
Abstract
Two carotenoid 1,2-hydratase (CrtC) genes from the photosynthetic bacteria Rubrivivax gelatinosus and Thiocapsa roseopersicina were cloned and expressed in Escherichia coli in an active form and purified by affinity chromatography. The biochemical properties of the recombinant enzymes and their substrate specificities were studied. The purified CrtCs catalyze cofactor independently the conversion of lycopene to 1-HO- and 1,1′-(HO)2-lycopene. The optimal pH and temperature for hydratase activity was 8.0 and 30°C, respectively. The apparent K m and V max values obtained for the hydration of lycopene were 24 μM and 0.31 nmol h−1 mg−1 for RgCrtC and 9.5 μM and 0.15 nmol h−1 mg−1 for TrCrtC, respectively. Sodium dodecyl sulfate polyacrylamide gel electrophoresis analysis revealed two protein bands of 44 and 38 kDa for TrCrtC, which indicate protein processing. Both hydratases are also able to convert the unnatural substrate geranylgeraniol (C20 substrate), which functionally resembles the natural substrate lycopene.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Optically pure tertiary alcohols are highly valuable building blocks for the synthesis of several bioactive natural products and pharmaceuticals (Kourist et al. 2008). However, the synthesis of optically pure tertiary alcohols in high yield without undesired side products is still a challenging task in traditional chemical synthesis (Cozzi et al. 2007). Much effort has therefore been devoted to the development of cleaner alternative technologies. The application of biocatalysts is recognized as a significant complement to the use of chemical reagents. Biocatalysts such as enzymes and whole microbial cells are increasingly being utilized for both environmental and economical reasons in a number of industries including agro-food, animal feed, detergent, textile, and specialty chemical industry. The market for enzymes has increased in an almost exponential manner from 1960 to 2000 (Fernandes 2010). This is due to the well-known benefits of enzymes. They are remarkable catalysts capable of accepting a wide array of complex substrates, are highly selective (enantio-, regio-, and chemoselective), and operate efficiently under mild conditions.
The possibility of using enzymes for the production of tertiary alcohols has generated our interest in the enzyme class of hydro-lyases (EC 4.2.1-), which catalyze the reversible addition of water to a carbon–carbon double bond. Although more than 100 hydro-lyases have been discovered to date, only a few examples have been used in industrial applications (Brady et al. 2004; Rzeznicka et al. 2010). For example, for the production of R-γ-dodeca-lactone, an essential flavor in whisky, oleate hydratase has been utilized, which catalyzes the conversion of oleic acid to form R-γ-hydroxy-stearate, which again is converted to the endproduct by baker’s yeast (Gocho et al. 1995; Wanikawa et al. 2000; Bevers et al. 2009)
Carotenoid 1,2-hydratase (CrtC), another member of the hydro-lyases group, occurs in the biosynthetic pathway of different acyclic carotenoids in photosynthetic bacteria. CrtC introduces a tertiary hydroxy group into a carotenoid molecule by addition of water to the carbon–carbon double bond at the C-1 position. Several carotenoid 1,2-hydratases have been identified in photosynthetic (Armstrong et al. 1989; Lang et al. 1995; Kovacs et al. 2003; Giraud et al. 2004; Frigaard et al. 2004) as well as in non-photosynthetic bacteria (Botella et al. 1995; Sun et al. 2009). Recently, a novel carotenoid 1,2-hydratase (CruF) has been described in the non-photosynthetic bacterium Deinococcus (Sun et al. 2009), which catalyzes C-1′,2′ hydration of γ-carotene. This enzyme though is evolutionarily distinct from the CrtC family in photosynthetic bacteria.
The CrtC from the purple non-sulfur photosynthetic bacterium Rubrivivax gelatinosus has been partially characterized and it was found to be a membrane-bound enzyme with a molecular weight of 44 kDa (Steiger et al. 2003). In vitro assay showed that the enzyme was able to hydrate the carbon–carbon double bond at the ψ-end group of several natural substrates such as neurosporene and lycopene to the corresponding products 1-HO- and 1,1′-(HO)2-neurosporene and 1-HO- and 1,1′-(HO)2-lycopene without the use of any cofactor. Through genetic analysis and characterization of the pigment biosynthesis genes in the purple sulfur photosynthetic bacterium Thiocapsa roseopersicina a putative protein was found that showed high identity to CrtC from R. gelatinosus (Kovacs et al. 2003). Gene cluster analysis of T. roseopersicina (Gammaproteobacteria) revealed a significant identity (55%) of the crtC gene product to the CrtC from R. gelatinosus (Betaproteobacteria), although the arrangement of the pigment biosynthesis gene cluster resembles more that of Rhodobacter species (Alphaproteobacteria; Igarashi et al. 2001). However, so far, the enzyme has not been isolated or characterized in any detail, which makes it a potential candidate for a hydro-lyase with new properties.
In order to make this group of enzymes more attractive for green hydration reactions in industrial applications, we have investigated parameters that could be of major importance to that field. Herein, we report on the detailed biochemical characterization of the two CrtCs from R. gelatinosus and T. roseopersicina. This provides an insight into their potential to be used as biocatalysts. The broad stability and activity profiles of both enzymes are promising for industrial biocatalysis.
Materials and methods
Construction of pET15b_CrtCRg and pET15b_CrtCTr expression vectors
The crtC Rg and crtC Tr genes were amplified with primers Rg_fw/Rg_rv (GGGAGTACCATATGCG-AGCAGCGGAGTC and ATACACTCGAGATGTATACGTCAAGCGCGG) and Tr_fw/Tr_rv (GGAGTAATCATATGCGAGCAGCGGGC and CCCTCGAGAACTATGTCTTCT-CAGCCGCC), respectively, containing restriction sites for NdeI (forward) and XhoI (reverse) (restriction sites are underlined). Amplification reactions were done under standard PCR conditions using plasmids pPQE30crtCRg and pTcrt3 respectively, as template (Supplementary Table 1). Using NdeI/XhoI restriction sites, the digested and purified fragment was ligated into the same sites of the pET15-b vector and transformed into E. coli TOP10 competent cells. The insertion of the crtC gene was verified by restriction analysis with NdeI/XhoI enzymes and DNA sequencing (BaseClear).
Expression and purification of recombinant proteins
E. coli BL21 (DE3) was the host for the pET15_CrtC plasmids. Cultures were grown at 37°C in Luria–Bertani broth with 100 μg ml−1 ampicillin until an OD600 value of 0.6–0.8 was reached. Protein expression was induced by addition of isopropyl-β-d-thiogalactopyranoside (IPTG) to a final concentration of 0.1 mM, followed by cultivation at 25°C overnight. The induced cells were harvested by centrifugation at 10,000 rpm for 10 min at 4°C (Sorvall), washed once with 50 mM Na2HPO4 buffer (pH 8.0), and suspended in the binding buffer (50 mM Na2HPO4, 300 mM NaCl, 20 mM imidazole, pH 8.0). Cell-free extract (CFE) was obtained after lysis of the cells with 1 mg ml−1 lysozyme for 1 h at 4°C followed by cell disruption at the pressure of 2.4 kBar (Constant systems, IUL instruments) and centrifugation at 10,000 rpm for 20 min at 4°C. The separation of the CFE into membrane fraction and supernatant was done by centrifugation at 45,000 rpm for 1 h at 4°C.
CFEs were filtered through 0.45 μm filter (Whatman, FP 30/0, 45 CA-S), and each extract was applied separately onto Ni-NTA HisTrap HP column (1.6 × 2.5 cm, 5 ml, GE Healthcare) previously equilibrated with binding buffer. The purification and the loading of the samples onto the column were performed with the high-performance liquid chromatography (HPLC) system in conjunction with the LCsolution software (Shimadzu). Unbound proteins were washed from the column with a gradient of 50–75 mM imidazole in washing buffer (50 mM Na2HPO4, 300 mM NaCl, pH 8.0). Then, the CrtC protein was eluted from the column with a gradient of 75–300 mM imidazole in elution buffer (50 mM Na2HPO4, 300 mM NaCl, pH 8.0). Enzyme fractions were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE; 10% Bis-Tris, BioRad) and visualized by staining with SimplyBlue SafeStain (Invitrogen). Fractions containing CrtC were combined and concentrated using Amicon Ultra-30 filters (Millipore). The concentrated sample was applied onto a PD-10 desalting column (GE Healthcare) previously equilibrated with 50 mM Na2HPO4 buffer (pH 8.0). The eluted enzyme sample was frozen in liquid nitrogen and stored in aliquots at −80°C.
Tandem MS analysis
The concentrated CrtC sample was further purified by SDS-PAGE. The protein band was excised from the gel and subjected to in-gel proteolytic digestion as previously described (Sevcenco et al. 2009).
CrtC activity assay and analysis of the products
Enzyme activity was determined either with the purified enzyme or with the CFE. The assay was performed with 50–100 μg enzyme in 200 μl 50 mM Na2HPO4 buffer (pH 8.0), containing 10 mg ml−1 L-α-phosphatidylcholine and 20 μM lycopene (Lanospharma Laboratories Co., Ltd) from a stock in acetone. After incubation at 28°C and shaking at 800 rpm in the dark, the substrate and products were extracted from the aqueous layer after a desired time interval. Prior to the extraction, 50 μl of saturated NaCl solution was added and the carotenoids extracted with one volume of dichloromethane. The mixtures were shaken for 5 min at 1,400 rpm and centrifuged for 1 min at 13,200 rpm (Eppendorf), and 150 μl of the dichloromethane phase was dried with a Speed Vac Concentrator (Thermo). The dried carotenoids were dissolved in 10 μl dichloromethane, diluted 1:10 with 100% acetonitrile, and analyzed with HPLC. Separation was performed with a Merck 4.6 × 50 mm Chromolith TM SpeedROD RP-18e 2 μm column with acetonitrile/water (95:5, v/v) as the eluent. Lycopene and the corresponding products were detected at 470 nm (SPD-20A, Shimadzu). Marker carotenoids were obtained as described by Steiger et al. (Steiger et al. 2000) and used for the identification of the reaction products.
The lycopene concentration in the assay was quantified from the calibration curve constructed by diluting a stock of lycopene in dichloromethane with acetone. A second calibration curve, which was used to quantify the reaction products, was constructed in the same way as the standard assay, including the extraction step.
For the determination of enzyme kinetic parameters, the purified enzyme was incubated for 4 h with different concentrations of lycopene (0.5–35 μM) in 50 mM Na2HPO4 buffer (pH 8.0), containing 10 mg ml−1 L-α-phosphatidylcholine. Each reaction was performed in duplicate. The affinity constant (K m) and the maximal velocity (V max) were calculated from the experimental data points using OriginPro 8 SR1 software.
Substrate specificity
Substrate specificity was assayed using the following acyclic alkenes: 2-methyl-2-butene (79 mM), 2-methyl-2-pentene (68 mM), farnesol (33 mM), and geranylgeraniol (14.3 mM), as substrates. Reactions were carried out using standard assay conditions. E. coli carrying the empty pET15-b vector was used as negative control reaction. Substrates and products were extracted from aqueous layer with one volume of ethyl acetate. The samples were dried with Na2SO4 prior to their injection. Separation and identification of the components was effected with a Shimadzu GC-MS coupled to a QP-2010S with a FactorFour VF-WAXms column (length, 30 m; diameter, 0.25 mm; and film thickness, 0.25 μm).
Effects of pH and temperature on CrtC activity
In order to investigate the pH effect on the CrtC activity, the reactions were carried out in buffers with varying pH values. The buffers used for pH test were sodium acetate (100 mM, pH 4.0–6.0), potassium phosphate (100 mM, pH 6.0–8.0), and Tris–HCl (50 mM, pH 8.0–9.0). The measurements were conducted at 28°C, and lycopene (20 μM) was used as substrate, as described in the section “CrtC activity assay and analysis of the products” under Materials and methods. The pH stability of the enzyme was performed by measuring the remaining activity at pH 8.0 after the enzyme had been incubated in the corresponding buffers for 30 min.
The optimum temperature for CrtC activity was determined by testing enzyme activity at temperatures ranging from 0°C to 50°C using the standard activity assay. The thermal stability was investigated by preincubating the enzyme at various temperatures (5–50°C) in the absence of substrate for 30 min, cooling the enzyme on ice, and then measuring the residual activity in a standard assay with lycopene as substrate.
Effects of inhibitors and metal ions on enzyme activity
The inhibitory effects on enzyme activity were investigated by performing activity assay under standard conditions in the presence of several metal ions (MgCl2, MnCl2, CoCl2, ZnCl2, CaCl2, and CuSO4) and chemicals (NAD+, NADH, protease inhibitor “Complete” Roche) with a final concentration of 1 mM. Lycopene (20 μM) was used as substrate, and the activity was measured as described above. Reaction mixture without any additive was used as control reaction and was designated as 100% activity.
Circular dichroism spectroscopy
The purified RgCrtC and TrCrtC were diluted to 0.04 and 0.03 mg ml−1, respectively, in 10 mM Na2HPO4, pH 8.0. Samples were incubated for 5 min at temperatures from 5°C to 90°C (5°C steps), and after each incubation, samples were scanned. Circular dichroism (CD) spectra were collected from 190 to 250 nm as an average of five spectra, with a data pitch of 1 nm. A band width of 1 nm was used with a detector response time of 0.25 s and scanning speed of 100 nm min−1. CD spectra were recorded on a Jasco J-810 spectrometer equipped with a Peltier temperature control unit in 0.1 cm path length cuvette (Chen et al. 1972).
Metal analysis using USN-ICP-OES
The metal content from purified protein sample and the buffer solution was measured using Perkin-Elmer 4300 dual view inductively coupled plasma (ICP) with optical emission spectroscopy (OES) spectrometer, coupled with ultrasonic nebulizer (USN) U-6000 AT, Cetac. Measurements were performed for different metals and at different wavelengths, as following: Co (228 and 238 nm), Fe (238 and 239 nm), Mo (202 and 203 nm), Ni (231 and 221 nm), and Zn (206 and 213 nm).
Results
Expression and purification of the carotenoid 1,2-hydratases
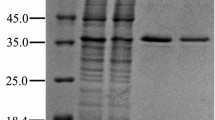
For biochemical characterization of the carotenoid 1,2-hydratases and comparison of their catalytic activities, the two crtC genes from R. gelatinosus and T. roseopersicina were cloned into the expression vector pET15-b. The constructed pET15b_CrtCRg and pET15b_CrtCTr plasmids were sequenced, and the results confirmed that the genes were successfully inserted in frame with the N-terminal His6-tag. In order to express the recombinant CrtCs, E. coli BL21 (DE3) was transformed with the expression plasmids. SDS-PAGE analysis revealed in both cases a 44 kDa band (Fig. 1), which is consistent with the value calculated from the deduced amino acid sequence. The expression level of RgCrtC was around two times higher than that of TrCrtC expressed under the same conditions. In the case of TrCrtC an additional faint band around 38 kDa was detected after induction (Fig. 1, lane 5) which is absent in the non-induced sample (Fig. 1, lane 4).

SDS-PAGE (10%) analysis of expression and IMAC purification for RgCrtC (lanes 1–3) and TrCrtC (lanes 4–6). M Precision plus protein standard; a whole cells before induction; b whole cells after induction with 0.1 mM IPTG and expression overnight at 25°C; c purified CrtCs
CrtCs were purified from CFEs by a single-step IMAC column and led to a nearly homogeneous band of 44 kDa in the case of RgCrtC and bands of 38 and 44 kDa in the case of TrCrtC. However, the larger band could not be detected again once the sample was stored at 20°C for a few days (Fig. 1, lane 6).
Hydratase activity
Activity measurements of the purified enzymes with lycopene as substrate demonstrated functional expression of the recombinant CrtCs in E. coli. The purified enzymes catalyze the conversion of lycopene into both 1-HO-lycopene and 1,1′-(HO)2-lycopene. For both CrtCs, the conversion rate was 30%, and the ratio between mono- and dihydroxylated product was 2:1. USN-ICP-OES metal analysis showed that the protein samples did not contain any significant amounts of iron, zinc, cobalt, nickel, or molybdenum (data not shown). Furthermore, it was observed that the addition of coenzymes NAD+/NADH or protease inhibitors had no detectable influence on enzyme activity. Although the effect of various metal ions on the hydratase activity was tested, no firm conclusion could be drawn from these data as the metals have a degrading effect on the substrate lycopene (Boon et al. 2009).
Enzyme kinetics
In order to compare the catalytic activities of the two expressed CrtCs, in vitro activity studies were performed. Since the conversion of lycopene to 1-HO-lycopene and 1,1′-(HO)2-lycopene with isolated enzyme was very slow, the reactions were carried out with CFEs (Fig.2). Kinetic parameters K m, V max, V max/K m, and k cat/K m were determined by activity assay using lycopene as substrate at 28°C (Table 1). The results are shown in a Michaelis–Menten plot (Fig. 3) as the reaction rate versus the substrate concentration. The affinity constant (K m) for recombinant RgCrtC and TrCrtC was calculated as 24 and 9.5 μM, respectively, and V max was 0.31 and 0.15 nmol h−1 mg−1, respectively. The substrate specificity values were calculated as V max/K m, and the results show a slightly higher specificity of TrCrtC with 1.6 × 102 compared to RgCrtC with 1.3 × 102 for lycopene. Furthermore, the catalytic efficiency values for TrCrtC (0.19 s−1 M−1) and RgCrtC (0.16 s−1 M−1) revealed no significant difference for lycopene hydration.

Reaction catalyzed by Rubrivivax gelatinosus and Thiocapsa roseopersicina carotenoid 1,2-hydratase; the conversion of lycopene into 1-HO-lycopene and 1,1′-(HO)2-lycopene (a). HPLC separation of carotenoids formed in vitro by Escherichia coli extract expressing the RgCrtC (solid line) and TrCrtC (dashed line). Peak 1, 1,1′-(HO)2-lycopene; peak 2, 1-HO-lycopene; peak 3, lycopene (b)

Michaelis–Menten plot of recombinant RgCrtC (filled circle) and TrCrtC (empty circle). The cell-free extracts were assayed with various lycopene concentrations (0.5–40 μM) in 50 mM Na2HPO4 sodium phosphate (pH 8.0) at 28°C for 4 h. The rates of product formation (1-HO-lycopene plus 1,1′-(HO)2-lycopene) are plotted against varying substrate concentrations. Kinetic constants are listed in Table
Substrate specificity
Substrate specificity was tested with acyclic alkenes of different chain length, which possess the same alkenyl functional group like lycopene, the natural substrate of CrtC (Supplementary Fig. 1). No activity was detected for the C5, C6, and C15 substrate using standard assay conditions. However, a product was detected with the C20 substrate geranylgeraniol for both RgCrtC and TrCrtC, which was absent in the control experiment. The conversion was very low, approximately 5% (Supplementary Fig. 2).
Effect of pH and temperature on hydratase activity and stability
The dependence of the activity of recombinant RgCrtC and TrCrtC at different pH values and temperatures was investigated using lycopene as substrate. The optimum pH for hydratase activity appeared to be pH 8.0 (Fig. 4a). While RgCrtC has a broader pH optimum ranging from pH 7.0–8.0, a significant decrease was observed for TrCrtC with only 50% activity at pH 7.0. No activity could be detected at pH 4.0–5.0 for both enzymes. At higher pH values, both enzymes showed rapid decrease in activity, although 50% of the relative activity was still detected at pH 9.0. Both enzymes retained much residual activity after 30 min incubation at pH 4.0–8.0, indicating that the CrtCs are stable in both slightly alkaline and acid environments (Supplementary Fig. 3a). However, compared with RgCrtC, the TrCrtC stability decreases in the range from pH 6.5–9.0 to only 56% of the residual activity, whereas RgCrtC still remains 95% activity at pH 9.0. Despite no detected activity at pH 4.0–5.0 (Fig. 4a), both enzymes seem to be stable at that pH range and still show 75–80% of residual activity.

Effect of pH (a) and temperature (b) on activity of RgCrtC (filled circle) and TrCrtC (empty circle). For pH effect, measurements were performed with lycopene under standard assay conditions using different buffers: 100 mM acetate (pH 4.0, 5.0 and 6.0), 100 mM potassium phosphate (pH 6.0, 7.0, and 8.0), and 50 mM Tris–HCl (pH 8.0, 8.6 and 9.0). For temperature effect, activity assays were performed with lycopene at various temperatures (1–50°C) under standard assay conditions
The effect of temperature on hydratase activity from 0°C to 50°C is depicted in Fig. 4b. The favorable temperature range was from 25°C to 35°C with an optimum at 30°C. Enzyme activity for RgCrtC and TrCrtC was significantly lower at 20°C (55% and 42%, respectively) and 40°C (47% and 31%, respectively). A negligible activity was found at 5°C (around 10%). Thermal stability was investigated by preincubating hydratases for 30 min at different temperatures and subsequently testing residual activity under standard assay conditions (Supplementary Fig. 3b). Maximum stability was recorded at 5°C. The enzymes did not show significant decrease of the activity up to 40°C. When preincubated at 45°C, they still showed relative high activity (around 50–60%). However, RgCrtC was extremely sensitive at 50°C, retaining only 6% activity after 30 min preincubation, while TrCrtC showed 30% residual activity at that temperature. Additionally, the temperature stability of RgCrtC and TrCrtC was studied using CD spectroscopy. It was not possible to obtain a good qualitative representation of the CD spectra from RgCrtC, in contrast to TrCrtC, which is shown in Fig. 5. TrCrtC was found to be relatively stable below 50°C. Significant change in the secondary structure was observed at temperatures above 50°C, which corresponds with the results obtained in the activity assay (Fig. 4b).

Enzyme stability of recombinant TrCrtC by CD spectroscopy. The purified CrtC was diluted to 0.03 mg/ml with 10 mM sodium phosphate, pH 8.0, and incubated for 5 min from 5°C to 90°C. CD assay was performed by wavelength scan from 190 to 250 nm
Discussion
This study reports on the purification and biochemical characterization of two heterologously expressed carotenoid 1,2-hydratases (CrtC) from photosynthetic bacteria, which are potential biocatalysts in the green hydration of carotenoid-like substrates. The two crtC genes from R. gelatinosus (1,221 bp) and T. roseopersicina (1,218 bp) were cloned, sequenced, and successfully expressed in E. coli BL21 (DE3). Many attempts have been made to optimize expression levels and to reduce formation of inclusion bodies (data not shown), as these enzymes are detected in the membrane fraction. Hydropathy plots, determined with Kyte and Doolittle (1982), did not reveal any putative transmembrane domain in the two hydratases. However, it was noticed that the first 45–55 amino acids of RgCrtC and TrCrtC showed a significantly high percentage of proline (13% and 16%, respectively), whereas the rest of the sequence has the usual proline amount of 9% and 8%, respectively. Ouchane et al. (1997) described this already for RgCrtC. Proline-rich regions in proteins are widely found in prokaryotes and eukaryotes (Williamson 1994). A non-repetitive (XP)n sequence like identified in RgCrtC (10×) and TrCrtC (9×) can have different functions as for instance stabilizing the enzyme by binding noncovalently to other proteins, binding to other hydrophobic structures like hydrophobic substrates or function as a “molecular trigger” passing signals to the inner membrane. Based on these findings, one may suggest that the hydrophobic N-terminus of the RgCrtC and TrCrtC could play a role in stabilizing the enzyme in the hydrophobic membrane area. This hypothesis is strengthened by the following data. On the nucleotide level, the two crtC sequences presented a relatively high identity of 70% (Altschul et al. 1990; Supplementary Fig. 4a). However, a significant difference was observed after the heterologous expression in E. coli. Although the gene sequence predicts a protein of 44 kDa (Supplementary Fig. 4b), the SDS-PAGE analysis of the expressed enzymes showed a second band of about 38 kDa for TrCrtC, which was absent in RgCrtC as well as in the empty vector control. Furthermore, membrane fractions with only a visible 38 kDa band showed good activity (data not shown), indicating that the lower band is active. MS data of this band revealed that the N-terminal proline rich part is missing, thereby supporting the hypothesis that this part is not important for biological activity or substrate binding but for membrane association. Moreover, analysis of amino acid sequence similarities of various known and putative CrtCs also shows that the first part of the sequence is missing in a number of the analyzed sequences (data not shown). As this phenomenon of protein processing is not known from literature to occur in the CrtC family, more experiments were performed. One approach currently under study that addresses our hypothesis involves construction of mutants, which lack the N-terminal part of the sequence. First results showed that the truncated CrtCs are fully functional and catalyze the conversion of lycopene to the corresponding products without any loss of activity (Supplementary Fig. 5). This observation confirms our hypothesis that the N-terminal part is not involved in catalytic reaction nor in substrate binding. Furthermore, this means that the truncated enzyme could be used in an industrial setting.
The RgCrtC and TrCrtC catalyze the conversion of lycopene with a V max of 0.31 and 0.15 nmol h−1 mg−1, respectively, and a K m of 24 and 9.5 μM, respectively, without the need of any cofactor. The lower value of K m observed for TrCrtC shows that this enzyme presents higher affinity for the substrate lycopene than RgCrtC. However, the catalytic efficiency values were similar for both enzymes, despite TrCrtC presenting twofold lower V max.
Maximum activity was detected for both enzymes at pH 8.0 and at the temperature of 30°C. Moreover, they also presented quite good activities at temperatures ranging from 25 to 35°C. However, temperatures above 50°C caused denaturation of the protein structure and therefore inactivation, which was confirmed by CD spectroscopy. Although both enzymes are rather similar in their pH and temperature profile, RgCrtC is more stable at higher pH, while TrCrtC is more stable at higher temperatures. This could be of importance when choosing the right enzyme for a biocatalytic process.
The substrate scope study of CrtCs is an important aspect, as no investigation has been made in this direction to date. Next to the substrate lycopene, activity measurements were reported in literature with two other natural substrates neurosporene and spheroidene, as demonstrated by Steiger et al. (2003). It was concluded that spheroidene, which possess a terminal methoxy functional group, serves as the best substrate for RgCrtC. Furthermore, this enzyme was also able to use monohydroxy carotenoids as substrates, which could not be observed for Rhodobacter capsulatus CrtC (Steiger et al. 2003). Our primary objective with the substrate specificity experiment was to investigate the possibility of using CrtC with unnatural substrates to produce highly valuable compounds for industry. Based on the observed activity with geranylgeraniol, we postulate that the minimum size of the substrates for both RgCrtC and TrCrtC is C20 (20 carbon) chain. However, the low conversion of about 5% clearly indicates that their substrate spectrum is limited. Since the crystal structure of CrtC has not yet been solved, one can only speculate about the size of the active site and the mechanism that is involved in the hydration of the substrates. Further structural and biochemical characterization is necessary to achieve a full understanding of this enzyme and its reaction mechanism.
In conclusion, both CrtCs are stable at a broad and suitable temperature and pH range and hydrate several long aliphatic substrates to give tertiary alcohols. Future studies will be directed at improving the activity of these hydratases.
References
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215:403–410
Armstrong GA, Alberti M, Leach F, Hearst JE (1989) Nucleotide-sequence, organization, and nature of the protein products of the carotenoid biosynthesis gene-cluster of Rhodobacter capsulatus. Mol Gen Genet 216:254–268
Bevers LE, Pinkse MWH, Verhaert P, Hagen WR (2009) Oleate hydratase catalyzes the hydration of a nonactivated carbon–carbon bond. J Bacteriol 191:5010–5012
Boon CS, McClements DJ, Weiss J, Decker EA (2009) Role of iron and hydroperoxides in the degradation of lycopene in oil-in-water emulsions. J Agric Food Chem 57:2993–2998
Botella JA, Murillo FJ, Ruizvazquez R (1995) A cluster of structural and regulatory genes for light-iduced carotenogenesis in Myxococcus xanthus. Eur J Biochem 233:238–248
Brady D, Beeton A, Zeevaart J, Kgaje C, van Rantwijk F, Sheldon RA (2004) Characterisation of nitrilase and nitrile hydratase biocatalytic systems. Appl Microbiol Biotechnol 64:76–85
Chen YH, Yang JT, Martinez HM (1972) Determination of the secondary structures of proteins by circular dichroism and optical rotatory dispersion. Biochemistry 11:4120–4131
Cozzi PG, Hilgraf R, Zimmermann N (2007) Enantioselective catalytic formation of quaternary stereogenic centers. Eur J Org Chem 2007:5969–5994
Fernandes P (2010) Miniaturization in biocatalysis. Int J Mol Sci 11:858–879
Frigaard NU, Maresca JA, Yunker CE, Jones AD, Bryant DA (2004) Genetic manipulation of carotenoid biosynthesis in the green sulfur bacterium Chlorobium tepidum. J Bacteriol 186:5210–5220
Giraud E, Hannibal L, Fardoux J, Jaubert M, Jourand P, Dreyfus B, Sturgis JN, Vermeglio A (2004) Two distinct crt gene clusters for two different functional classes of carotenoid in Bradyrhizobium. J Biol Chem 279:15076–15083
Gocho S, Tabogami N, Inagaki M, Kawabata C, Komai T (1995) Biotransformation of oleic acid to optically active gamma-dodecalactone. Biosci Biotechnol Biochem 59:1571–1572
Igarashi N, Harada J, Nagashima S, Matsuura K, Shimada K, Nagashima KVP (2001) Horizontal transfer of the photosynthesis gene cluster and operon rearrangement in purple bacteria. J Mol Evol 52:333–341
Kourist R, de Maria PD, Bornscheuer UT (2008) Enzymatic synthesis of optically active tertiary alcohols: expanding the biocatalysis toolbox. Chembiochem 9:491–498
Kovacs AT, Rakhely G, Kovacs KL (2003) Genes involved in the biosynthesis of photosynthetic pigments in the purple sulfur photosynthetic bacterium Thiocapsa roseopersicina. Appl Environ Microbiol 69:3093–3102
Kyte J, Doolittle RF (1982) A simple method for displaying the hydropathic character of a protein. J Mol Biol 157:105–132
Lang HP, Cogdell RJ, Takaichi S, Hunter CN (1995) Complete DNA-sequence, specific tn5 insertion map, and gene assignment of the carotenoid biosynthesis pathway of Rhodobacter sphaeroides. J Bacteriol 177:2064–2073
Ouchane S, Picaud M, Vernotte C, ReissHusson F, Astier C (1997) Pleiotropic effects of puf interposon mutagenesis on carotenoid biosynthesis in Rubrivivax gelatinosus—a new gene organization in purple bacteria. J Biol Chem 272:1670–1676
Rzeznicka K, Schatzle S, Bottcher D, Klein J, Bornscheuer UT (2010) Cloning and functional expression of a nitrile hydratase (nhase) from Rhodococcus equi tg328-2 in Escherichia coli, its purification and biochemical characterisation. Appl Microbiol Biotechnol 85:1417–1425
Sevcenco AM, Krijger GC, Pinkse MWH, Verhaert P, Hagen WR, Hagedoorn PL (2009) Development of a generic approach to native metalloproteomics: application to the quantitative identification of soluble copper proteins in Escherichia coli. J Biol Inorg Chem 14:631–640
Steiger S, Astier C, Sandmann G (2000) Substrate specificity of the expressed carotenoid 3,4-desaturase from Rubrivivax gelatinosus reveals the detailed reaction sequence to spheroidene and spirilloxanthin. Biochem J 349:635–640
Steiger S, Mazet A, Sandmann G (2003) Heterologous expression, purification, and enzymatic characterization of the acyclic carotenoid 1,2-hydratase from Rubrivivax gelatinosus. Arch Biochem Biophys 414:51–58
Sun ZT, Shen SC, Wang C, Wang H, Hu YP, Jiao JD, Ma TT, Tian B, Hua YJ (2009) A novel carotenoid 1,2-hydratase (cruf) from two species of the non-photosynthetic bacterium Deinococcus. Microbiology-Sgm 155:2775–2783
Wanikawa A, Hosoi K, Takise I, Kato T (2000) Detection of gamma-lactones in malt whisky. J Inst Brew 106:39–43
Williamson MP (1994) The structure and function of proline-rich regions in proteins. Biochem J 297:249–260
Acknowledgments
We thank Prof. Dr. Gerhard Sandmann and Prof. Dr. Kornél L. Kovács for providing the plasmids. This project is financially supported by The Netherlands Ministry of Economic Affairs and the B-Basic partner organizations (http://www.b-basic.nl) through B-Basic, a public-private NWO-ACTS programme [Advanced Chemical Technologies for Sustainability (ACTS)].
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 1896 kb)
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Hiseni, A., Arends, I.W.C.E. & Otten, L.G. Biochemical characterization of the carotenoid 1,2-hydratases (CrtC) from Rubrivivax gelatinosus and Thiocapsa roseopersicina . Appl Microbiol Biotechnol 91, 1029–1036 (2011). https://doi.org/10.1007/s00253-011-3324-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-011-3324-1