
Abstract
Pantoea ananatis AJ13355 is a newly identified member of the Enterobacteriaceae family with promising biotechnological applications. This bacterium is able to grow at an acidic pH and is resistant to saturating concentrations of L-glutamic acid, making this organism a suitable host for the production of L-glutamate. In the current study, the complete genomic sequence of P. ananatis AJ13355 was determined. The genome was found to consist of a single circular chromosome consisting of 4,555,536 bp [DDBJ: AP012032] and a circular plasmid, pEA320, of 321,744 bp [DDBJ: AP012033]. After automated annotation, 4,071 protein-coding sequences were identified in the P. ananatis AJ13355 genome. For 4,025 of these genes, functions were assigned based on homologies to known proteins. A high level of nucleotide sequence identity (99%) was revealed between the genome of P. ananatis AJ13355 and the previously published genome of P. ananatis LMG 20103. Short colinear regions, which are identical to DNA sequences in the Escherichia coli MG1655 chromosome, were found to be widely dispersed along the P. ananatis AJ13355 genome. Conjugal gene transfer from E. coli to P. ananatis, mediated by homologous recombination between short identical sequences, was also experimentally demonstrated. The determination of the genome sequence has paved the way for the directed metabolic engineering of P. ananatis to produce biotechnologically relevant compounds.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
In the mid-1990s, specialists from the Ajinomoto Co., Inc. (Kawasaki, Japan), collected a gram-negative acidophilic bacterium from the soil of a tea plantation in Iwata-shi (Shizuoka, Japan), which was designated as strain AJ13355. This bacterium has proven to be capable of growing on a variety of sugars and organic acids at acidic and neutral pH values, and it is resistant to high concentrations of l-glutamic acid (l-Glu) (Moriya et al. 1999). According to standard microbiological tests on its bacteriological properties and nucleotide sequencing of its 16S rRNA (Kwon et al. 1997), this strain was identified as Pantoea ananatis (Takahashi et al. 2008).
The natural resistance of the AJ13355 strain to saturating l-Glu concentrations was used to pinpoint promising l-Glu producers using metabolically engineered P. ananatis strains and to develop an l-Glu fermentation process at an acidic pH accompanied by product precipitation (Izui et al. 2006). Other studies have indicated the potential for exploiting AJ13355-based engineered strains for overproduction of l-aspartic acid (Mokhova et al. 2010) and the other strains belonging to the Pantoea genus to produce cytotoxic and antiproliferative compounds (Kamimura et al. 1997; Page et al. 1999), 2,3-butanediol (Jiang 2007), pyrogallol (Zeida et al. 1998), glycerol (Dodge and Valle 2009), l-DOPA (Kumagai et al. 2008), such vitamins as carotenoids and vitamin E (Yoon et al. 2007; Albermann et al. 2008), pyrroloquinoline quinone (PQQ) (Andreeva et al. 2011), and ascorbic acid intermediates (Dodge and Valle 2009).
These findings encourage a practical interest in basic research on the structure of the P. ananatis AJ13355 genome, the metabolic features of this strain, and tools for genetically manipulating the bacterium.
Previously, a set of high-performance methods for the target modification of P. ananatis chromosome (such as deletion or insertions of genetic material, nucleotide change, modification of regulatory region, and construction of unmarked mutations) based on the site-specific and homologous recombination of phage λ, together with the possible transfer of marked mutations by electroporation of the chromosomal DNA, were adapted to genetically engineer P. ananatis AJ13355 and its derivatives (Katashkina et al. 2009). Recently, the method for in vivo cloning of large fragments of P. ananatis chromosome followed by it amplification via “Dual In/Out technology” (Minaeva et al. 2008) was developed (see, for more details, Andreeva et al. 2011). In the current study, the P. ananatis AJ13355 genome was sequenced and annotated.
Materials and Methods
Bacterial strains and plasmids
The strains and plasmids used in the present study are shown in Table 1.
Recombinant DNA techniques
All DNA manipulations were performed according to standard procedures (Sambrook and Russell 2001). λRed-driven modifications of the P. ananatis AJ13355 chromosome were completed according to previously developed methods (Katashkina et al. 2009).
Genome sequencing and annotation
Bacterial cultures of P. ananatis AJ13355 were grown in Luria–Bertani (LB) medium and were subsequently used to isolate genomic DNA using the Qiagen Genomic-Kit (Qiagen K.K., Tokyo, Japan).
Small-insert and large-insert P. ananatis AJ13355 DNA libraries were constructed after cloning fragments of 1–3 or 30–40 kb in length, which were obtained by partial digestion with Sau3A and purified by gel electrophoresis using low-melting agarose into the pUC18 plasmid or SuperCos 1 cosmid vector (Stratagene, La Jolla, CA), respectively. Sequencing was performed using either a DYEnamic ET Terminator Cycle Sequencing Kit (Amersham Pharmacia Biotech UK, Buckinghamshire, UK) or a BigDye Terminators with an ABI Prism 3700 DNA Analyzer (Applied Biosystems Japan, Tokyo, Japan). Obtained sequences were assembled into contigs using Clustering and Alignment Tool (CAT) software (Hitachi, Ltd., Tokyo, Japan).
Based on the end-sequence data from the obtained libraries, direct linkages between the contigs were estimated using CAT software, and linking cosmid clones were selected from the large-insert library. To fill the gaps between the contigs, sequences of the inserted fragments of the selected clones were determined using the primer-walking method. To determine the nucleotide sequences of the remaining gaps between the contigs, P. ananatis genomic DNA was amplified by polymerase chain reaction (PCR) using primers designed from the end sequences of the contigs, and the amplified products were directly sequenced by primer walking.
The edited P. ananatis AJ13355 genome sequences were uploaded in FASTA format and annotated.
Protein-coding sequences (CDSs) were predicted using GLIMMER software (Kasif et al. 1999), with an initial construction of the training model by sequence alignment of the genomes of P. ananatis and the reference strain Escherichia coli MG1655 using MAUVE software (Darling et al. 2010). The gene positions and their functional annotations were transferred from the reference to the P. ananatis AJ13355 genome based on strongly homologous regions to create the training model. Identified CDSs were compared against a nonredundant protein sequence database using TBLASTN (http://ncbi.nlm.nih.gov) software or using SwissProt database (http://www.uniprot.org) in cases of a low level of homology or P. ananatis-specific regions. Only the hits with an e value of less than 10−10 and a protein-length coverage of more than 75% were used. For each homologous region, the best hits were considered to be the sources of the functional annotations. Noncoding RNA predictions were performed using a framework for noncoding RNA detection (Raasch et al. 2010). Finally, all predictions were combined to produce a genome annotation and were checked by visual inspection to correct for gene starts and to reject false-positive predicted genes.
Comparative genome analysis
MAUVE software, a dot-matcher tool from the EMBOSS package (Rice et al. 2000), BLAST software (http://ncbi.nlm.nih.gov), and homemade Perl scripts were used for genome comparison and finding orthologous CDSs. The plasmids were linked to the end of the linearized chromosome of a corresponding Pantoea strain for easy understanding of the differences in the genomic structures.
For CDSs, found orthologues were linked to their genome position to evaluate synteny (i.e., a conserved order of orthologous CDSs between the compared genomes). For the generation of a Venn diagram, CDSs possessing an amino acid identity higher than 30% were defined as orthologues.
Construction of the E. coli Hfr strain
An oriT site was introduced into the E. coli MG1655 chromosome in the abrB gene, which is located approximately 10 kb upstream of the sucA gene. To achieve this insertion, a DNA fragment from the E. coli abrB gene, flanked by the EcoRI and SmaI restriction sites, was amplified by PCR using the E. coli MG1655 chromosome as a template and using the primers
-
P1: 5′-gatccccggggatacaccaaagagaaaataatgagggagc-3′ and
-
P2: 5′-gatcgaattccgcttcggcgcataggttgaaataaaccgc-3′,
the SmaI and EcoRI sites are underlined. The amplified DNA fragment was inserted between the EcoRI and SmaI sites in the mobilizable, conditionally replicated (pir +-dependent) plasmid pUT399 (CmR), with an origin of replication of γR6K and Mob+ locus from RP4 (Zhang and Meyer 1997) to generate pUT399-abrB [DDBJ: AB610285]. The E. coli MG1655 strain was subsequently transformed by pUT399-abrB, and an integrant, MG1655abrB::oriT, was obtained due to homologous recombination.
Conjugative crossing between E. coli and P. ananatis
The E. coli donor strain (MG1655abrB::oriT/pRK2013) carrying the conjugative pRK2013 (KmR) plasmid was cultivated at 37 °C on an LB agar plate. The TcR-plasmid carrier P. ananatis strain (IZ130/pMW118-(λattL-tetA-tetR-λattR), used as a recipient, was cultivated at 34 °C on an LB agar plate supplemented with 0.5% glucose and 1× M9 salt. Cells of both strains were collected from one fourth of a plate, washed twice with 1 ml of double-distilled water and resuspended in 300 μl of double-distilled water. Next, a 25-μl suspension of each strain was mixed and plated on an LB agar plate. After 3 h of incubation at room temperature, the cells were collected, plated on selective medium (M9 containing l-Glu [2 g/L] as the sole carbon source and 12.5 mg/l tetracycline [Tc]) and cultivated at 34 °C. After 1 week of cultivation, l-Glu-assimilating TcR-clones were randomly selected, and their chromosome structures were preliminarily checked by PCR using the primers P3: 5′-tgggacgaagagtacccgaacaaag-3′ and
-
P4: 5′-cgctgacaagctggagcaggaaaaa-3′.
Results
Assembly and annotation of the P. ananatis AJ13355 genome sequence
The genome of P. ananatis AJ13355 was sequenced using the shotgun method. One small-insert plasmid library (1–3 kb) and one cosmid library (30–40 kb) were constructed through the digestion and cloning of the genomic DNA.
Approximately 76,000 samples from the plasmid-based library (corresponding to more than 8.9-fold coverage of the genome size) were sequenced and were assembled using CAT software, which yielded 215 contigs with 96.3% coverage. End sequences from 600 samples of the cosmid-based library (corresponding to a coverage of more than fivefold of the genome size) were assembled using CAT software, which yielded nine contigs with 97.7% coverage. All the sequence and physical gaps were then closed by editing the ends of the sequence traces, primer walking on plasmid clones, and combinatorial PCR, which was followed by sequencing of the PCR product. As a result, two circular DNA molecules were obtained: a 4,555,536-bp chromosome [DDBJ: AP012032] and a 321,744-bp molecule, corresponding to the pEA320 plasmid [DDBJ: AP012033]. Previously, the autonomous replication of pEA320 in P. ananatis AJ13355 had been observed experimentally (Hara et al. 2007). General features of the P. ananatis genome are summarized in Table 2.
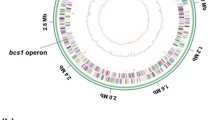
The GC-skew analysis of the P. ananatis AJ13355 chromosome, which is generally applicable to the identification of the leading and lagging strands in DNA replication (Grigoriev 1998), indicated bidirectional replication that starts at the proposed oriC sequence, near the mioC and gidA genes (with coordinates 3,803,243–3,803,475 in the sequenced chromosome), and ends close to the calculated replication terminus, around 1.5 Mb (Fig. 1). The 22 genes of the ribosomal RNAs identified in the P. ananatis chromosome were grouped into seven rrn operons. These operons are located in the two oppositely replicating halves (replichores) of the chromosome and are transcribed in the direction of replication, as previously demonstrated for several bacterial chromosomes (Blattner et al. 1997; Kunst et al. 1997; Veith et al. 2004).

Circular representation of the P. ananatis AJ13355 chromosome. Circles are numbered from 1 (outermost) to 8 (innermost). Circles 2 and 3 show the locations of predicted CDSs on the + and − strands, respectively. Circles 1, 4, and 5 show synteny with the E. coli MG1655 chromosome, as follows: circle 1, locations of orthologous CDSs on the + strand; circle 4, locations of orthologous CDSs on the − strand; and circle 5, locations of all P. ananatis CDSs orthologous to E. coli proteins. Circle 6 shows a G + C content greater and less than the average (0.538) outside and inside the corresponding circle, respectively. Circle 7 shows the positions and direction of transcription of seven P. ananatis rrn operons. Circle 8 depicts the GC skew (G−C/G + C)
GLIMMER and TBLASTN software were used to annotate 3,789 CDSs for the P. ananatis AJ13355 chromosome. Among these, 3,753 CDSs were homologous to previously annotated proteins.
According to the sequence data, the overall G + C content of the P. ananatis AJ13355 chromosome is 53.8% (Table 1), whereas the pEA320 megaplasmid exhibits a G + C content of 53.4%. These values are similar, suggesting that acquisition of the plasmid by P. ananatis AJ13355 was not a recent event, unless it was acquired by horizontal transfer from an organism with a similar G + C content.
According to predictions based on GLIMMER and TBLASTN software analysis, the pEA320 plasmid contains 282 CDSs. Among these CDSs, 39% are recognized as hypothetical proteins. GC-skew calculations predicted the location of the pEA320 origin of replication to be around 107 kb. This region contains the repA gene, coding for a homologue (64.7%) of the RepFIB replication initiation protein, which is specific for incompatibility group FI plasmids. Putative parAB genes were identified upstream of the repA gene. It is known that ParAB proteins are involved in the mechanisms of stable inheritance of large, low-copy-number plasmids (Velmurugan et al. 2003).
Comparison of the P. ananatis AJ13355 genome sequence with that of other members of the Pantoea genera
Over the last 3 years, the genomes of three Pantoea strains have been sequenced and annotated: P. ananatis LMG20103 (De Maayer et al. 2010), P. vagans C9-1 (Smits et al. 2010a), and Pantoea sp. At-9b (chromosome–NC_014837.1; plasmids—from NC_014838.1 to NC_014842.1). Figure 2 indicates the segments of pronounced colinear similarity between the compared genome pairs at the nucleotide level and suggests that patches of external DNA have been added to a conserved genomic core. Indeed, the genome of P. ananatis LMG20103, consisting of a single circular chromosome (4,703,373 bp in length), and the bacterial chromosome of P. ananatis AJ13355 share a common “backbone” sequence that is highly colinear, except for one extended region (about 350 kb) that is absent from the chromosome of the latter strain. However, this region of the P. ananatis LMG20103 chromosome is highly homologous to the entire autonomously replicated pEA320 plasmid from strain AJ13355 (Fig. 2b). Several homologous regions (about 10 kb long) were also found between pEA320 and chromosome Pantoea sp. At-9b and a chromosome of P. vagans C9-1 (data not shown).

Two-dimensional similarity plots comparing the nucleotide sequences of the Pantoea genomes. The comparison was conducted via BLASTN software using a discontiguous megablast algorithm with default parameters. The plots are as follows: P. ananatis AJ13355 versus Pantoea sp. At-9b a, P. ananatis AJ13355 versus P. ananatis LMG 20103 b, P. ananatis AJ13355 versus P. vagans C9-1 c, and plasmid pEA320 from P. ananatis AJ13355 versus plasmid pPAG from P. vagans C9-1 d
P. vagans C9-1 and Pantoea sp. At-9b comprise three and five megaplasmids, respectively, in addition to their chromosomal DNA (Table 3). Plasmid pPAG3 from P. vagans C9-1 carries at least seven extended fragments (more than 10 kb in length) homologous to pEA320 (Fig. 2d). These homologous regions contain genes and operons that were verified as being expressed from the pPAG3 plasmid and that provide phenotypic features to P. vagans C9-1 (Smits et al. 2010b). These features include thiamine and carotenoid biosynthesis and the phosphoenolpyruvate transport system for cellobiose, arbutin, and salicin. The same regions of pPAG3 were found by BLAST-mediated computer search to be homologous to the P. ananatis LMG20103 chromosome (data not shown).
An automatic comparison of P. ananatis AJ13355 and LMG20103 and P. vagans C9-1 was performed at the CDS level, the results of which are summarized in Fig. 3. All three organisms have a core genome of 3140 orthologous CDSs in common. There are 224 CDSs unique to P. ananatis AJ13355, 278 CDSs unique to LMG20103, and 1197 CDSs unique to P. vagans C9-1. P. ananatis AJ13355 shares 634 CDSs with LMG20103 and only 69 CDSs with P. vagans C9-1.

Protein content comparison of the P. ananatis AJ13355, P. ananatis LMG 20103, and P. vagans C9-1 strains. The Venn diagram shows the number of CDSs shared between two or all three strains by a corresponding intersection of the circles, which represent each organism. CDSs showing more than 30% identity were considered to be orthologues
Homology between the chromosomes of P. ananatis AJ13355 and E. coli MG1655
The P. ananatis AJ13355 genome was compared with that of E. coli, the most examined member of the Enterobacteriaceae family. The total identity of the P. ananatis AJ13355 and E. coli MG1655 chromosomes at the nucleotide level, as evaluated by BLASTN software, did not exceed 35%. At the same time, this comparative analysis indicated segments of pronounced similarity, which harbor regions of full nucleotide identity for lengths of up to 50 bp. These regions were mainly colinear, except for two extended inversions. Meanwhile, at the nucleotide level, the pEA320 plasmid did not possess any significant homology with the E. coli MG1655 chromosome.
A comparative study was undertaken to determine the putative orthologous CDSs between the P. ananatis AJ13355 and E. coli MG1655 strains. The 702 and 1,723 orthologous CDSs with ≥80% and 60%–80% amino acid identity, respectively, between proteins were found based on BLAST and MAUVE software-mediated analysis. There were also 1,146 P. ananatis CDSs exhibiting a lower level of homology to E. coli proteins (30%–60%). In addition, 219 CDSs from the P. ananatis AJ13355 chromosome were not linked to any homologues in the E. coli chromosome (amino acid identity less than 30%).
Although the presence of functional genes tends to be more conserved than the genes positions in different genomes (Huynen and Bork 1998; Lathe et al. 2000), the detected gene content homology also represents synteny between the compared chromosomes of the P. ananatis AJ13355 and E. coli MG1655 (Fig. 4) strains. In the P. ananatis AJ13355 chromosome, 2,285 of 2,425 CDSs, with a level of homology of more than 60% relative to E. coli proteins, are syntenic when placed alongside the MG1655 chromosome. The residual 140 orthologous CDSs are located in two regions of the P. ananatis chromosome that appear as inversions when compared with the E. coli genome.

Synteny between the chromosomes of P. ananatis AJ13355 and E. coli MG1655. The graph represents an X–Y plot of dots forming syntenic regions between the bacterial chromosomes. Each dot represents a P. ananatis CDS with an orthologue in the E. coli genome, with coordinates corresponding to the position of the respective region in each chromosome. Orthologous CDSs were detected by reciprocal best BLASTP matches. The color code indicates the amino acid identity in the orthologous CDSs as follows: red, ≥80%; green, 60%–80%; blue, 30%–60%; and black, <30% (nonhomologous proteins)
The general synteny in gene content homology and the dispersed regions of full nucleotide identity along the genomes of E. coli and P. ananatis suggested a possible homologous recombination-mediated transfer of large blocks of genes in conjugative interspecific matings, as addressed below.
Conjugative gene transfer between E. coli and P. ananatis
The P. ananatis IZ130 strain could not utilize l-Glu as its sole carbon source due to ΔsucA-mediated disruption of the tricarboxylic acid cycle. The conjugative-plasmid carrier strain E. coli MG1655abrB::oriT/pRK2013 was able to transfer the sucA gene of the wild type linked with oriT. The latter strain, as a donor, and a TcR-carrier plasmid strain IZ130/pMW118-(λattL-tetA-tetR-λattR), as a recipient, were used in a biparental mating experiment. The presence of the TcR plasmid in the recipient P. ananatis strain provided selection against the donor E. coli strain on media containing Tc.
SucA genes in the P. ananatis AJ13355 and E. coli MG1655 chromosomes are located among syntenic sdh-suc CDSs that manifest an additional high level of homology at the level of the nucleotide sequences (Fig. 5a).

A short stretch of nucleotide identity provides homologous recombination between the P. ananatis AJ13355 and E. coli MG1655 chromosomes. a Dot plot of nucleotide similarity between the sdh-suc chromosomal regions of the P. ananatis AJ13355 and E. coli MG1655 strains. The gene order in the corresponding regions of the chromosomes is indicated. Black arrows mark genes located in regions of nucleotide homology, whereas empty arrows indicate genes located in the nonhomologous regions. Numbers (1) and (2) indicate regions of the sdhD and sucD genes where recombination likely occurred during conjugation-mediated sucA gene transfer. b Alignment of nucleotide sequences at the junction region of the recipient Pantoea strain, the donor E. coli strain, and the resulting recombinant Pantoea strain. Vertical lines indicate nucleotides identical to that of the recipient strain sequence, and letters indicate corresponding nucleotide changes
Biparental mating followed by selection of the TcR-bacteria on minimal medium with l-Glu as the sole carbon source resulted in recombinant SucA+ P. ananatis clones at a frequency of approximately 10−7–10−8. The homologous recombination-mediated replacement of the mutant P. ananatis loci by the DNA fragment from the mobilizable E. coli chromosome was initially established by PCR analysis and was confirmed by direct DNA sequencing of the 10-kb chromosome region surrounding sucA from one of the recombinant clones. According to the data presented in Fig. 5b, the recombination event that resulted in the tested strain had occurred with junctions in the sdhD and sucD genes of E. coli and P. ananatis. A nucleotide sequence identity of 11 bp was found between the recombineered chromosomes at the junction points.
Discussion
The determination and annotation of the complete genome sequence of P. ananatis AJ13355 have broadened the opportunities for basic research on and goal-directed metabolic engineering of this biotechnologically relevant organism. The present study has established a foundation for proteome and transcriptome analysis, genome-scale reconstruction of metabolic networks based on genome annotation, and valid flux analysis. Together with previously described and applied genetic engineering techniques, the establishment of this organism’s annotated genome sequence makes P. ananatis AJ13355 an attractive platform organism for the industrial production of organic acids, such as l-Glu and other biotechnologically relevant compounds.
A comparison between P. ananatis AJ13355 and earlier sequenced Pantoea genomes revealed high structural homology of their nucleotide sequences (Fig. 2) and a large number of shared proteins (Fig. 3). The main difference is the location of part of these genomes: on the chromosome for P. ananatis LMG 20103 and on megaplasmids for P. ananatis AJ13355 and P. vagans C9-1. P. ananatis LMG 20103 (De Maayer et al. 2010) is a plant pathogen that was isolated from diseased Eucalyptus in South Africa. In contrast, P. vagans is a gram-negative enterobacterial plant epiphyte from a broad range of plants. The latter is commercially registered for the biological control of fire blight, a disease of pear and apple trees caused by Erwinia amylovora (Smits et al. 2010a; 2010b). Meanwhile, the P. ananatis AJ13355 strain, isolated from soil, has passed all tests required for the industrialization of bacterial strains and has been recognized as a strain of Biosafety Level 1. The differences in the genome structures described above, specifically the autonomous replication of the part of the P. ananatis LMG 20103 chromosome in the two nonpathogenic Pantoea strains, and the presence of unique CDSs in each of the compared genomes (Fig. 3), could elucidate the genetic nature of the pathogenicity of P. ananatis LMG 20103.
The P. ananatis AJ13355 chromosome possesses a relatively high level of orthologous CDS synteny with the E. coli genome. The E. coli metabolic network is the most studied in the Enterobacteriaceae family. A comparison between the P. ananatis AJ13355 and E. coli CDSs encoding proteins involved in central metabolic pathways has revealed mainly coincidences but also the presence of several differences (Fig. 6) that could influence the strain-specific metabolic behavior. The dissimilarities are as follows:

Proposed similarities and differences in the maps of E. coli MG1655 and P. ananatis AJ13355 central carbon and ammonia metabolism based on the presence and absence of orthologous CDSs. The following color code was used: blue boxes, homologous CDSs; yellow boxes, CDSs in the P. ananatis genome that are not homologous in E. coli; orange box, CDS product that is active, due to the existence of the cofactor biosynthetic pathway in the P. ananatis genome, but not in E. coli K12; and white boxes, E. coli genes not found in the P. ananatis AJ13355 genome. Used abbreviations for metabolites are included as supplementary material (Table S1). The reaction directions are indicated accordingly to previously published E. coli genome-scale metabolic reconstruction (Feist et al. 2007)
-
(1) The functionally active PQQ biosynthetic operon, pqqABCDEF, which is structurally homologous to a similar genetic element in the Klebsiella pneumoniae chromosome (Meulenberg et al. 1992), was found and experimentally tested in the genome of P. ananatis AJ13355 (Andreeva et al. 2011). These bacteria provided glucose oxidation into gluconic acid by the PQQ-linked, membrane-bound glucose dehydrogenase (mGlcDH EC 1.1.99.17), which is orthologous to the corresponding E. coli protein (Cleton-Jansen et al. 1990). Meanwhile, in E. coli, only the inactive apoform of mGlcDH, the protein product of the gcd gene, is expressed without the formation of PQQ as a cofactor (Matsushita et al. 1997).
-
(2) A lack of the edd gene leads to disruption of the Entner–Doudoroff pathway in P. ananatis, which could be restored after the introduction of the E. coli edd-eda operon (Hara et al. 2003).
-
(3) In E. coli, NADPH-dependent glutamate synthase (GOGAT) and ATP-dependent glutamine synthetase (the products of gltBD and glnA, respectively) and NADPH-dependent glutamate dehydrogenase (GluDH) (encoded by the gdhA gene) form two alternative pathways for ammonia assimilation through the amination of α-ketoglutarate to l-Glu (Reitzer 1996). The P. ananatis AJ13355 genome does not contain CDSs coding for close homologues of the E. coli GOGAT or GluDH. Instead, it contains a putative gltB gene (CDS 001_1015 located in the chromosome) encoding a close homologue of the large subunit of GOGAT from Agrobacterium tumefaciens and a putative gdhA gene (CDS 001_0033 located in the megaplasmid pEA320) encoding a close homologue of putative NADH-dependent GluDH from Salmonella typhimurium. Until now, the dependence of P. ananatis GluDH on NADH as a cofactor has been experimentally confirmed (Mokhova et al., in preparation). NADPH-dependent GOGAT and GluDH are among the main consumers of NADPH in E.coli cells growing in medium containing ammonia as a nitrogen source (Pramanik and Keasling 1997; Reitzer 1996). The corresponding reactions function mainly to deliver the amino group of the provided l-Glu and glutamine (l-Gln) via transamination to cell mass synthesis and, to a lesser extent, to provide l-Glu and l-Gln for protein synthesis (Reitzer 1996). If the existent NADH-dependent GluDH significantly contributes to ammonia assimilation by P. ananatis cells under certain conditions, the carbon flux distribution among the pathways could significantly differ from the flux distribution in the closely related E. coli metabolic network with functional NADPH-dependent GluDH. The latter proposal is based on experimental results obtained for Corynebacteria strains that differed in the expression of GluDH with NADH or NADPH cofactor specificities (Marx et al. 1999).
Thus, despite a similar set of metabolic genes present in P. ananatis and E. coli, several CDSs are unique in these genomes. After being transferred to a heterologous host, the latter CDSs could be useful in improving the performance of metabolically engineered strains based on these closely related, but notably different, bacteria. In addition, these interspecies gene exchanges, probably, could be realized due to the experimentally confirmed conjugative transfer, which is considered as “self-cloning” that is important for the industrial applications of the engineered strains expressing heterologous genes.
References
Albermann C, Ghanegaonkar S, Lemuth K, Vallon T, Reuss M, Armbruster W, Sprenger GA (2008) Biosynthesis of the vitamin E compound delta-tocotrienol in recombinant Escherichia coli cells. Chembiochem 9:2524–2533
Andreeva IG, Golubeva LI, Kuvaeva TM, Gak ER, Katashkina JI, Mashko SV (2011) Identification of Pantoea ananatis gene encoding membrane pyrroloquinoline quinone (PQQ)-dependent glucose dehydrogenase and pqqABCDEF operon essential for PQQ biosynthesis. FEMS Microbiol Lett 318:55–60
Blattner FR, Plunkett G 3rd, Bloch CA, Perna NT, Burland V, Riley M, Collado-Vides J, Glasner JD, Rode CK, Mayhew GF, Gregor J, Davis NW, Kirkpatrick HA, Goeden MA, Rose DJ, Mau B, Shao Y (1997) The complete genome sequence of Escherichia coli K-12. Science 277:1453–1462
Cleton-Jansen AM, Goosen N, Fayet O, van de Putte (1990) Cloning, mapping, and sequencing of the gene encoding Escherichia coli quinoprotein glucose dehydrogenase. J Bacteriol 172:6308–6315
Darling AE, Mau B, Perna NT (2010) Progressive Mauve: multiple genome alignment with gene gain, loss, and rearrangement. PLoS One 5:e11147
de Lorenzo V, Timmis KN (1994) Analysis and construction of stable phenotypes in gram-negative bacteria with Tn5- and Tn10-derived minitransposons. Methods Enzymol 235:386–405
De Maayer P, Chan WY, Venter SN, Toth IK, Birch PR, Joubert F, Coutinho TA (2010) Genome sequence of Pantoea ananatis LMG20103, the causative agent of Eucalyptus blight and dieback. J Bacteriol 192:2936–2937
Dodge T, Valle F (2009) Uncoupled productive and catabolic host cell pathways. European patent 2055773(B1)
Feist AM, Henry CS, Reed JL, Krummenacker M, Joyce AR, Karp PD, Broadbelt LJ, Hatzimanikatis V, Palsson BO (2007) A genome-scale metabolic reconstruction for Escherichia coli K-12 MG1655 that accounts for 1260 ORFs and thermodynamic information. Mol Syst Biol 3:121
Figurski DH, Helinski DR (1979) Replication of an origin-containing derivative of plasmid RK2 dependent on a plasmid function provided in trans. Proc Natl Acad Sci USA 76:1648–1652
Grigoriev A (1998) Analyzing genomes with cumulative skew diagrams. Nucleic Acids Res 26:2286–2290
Hara Y, Izui Hiroshi, Asano T, Watanabe Y, Nakamatsu T (2003) Method for producing l-amino acid. European patent 1352966(A2)
Hara Y, Izui H, Noguchi H (2007) Novel plasmid autonomously replicable in Enterobacteriaceae family. European patent 1853621
Huynen MA, Bork P (1998) Measuring genome evolution. Proc Natl Acad Sci USA 95:5849–5856
Izui H, Hara Y, Sato M, Akiyoshi N (2003) Method for producing l-glutamic acid. US patent 6596517
Izui H, Moriya M, Hirano S, Hara Y, Ito H, Matsui K (2006) Method for producing l-glutamic acid by fermentation accompanied by precipitation. US patent 7015010
Jiang XW (2007) Using Pantoea agglomerans KFS-9 fungus to produce 2,3-butanediol, and application in cosmetic. Patent application CN1958785(A)
Kamimura D, Kuramoto M, Yamada K, Yazawa K, Kano M (1997) Oligomycin SC compounds of Pantoea agglomerans as anticancer agents. Patent application JP9208587(A)
Kasif S, White O, Salzberg SL (1999) Improved microbial gene identification with GLIMMER. Nucleic Acids Res 27:4636–4641
Katashkina JI, Hara Y, Golubeva LI, Andreeva IG, Kuvaeva TM, Mashko SV (2009) Use of the λRed-recombineering method for genetic engineering of Pantoea ananatis. BMC Mol Biol 10:34. doi:10.1186/1471-2199-10-34
Kumagai H, Suzuki H, Katayama T, Nawata M, Nakazawa H (2008) Mutant tyrosine repressor, a gene encoding the same, and a method for producing l-DOPA. US patent 7365161
Kunst F, Ogasawara N, Moszer I, Albertini AM, Alloni G, Azevedo V, Bertero MG, Bessières P, Bolotin A, Borchert S, Borriss R, Boursier L, Brans A, Braun M, Brignell SC, Bron S, Brouillet S, Bruschi CV, Caldwell B, Capuano V, Carter NM, Choi S-K, Codani J-J, Connerton IF, Cummings NJ, Daniel RA, Denizot F, Devine KM, Düsterhöft A, Ehrlich SD, Emmerson PT, Entian KD, Errington J, Fabret C, Ferrari E, Foulger D, Fritz C, Fujita M, Fujita Y, Fuma S, Galizzi A, Galleron N, Ghim S-Y, Glaser P, Goffeau A, Golightly EJ, Grandi G, Guiseppi G, Guy BG, Haga K, Haiech J, Harwood CR, Hénaut A, Hilbert H, Holsappel S, Hosono S, Hullo M-F, Itaya M, Jones L, Joris B, Karamata D, Kasahara Y, Klaerr-Blanchard M, Klein C, Kobayashi Y, Koetter P, Koningstein G, Krogh S, Kumano M, Kurita K, Lapidus A, Lardinois S, Lauber J, Lazarevic V, Lee S-M, Levine A, Liu H, Masuda S, Mauël C, Médigue C, Medina N, Mellado RP, Mizuno M, Moestl D, Nakai S, Noback M, Noone D, O’Reilly M, Ogawa K, Ogiwara K, Oudega B, Park S-H, Parro V, Pohl TM, Portetelle D, Porwollik S, Prescott AM, Presecan E, Pujic P, Purnelle B, Rapoport G, Rey M, Reynolds S, Rieger M, Rivolta C, Rocha E, Roche B, Rose M, Sadaie Y, Sato T, Scanlan E, Schleich S, Schroeter R, Scoffone F, Sekiguchi J, Sekowska A, Seror SJ, Serror P, Shin B-S, Soldo B, Sorokin A, Tacconi E, Takagi T, Takahashi H, Takemaru K, Takeuchi M, Tamakoshi A, Tanaka T, Terpstra P, Tognoni A, Tosato V, Uchiyama S, Vandenbol M, Vannier F, Vassarotti A, Viari A, Wambutt R, Wedler E, Wedler H, Weitzenegger T, Winters P, Wipat A, Yamamoto H, Yamane K, Yasumoto K, Yata K, Yoshida K, Yoshikawa H-F, Zumstein E, Yoshikawa H, Danchin (1997) The complete genome sequence of the gram-positive bacterium Bacillus subtilis. Nature 390:249–256
Kwon SW, Go SJ, Kang HW, Ryu JC, Jo JK (1997) Phylogenetic analysis of Erwinia species based on 16S rRNA gene sequences. Int J Syst Bacteriol 47:1061–1067
Lathe WC 3rd, Snel B, Bork P (2000) Gene context conservation of a higher order than operons. Trends Biochem Sci 25:474–479
Marx A, Eikmanns BJ, Sahm H, de Graaf AA, Eggeling L (1999) Response of the central metabolism in Corynebacterium glutamicum to the use of an NADH-dependent glutamate dehydrogenase. Metab Eng 1:35–48
Matsushita K, Arents JC, Bader R, Yamada M, Adachi O, Postma PW (1997) Escherichia coli is unable to produce pyrroloquinoline quinone (PQQ). Microbiology 143:3149–3156
Meulenberg JJ, Sellink E, Riegman NH, Postma PW (1992) Nucleotide sequence and structure of the Klebsiella pneumoniae pqq operon. Mol Gen Genet 232:284–294
Minaeva NI, Gak ER, Zimenkov DV, Skorokhodova AYu, Biryukova IV, Mashko SV (2008) Dual-in/out strategy for genes integration into bacterial chromosome: a novel approach to step-by-step construction of plasmid-less marker-less recombinant E. coli strains with predesigned genome structure. BMC Biotechnol 8:63 doi:10.1186/1472-6750-8-63
Mokhova ON, Kuvaeva TM, Golubeva LI, Kolokolova AV, Katashkina JY (2010) A bacterium belonging to the genus Pantoea producing an l-aspartic acid or l-aspartic acid-derived metabolites. Patent application WO2010038905(A1)
Moriya M, Izui H, Ono E, Matsui K, Ito H, Hara Y (1999) l-glutamic acid-producing bacterium and method for producing l-glutamic acid. US patent 6331419
Page M, Landry N, Boissinot M, Helie M-C, Harvey M, Gagne M (1999) Bacterial mass production of taxanes and paclitaxel. Patent application WO9932651(A1)
Pramanik J, Keasling JD (1997) Stoichiometric model of Escherichia coli metabolism: incorporation of growth-rate dependent biomass composition and mechanistic energy requirements. Biotechnol Bioeng 56:398–421
Raasch P, Schmitz U, Patenge N, Vera J, Kreikemeyer B, Wolkenhauer O (2010) Non-coding RNA detection methods combined to improve usability, reproducibility and precision. BMC Bioinformatics 11:491
Reitzer LJ (1996) Ammonia assimilation and biosynthesis of glutamine, glutamate, aspartate, asparagines, l-alanine, and d-alanine. In: Neidhardt FC (ed) Escherichia coli and Salmonella: cellular and molecular biology, 2nd edn. ASM, Washington, pp 391–407
Rice P, Longden I, Bleasby A (2000) EMBOSS: the European molecular biology open software suite. Trends in Genetics 16:276–277
Sambrook J, Russell DW (2001) Molecular cloning: a laboratory manual, 3rd edn. Cold Spring Harbor laboratory, Cold Spring Harbor
Smits TH, Rezzonico F, Kamber T, Goesmann A, Ishimaru CA, Stockwell VO, Frey JE, Duffy B (2010a) Genome sequence of the biocontrol agent Pantoea vagans strain C9-1. J Bacteriol 192:6486–6487
Smits TH, Rezzonico F, Pelludat C, Goesmann A, Frey JE, Duffy B (2010b) Genomic and phenotypic characterization of a nonpigmented variant of Pantoea vagans biocontrol strain C9-1 lacking the 530-kb megaplasmid pPag3. FEMS Microbiol Lett 308:48–54
TakahashiY, Tateyama Y, Sato M (2008) Process for producing l-glutamic acid US patent 7354744(B2)
Veith B, Herzberg C, Steckel S, Feesche J, Maurer KH, Ehrenreich P, Bäumer S, Henne A, Liesegang H, Merkl R, Ehrenreich A, Gottschalk GL (2004) The complete genome sequence of Bacillus licheniformis DSM13, an organism with great industrial potential. J Mol Microbiol Biotechnol 7:204–211
Velmurugan S, Mehta S, Uzri D, Jayaram M (2003) Stable propagation of ‘selfish’ genetic elements. J Biosci 28:623–636
Yoon SH, Kim JE, Lee SH, Park HM, Choi MS, Kim JY, Lee SH, Shin YC, Keasling JD, Kim SW (2007) Engineering the lycopene synthetic pathway in E. coli by comparison of the carotenoid genes of Pantoea agglomerans and Pantoea ananatis. Appl Microbiol Biotechnol 74:131–139
Zeida M, Wieser M, Yoshida T, Sugio T, Nagasawa T (1998) Purification and characterization of gallic acid decarboxylase from Pantoea agglomerans T71. Appl Environ Microbiol 64:4743–4747
Zhang S, Meyer R (1997) The relaxosome protein MobC promotes conjugal plasmid mobilization by extending DNA strand separation to the nick site at the origin of transfer. Mol Microbiol 25:509–516
Acknowledgments
This work was funded by Ajinomoto Co. The authors acknowledge Prof. Andrey A. Mironov for supplying the computer software used in the initial step of the work. They thank Prof. Rustem S. Shakulov for fruitful discussions. A special thanks also goes to Drs. Kiyoshi Matsuno and Shintaro Iwatani for their invaluable suggestions and managerial help during the manuscript preparation.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
Yurii I. Kozlov is deceased.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Table S1
Abbreviations for metabolites (DOC 51 kb)
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Hara, Y., Kadotani, N., Izui, H. et al. The complete genome sequence of Pantoea ananatis AJ13355, an organism with great biotechnological potential. Appl Microbiol Biotechnol 93, 331–341 (2012). https://doi.org/10.1007/s00253-011-3713-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-011-3713-5