growth performance between Cap1-233-vaccinated and unimmunized pigs.
In conclusion, a full-length authentic Cap protein of PCV2 has been successfully expressed using a modified E. coli expression system, and the expressed Cap was able to self-assemble into VLPs. Further characterizing variant Cap recombinant proteins reveals that the region in the N-terminal 103 residues containing the NLS and the likely dimerization domains are required for self-assembly of stable VLPs, and the sole Cys108 plays an important role in the integrity of VLPs. The PCV2 VLP-immunized pigs could elicit specific antibody responses and were prevented from PCV2 challenge, thus suggesting its potential as an alternative PCV2 subunit vaccine candidate with advantages of easy preparation and low cost.
References
Allan GM, Ellis JA (2000) Porcine circoviruses: a review. J Vet Diagn Invest 12:3–14
Allen GM, McNeilly F, Cassidy JP, Reilly GAC, Adair B, Ellis WA, McNulty MS (1995) Pathogenesis of porcine circovirus; experimental infections of colostrum deprived piglets and examination of pig foetal material. Vet Microbiol 44:49–64
Antonis AF, Bruschke CJ, Rueda P, Maranga L, Casal JI, Vela C, Hilgers LA, Belt PB, Weerdmeester K, Carrondo MJ, Langeveld JP (2006) A novel recombinant virus-like particle vaccine for prevention of porcine parvovirus-induced reproductive failure. Vaccine 24:5481–5490
Beach NM, Meng XJ (2011) Efficacy and future prospects of commercially available and experimental vaccines against porcine circovirus type 2 (PCV2). Virus Res. doi:10.1016/j.virusres.2011.09.041
Chae C (2005) A review of porcine circovirus 2-associated syndromes and diseases. Vet J 169:326–336
Crowther RA, Berriman JA, Curran WL, Allan GM, Todd D (2003) Comparison of the structures of three circoviruses: chicken anemia virus, porcine circovirus type 2, and beak and feather disease virus. J Virol 77:13036–13041
Ellis J, Clark E, Haines D, West K, Krakowka S, Kennedy S, Allan GM (2004) Porcine circovirus-2 and concurrent infections in the field. Vet Microbiol 98:159–163
Grgacic EV, Anderson DA (2006) Virus-like particles: passport to immune recognition. Methods 40:60–65
Khayat R, Brunn N, Speir JA, Hardham JM, Ankenbauer RG, Schneemann A, Johnson JE (2011) The 2.3 Angstrom structure of porcine circovirus 2. J Virol 85:7856–7862
Lekcharoensuk P, Morozov I, Paul PS, Thangthumniyom N, Wajjawalku W, Meng XJ (2004) Epitope mapping of the major capsid protein of type 2 porcine circovirus (PCV2) by using chimeric PCV1 and PCV2. J Virol 78:8135–8145
Liu Q, Tikoo KS, Babiuk LA (2001) Nuclear localization of the ORF2 protein encoded by porcine circovirus type 2. Virology 285:91–99
Liu C, Ihara T, Nunoya T, Ueda S (2004) Development of an ELISA based on the baculovirus-expressed capsid protein of porcine circovirus type 2 as antigen. J Vet Med Sci 66:237–242
Liu J, Chen I, Kwang J (2005) Characterization of a previously unidentified viral protein in porcine circovirus type 2-infected cells and its role in virus-induced apoptosis. J Virol 79:8262–8274
Ludwig C, Wagner R (2007) Virus-like particles-universal molecular toolboxes. Curr Opin Biotechnol 18:537–545
Mankertz A, Mankertz J, Wolf K, Buhk HL (1998) Identification of a protein essential for replication of porcine circovirus. J Gen Virol 79:381–384
Marcekova Z, Psikal I, Kosinova E, Benada O, Sebo P, Bumba L (2009) Heterologous expression of full-length capsid protein of porcine circovirus 2 in Escherichia coli and its potential use for detection of antibodies. J Virol Methods 162:133–141
Meehan BM, McNeilly F, Todd D, Kennedy S, Jewhurst VA, Ellis JA, Hassard LE, Clark EG, Haines DM, Allan GM (1998) Characterization of novel circovirus DNAs associated with wasting syndromes in pigs. J Gen Virol 79:2171–2179
Nawagitgul P, Morozov I, Bolin SR, Harms PA, Sorden SD, Paul PS (2000) Open reading frame 2 of porcine circovirus type 2 encodes a major capsid protein. J Gen Virol 81:2281–2287
Nawagitgul P, Harms PA, Morozov I, Thacker BJ, Sorden SD, Lekcharoensuk C, Paul PS (2002) Modified indirect porcine circovirus (PCV) type 2-based and recombinant capsid protein (ORF2)-based enzyme-linked immunosorbent assays for detection of antibodies to PCV. Clin Diagn Lab Immunol 9:33–40
Noad R, Roy P (2003) Virus-like particles as immunogens. Trends Microbiol 11:438–444
Tischer I, Mields W, Wolff D, Vagt M, Griem W (1986) Studies on epidemiology and pathogenicity of porcine circovirus. Arch Virol 91:271–276
Trundova M, Celer V (2007) Expression of porcine circovirus 2 ORF2 gene requires codon optimized E. coli cells. Virus Genes 34:199–204
Truong C, Mahe D, Blanchard P, Le Dimna M, Madec F, Jestin A, Albina E (2001) Identification of an immunorelevant ORF2 epitope from porcine circovirus type 2 as a serological marker for experimental and natural infection. Arch Virol 146:1197–1211
Wu PC, Chien MS, Tseng YY, Lin J, Lin WL, Yang CY, Huang C (2008) Expression of the porcine circovirus type 2 capsid protein subunits and application to an indirect ELISA. J Biotechnol 133:58–64
Yin S, Sun S, Yang S, Shang Y, Cai X, Liu X (2010) Self-assembly of virus-like particles of porcine circovirus type 2 capsid protein expressed from Escherichia coli. Virol J 7:166–170
Zhou S, Standring DN (1992a) Hepatitis B virus capsid particles are assembled from core-protein dimer precursors. Proc Natl Acad Sci U S A 89:10046–10050
Zhou S, Standring DN (1992b) Cys residues of the hepatitis B virus capsid protein are not essential for the assembly of viral core particles but can influence their stability. J Virol 66:5393–5398
Zhou JY, Shang SB, Gong H, Chen QX, Wu JX, Shen HG, Chen TF, Guo JQ (2005) In vitro expression, monoclonal antibody and bioactivity for capsid protein of porcine circovirus type II without nuclear localization signal. J Biotechnol 118:201–211
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Wu, PC., Lin, WL., Wu, CM. et al. Characterization of porcine circovirus type 2 (PCV2) capsid particle assembly and its application to virus-like particle vaccine development. Appl Microbiol Biotechnol 95, 1501–1507 (2012). https://doi.org/10.1007/s00253-012-4015-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-012-4015-2