
Abstract
Bacteriophage KP34 is a novel virus belonging to the subfamily Autographivirinae lytic for extended-spectrum β-lactamase-producing Klebsiella pneumoniae strains. Its biological features, morphology, susceptibility to chemical and physical agents, burst size, host specificity and activity spectrum were determined. As a potential antibacterial agent used in therapy, KP34 molecular features including genome sequence and protein composition were examined. Phylogenetic analyses and clustering of KP34 phage genome sequences revealed its clear relationships with “phiKMV-like viruses”. Simultaneously, whole-genome analyses permitted clustering and classification of all phages, with completely sequenced genomes, belonging to the Podoviridae.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Most of the publications during the last decades regarding bacteriophages lytic for enteric rods described their application in phage typing (Ślopek et al. 1972; Dąbrowski et al. 1988) or phage therapy (Ślopek et al. 1981a, b, 1984; Sakandelidze and Meipariani 1974; Zhukov-Verezhnikov et al. 1978; Richardson et al. 1999; Weber-Dąbrowska et al. 2000, 2001, 2003; Sulakvelidze et al. 2001). Data concerning newly isolated Klebsiella phages focus on biological characteristics or biodistribution and therapeutic efficacy in animal models (Bogovazova et al. 1991; Kumari et al. 2009; 2010a, b; Verma et al. 2009). None of the above described Klebsiella phages was tested regarding genome organisation or sequence similarity to already known viruses. Although fast and efficient techniques of DNA sequencing and proteomics are available today and the detailed analysis of genome structure and organisation is possible, there is not much molecular data about Klebsiella virulent bacteriophages. As of today, only four complete genomes of Klebsiella-specific lytic viruses have been deposited in GenBank, and three of them (KP15, KP32, KP34) were sequenced by our team. Recognition and analysis of genome structure and genes function is the required step before bacteriophages can be approved as therapeutic agents. The knowledge of protein composition allows one to definitively establish the absence of possible toxins potentially dangerous during phage application. The present study focused on novel “phiKMV-like virus” KP34 propagated on a multidrug-resistant Klebsiella pneumoniae strain producing an extended-spectrum β-lactamase (ESBL). Phage KP34 was fully characterised regarding its host specificity, activity spectrum and also morphological, biological, molecular and genomic features. This bacteriophage, which is a new member of the Podoviridae, is being considered for potential use in phage therapy.
Materials and methods
Isolation and purification of phages
K. pneumoniae-specific bacteriophage KP34 was isolated from a sewage sample by enrichment. A sewage sample was centrifuged (15,000×g/15 min) and the supernatant filtered through a 0.22-μm Millipore filter. Phage propagation followed the modified method of Ślopek et al. (1972). One millilitre of filtered sewage sample and 0.5 ml of K. pneumoniae 77 ESBL (+), grown overnight in Mueller Hinton Broth (MHB; bioMérieux Polska, Warsaw, Poland), were added to 10 ml of MHB and incubated at 37°C until complete lysis appeared (approximately 4–6 h). The suspension was then filtered through a 0.22-μm Millipore filter. The procedure was repeated three times to eliminated bactericidal activity of some chemical compounds probably presented in the sewage sample. The bacteriophage titer in the filtrate was assessed using the double-agar layer technique according to Adams (1959). The isolated phage was named KP34, with the first two letters indicating the host genus and the species name. This virus has been deposited in the Polish Collection of Microorganisms (Institute of Immunology and Experimental Therapy, Polish Academy of Sciences, Wrocław, Poland) under accession number F/00067.
Electron microscopy
A high-titer phage lysate previously filtered through a 0.22-μm Millipore filter was centrifuged at 25,000×g for 60 min, and the pellet was washed twice in ammonium acetate (0.1 M, pH 7.0). A portion of the resuspended sediment was deposited on carbon-coated Formvar films, stained with 2% uranyl acetate and examined in a JEM 100C (Joel LTD, Tokyo, Japan) transmission electron microscope at 80 kV with magnification of ×66,000. The phage size was determined from the average of five to seven independent measurements using T4 phage tail (114 nm) as the magnification control.
Phage adsorption procedure
The adsorption of phages to bacterial host cells was examined using previously described by (Adams 1959; Ackermann and DuBow 1987; Roncero et al. 1990; Gallet et al. 2009) with slight modifications introduced in our laboratory. Overnight cultures of K. pneumoniae 77 ESBL (+) grown on Mueller Hinton Agar (MHA; bioMérieux Polska, Warsaw, Poland) were used. Cells from the agar medium were suspended in enrichment broth (MHB) to an optical density at 600 nm of approximately 0.9–1.0. An equal volume of bacterial suspension and phage diluted to 105–106 pfu/ml were incubated at 37°C for 5 min. After incubation, the culture was filtered (0.22 μm) and the number of free phages was determined, in duplicate, in the filtrate using the double-agar-layer method. The reduction in phage titer was the number of phages adsorbed to the cells. No reduction in phage titer in control filtration (0.22 μm Millipore filters) was observed.
Burst size experiments
A one-step growth curve of KP34 was performed according to the method of Pajunen et al. (2000) with modifications. The density of a mid-exponential bacterial culture (MHB) was adjusted to 2 × 108 cfu/ml. To 0.9 ml of this cell suspension was added 0.1 ml of bacteriophage in order to achieve a multiplicity of infection of 0.005. Phages were allowed to adsorb for 5 min at 37°C, after which time the mixture was diluted to 10−5, and samples, in triplicate, were taken at 5-min intervals for titration. Experiment was performed on three different occasions, and values depict the mean of three observations ± standard deviation.
Storage stability of phage culture
The stability of KP34 preparation, neat at −70°C, −20°C, 4°C and 20°C or in the presence of 25% (v/v) glycerol at −70°C and −20°C was determined after 3 months of storage using the double-agar-layer technique according to Adams (1959).
Sensitivity of phage particles to temperature, chloroform and pH
A filter-sterilized bacteriophage preparation at 107 pfu/ml was incubated at 60°C for 10 min with intermittent shaking. An equal volume of bacteriophage (107 pfu/ml) was mixed with chloroform and incubated for 2 and 24 h at room temperature with intermittent shaking. Further preparations of KP34 (107 pfu/ml) was incubated at pH 2, 4, 5, 6, 8 and 10 for 1 and 5 h at room temperature and at 37°C also with intermittent shaking. After all these experiments, the bacteriophage titer was assessed using the double-agar-layer technique (Adams 1959).
Determination of KP34 phage bacterial host range
The strains used in this study are listed in Table S1 (in electronic supplementary material). Bacteria were stored at −70°C in Trypticase Soy Broth (Becton Dickinson and Company, Cockeysville, MD, USA) supplemented with 20% glycerol. Prior to phage sensitivity testing, bacteria were subcultured at least twice in Trypticase Soy Broth. Unless otherwise stated in all phage experiments, 4–6-h bacterial cultures were used. To determine bacterial susceptibility to KP34 phage-mediated lysis, bacteria grown in liquid MHB medium were transferred onto MHA agar plates (bioMérieux). After drying, a drop of the phage suspension (108 pfu/ml) was placed on the bacterial layer and incubated at 37°C. The plates were checked 4–6 h and again 18 h later for the presence of bacterial lysis. Spot testing is a rapid and efficient method for determining the host range in large collection of bacteria (Clokie and Kropinski 2009).
Bacteriophage structural protein analysis by SDS-PAGE
Phage particles were partially purified by PEG precipitation (Sambrook and Russell 2001). After centrifugation, the pellets were suspended in 100 mM NaCl, 100 mM Tris–HCl (pH 7.5) and 25 mM EDTA buffer. Further purification was carried out by extraction with chloroform (1:1 v/v) followed by centrifugation. The concentrated phage particles were collected from the aqueous phase, mixed with the sample buffer (62.5 mM Tris HCl, 2% (w/v) SDS, 10% (w/v) glycerol, 5% (v/v) 2-mercaptoethanol, 0.001% (w/v) bromophenol blue) and heated in a boiling water bath for 5 min. Polyacrylamide gel electrophoresis was performed as described by Laemmli (1970). Discontinuous sodium dodecyl sulphate (SDS) gel electrophoresis was carried out on slabs of 10% acrylamide. After electrophoresis, the gels were stained with Bio-Safe Coomassie Stain (Bio-Rad, Hercules, CA, USA). Bio-Rad Quantity One software was used for the molecular analysis of the phage structural proteins based upon a Sigma-Aldrich wide-range molecular weight marker S8445 protein standard. Molecular mass of structural proteins (kilodalton) of identified open reading frames (ORFs) products were estimated on the basis of amino acid sequence composition (http://izoelektryczny.ovh.org/).
DNA isolation and restriction endonuclease analysis
Bacteriophage DNA was extracted and purified from phage lysate using a QIAGEN® Lambda Midi Kit (QIAGEN Inc., Valencia, CA, USA) and following the manufacturer’s protocol. Phage DNA was digested with the restriction endonucleases (EcoRV, EcoRI, HindIII, NsiI, NcoI, PaeI, Fermentas Life Science, Vilnius, Lithuania) according to the supplier’s recommendations. DNA fragments were separated by electrophoresis in 0.6% agarose gel containing ethidium bromide (0.5 μg/ml) in Tris–boric acid–EDTA buffer, at 90 V in a Bio-Rad agarose gel electrophoresis system. Restriction digestions were carried out in triplicate.
Nucleotide sequence
KP34 DNA was sequenced by a commercial company, Genomed Ltd. (Warsaw, Poland), and its annotated sequence has been deposited in GenBank under accession number GQ413938.
Clustering of phage genome sequences
Ninety-two complete Podoviridae genomic sequences were downloaded from GenBank (http://www.ncbi.nlm.nih.gov; see Table S2 in electronic supplementary material). Information about the classification of these phages was taken from the GenBank annotations and from Lavigne et al. (2008). To cluster phages based on their genome sequences, cluster analysis of sequences (CLANS) was applied, which uses a version of the Fruchterman–Reingold graph layout algorithm to visualize pairwise sequence similarities in either two-dimensional or three-dimensional space (Frickey and Lupas 2004). The program performs all-against-all BLAST searches and calculates pairwise attraction values based on P values of high scoring segment pairs (HSPs). Analysed sequences are represented in the graph by vertices which are connected by edges reflecting attractive forces proportional to the negative logarithm of the HSP’s P value. For the whole set of 92 Podoviridae genome sequences, we performed all-against-all TBLASTX searches assuming a word-size = 2. The pairwise comparison of 36 Autographivirinae genome sequences was made by BLASTN assuming a word-size = 7 to increase sensitivity.
The applied method differs from the approach developed by Lima-Mendez et al. (2008) for a reticulate classification of phages, which is based on gene content. Starting from gene families, the authors built a weighted graph, where nodes represented phages and edges represented phage–phage similarities in terms of shared genes.
Phylogenetic analyses
Homologous sequences of four selected Klebsiella phage KP34 proteins—capsid protein (accession number ACY66716.1), putative internal core protein (ACY66722.1) and tail tubular proteins A (ACY66718.1) and B (ACY66719.1)—were obtained through a thorough search of GenBank using BLAST. To verify the BLAST results and determine domain content, we searched Conserved Domain Database (Marchler-Bauer et al. 2005). Only sequences that showed significant hits to domains present in the phage KP34 proteins were included in final analyses. Amino acid alignments were obtained in MAFFT program using slow and accurate algorithm L-INS-i with 1,000 cycles of iterative refinement (Katoh et al. 2005). Sites suitable for further phylogenetic analyses were extracted from the alignments with Gblocks 0.91b assuming less stringent criteria (Castresana 2000). Phylogenetic trees were inferred by the maximum likelihood method in Phyml (Guindon and Gascuel 2003) and by a Bayesian approach in PhyloBayes (Lartillot and Philippe 2004) software. In Phyml, we used amino acid substitution models as proposed by ProtTest program 2.4 (Abascal et al. 2005): LG + Γ (for capsid protein), LG + Γ + F (for core protein) and LG + I + Γ + F (for tail tubular proteins A and B). We assumed five discrete categories for gamma distributed rates and the best heuristic search algorithms in Phyml, i.e. NNI and SPR. Edge support was assessed by the bootstrap analysis with 1,000 replicates and by the approximate likelihood ratio test based on χ 2 and Shimodaira–Hasegawa-like procedure. The minimum of these two support values was shown at nodes in presented trees. In PhyloBayes analysis, two independent Markov chains were run for 200,000 cycles assuming the CAT-Poisson+ Γ model with number of components, weights and profiles inferred from the data and five discrete categories for gamma distributed rates. After getting a convergence, the last 50,000 trees from each chain were collected to compute posterior consensus.
Statistical analysis
A comparison of differences in the properties of phage susceptible strains basing on the ESBL carrying plasmid was performed by χ 2 test with Yates’ correction. Statistical analysis of the data was done using StatSoft’s (Tulsa, OK, USA) statistical package STATISTICA9, and the differences were considered significant at P ≤ 0.05.
Results
Isolation and physicochemical properties of KP34
During this study, six different sewage sources were screened separately for phage presence using clinical K. pneumoniae strain 77 as the host, but in only a sample collected from a wastewater treatment plant located near Wrocław, Poland was lytic activity detected. Phage KP34 was purified by single plaque picking, dilution and titration. In the double-agar-layer technique, this virus produces plaques 5 mm in diameter with small clear centre surrounded by hazy ring (halo). The presence of the halo might suggest the production of the soluble phage enzymes, for example polysaccharide depolymerases, as suggested by Hughes et al. (1998). Phage KP34 was negatively stained with uranyl acetate and observed by electron microscopy (Fig. 1). It possesses an icosahedral head approximately 57 × 63 nm connected to short (15 nm) tail common to members of the Podoviridae family, morphotype C1 (Ackermann and DuBow 1987). These dimensions are consistent with T7 type phages.

Electron micrograph of phage KP34 negatively stained with uranyl acetate. The bar indicates 100 nm
Determination of KP34 phage bacterial host range and lytic potential
A high percentage (99.7%) of the KP34 particles adsorbed to K. pneumoniae 77 cells after 5 min of incubation. The one-step growth curve of KP34 indicated that the latent period was short (15 min) and the estimated burst size was ∼40–50 phage particles per infected bacterial cell. The lytic activity of KP34 tested against the 332 bacterial strains (Table S1) was limited only to K. pneumoniae subsp. pneumoniae strains. All of 55 Enterobacter sp., 100 Escherichia coli and 72 Klebsiella oxytoca strains were found to be resistant to this phage. Among the 101 K. pneumoniae subsp. pneumoniae isolates 42 (41.6%) were susceptible to KP34, but none of the K. pneumoniae subsp. ozaenae or K. pneumoniae subsp. rhinoscleromatis strains was sensitive. To verify specificity of KP34 to ESBL(+) and ESBL(−) K. pneumoniae subsp. pneumoniae strains the NMI collection was analysed. In that group, all 50 ESBL(+) and 50 ESBL(−) isolates were recognised in typing by the randomly amplified polymorphic DNA technique as separated clones. It turned out that 47.1% of ESBL(+) versus 36% of ESBL(−) strains were susceptible to KP34 phage. However, KP34 phage exhibited lytic activity against ESBL(+) strains much faster (after 6 h of incubation) in comparison to ESBL(−) strains where visible plaques were better seen after 18 h of incubation.
Stability of phage particles
Storage stability is the one of the important parameters of phage application in therapy. A vital property of industrial phage preparations is low susceptibility to temperature fluctuations and possibility of storage under different temperature conditions. KP34 high-titer phage lysates were stored in different ways: at room temperature, 4°C, −20°C and −70°C and in 25% glycerol at −20°C and −70°C. After 3 month storage, there was no significant loss of active phage particles at all of the tested temperatures, except at room temperature, where the number of plaque-forming unit was reduced by three orders of magnitude. KP34 was found to be relatively sensitive to high temperature, with a 100-fold decrease in titer observed after 10 min at 60°C. KP34 was unaffected by chloroform treatment for 2 and 24 h. The susceptibility of KP34 to different pH conditions showed that the phage was totally destroyed at pH 2 after 1 h of incubation at both temperatures. Incubation at pH 4 and 37°C caused a three and seven log decrease in phage titer after 1 and 5 h of incubation, respectively. The same acidity reduced plaque-forming units per millilitre 10-fold at room temperature after 5 h. KP34 was stable within a pH range 5–10.
Genome analysis
KP34 DNA was sensitive to digestion by a variety of restriction endonucleases (EcoRI, EcoRV, HindIII, NsiI, NcoI, PaeI) indicated that it is unmodified double-stranded DNA. The sequence of phage KP34 consisted of 43,809 bp (G + C content 54%) which is close to the genome size of other members of the Autographivirinae (Sillankorva et al. 2008). A total of 57 ORFs were predicted in the KP34 phage genome (Table 1), talking up a total of 93% of the coding capacity of this virus. The predicted ORFs encoded hypothetical proteins from 36 to 1,232 amino acid residues. Twenty-two of the KP34 potential gene products showed significant sequence similarity to proteins from Vibrio phage VP93 classified to the “phiKMV-like viruses” (Bastías et al. 2010). Nine KP34 potential gene products showed sequence similarity to proteins from different viruses infecting Escherichia, Burkholderia, Yersinia, and Iodobacteria strains (Table 1). Fifty-three of the KP34 ORFs start with an AUG codon, while four begin with UUG. Among the stop codons, UAA was the most commonly used (32 ORFs). The remainder ended with UGA (17 occurrences) or UAG (eight).
Structural protein analysis
Analysis of genome sequence revealed presence of eight genes that coded for structural proteins (Table 1). All of these identified gene products showed identity with homologous proteins found in Vibrio phage VP93. The highest sequence similarity was detected for the capsid protein (73%) which possesses an estimated molecular mass of 37.8 kDa. Comparative proteomic analyses, using data on φKMV (Lavigne et al. 2006), allowed us to postulate that the product of gene 42 (locus lag KP-KP34p42) which is described as a conserved hypothetical protein could be additional structural protein (Fig. 3). In case of the Vibrio VP93, the predicted function of the protein homologous to KP34 gp42 is also designated as unknown. For further characteristics of KP34, its structural protein composition was analysed by SDS-PAGE (Fig. 2). At least ten different protein bands were detected in the gel ranging from approximately 20 to 97 kDa. According to genome analysis, nine structural proteins could be expected. The two most predominant peptide bands were approximately 42 and 39 kDa. The abundance and estimated mass of gp42, along with its homology to φKMV, led us to conclude that the 39-kDa band is the capsid protein.

SDS-PAGE analysis of purified KP34 virus particles with Sigma-Aldrich wide-range molecular weight markers in the left lane
Clustering of phage genome sequences
We used the CLANS software package to reveal the relationship of Klebsiella phage KP34 genome to complete genome sequences of other bacteriophages. The graph presented in Fig. S1 (in electronic supplementary material) reflects global similarity of 92 Podoviridae genomic sequences at the amino acid level. In Table S2 (in electronic supplementary material), we list all analysed members of the Podoviridae with proposed taxonomic classification. Almost all phages clustered according to their taxonomic affiliation proposed by Lavigne et al. (2008). Interestingly, in the scale of the whole graph, a compact cluster is formed by BPP-1-, P22- and “ε15-like viruses” together with 13 currently unclassified phages (see Table S2).
In our analyses, Lactococcus phage asccφ28 shows a close relationship with the Picovirinae which is in accord with the findings of Kotsonis et al. (2008). On the other hand, the connections of Lactococcus phage KSY1 to the “AHJD-like viruses” seen in the figure result from significant hits of protein KSY1p075 (tail protein) and KSY1p076 (hypothetical protein) to the corresponding homologues. Their genes were probably acquired by the Lactococcus phage KSY1 genome via horizontal gene transfer. Lactococcus phage KSY1 shows also quite strong attraction to Lactococcus phage asccφ28 in the graph. This affiliation results from several significant hits from the lysin (amidase), which was also found in Lactobacillus casei and Lactobacillus rhamosus and in phages classified to Siphoviridae: Lactobacillus phages A2 and PL-1 and Lactococcus phage r1t. This suggests that the genomic region coding the lysin was probably subjected to horizontal gene transfer similarly to other region encoding components of the distal tail structure (Chopin et al. 2007). The most distant position in the graph is occupied by two “119X-like viruses” which do not connect with any Picovirinae viruses which in turn show moderate affiliation to the rest of viruses. “LUZ24-viruses”, Enterobacteria phage phiEco32 and Roseobacter phage SIO1 show the strongest attraction to Autographivirinae.
Since Klebsiella phage KP34 showed the closest affiliation to the Autographivirinae subfamily, we decided to carry out a separate whole-genome comparison of this subfamily using BLASTN to analyse this relationship in details (Fig. 3). This analysis revealed four distinct clusters corresponding to phiKMV-, P60-, SP6- and “T7-like viruses”. Klebsiella phage KP34 belongs to the cluster of “phiKMV-like viruses” and is the most closely related to the recently sequenced Vibrio phage VP93 also classified to this genus (Bastías et al. 2010). Among “phiKMV-like viruses”, six Pseudomonas and two Enterobacteria phages form the most compact grouping. The members of SP6-like viruses appear close relatives to “T7-like viruses” through the CLANS analysis. This agrees with genomic analysis of Enterobacteria phages SP6 and K1-5 which are considered an estranged subgroup of the T7-like viruses (Scholl et al. 2004).

Graphic layout made in CLANS software for 36 Autographivirinae genome sequences using BLASTN searches. Analysed sequences are represented by vertices connected by edges reflecting attractive forces proportional to the negative logarithm of the HSP’s P value. The greyness intensity of the connections is proportional to these forces. The not shown phages names forming compact clusters are fully listed in Table S2 in electronic supplementary material
Phylogenetic analyses
Phylogenetic analyses were performed on four selected Klebsiella phage KP34 proteins: capsid protein, putative internal core protein KP-KP34p48 (gene 48; Fig. S2A, B in electronic supplementary material), tail tubular protein A, and tail tubular protein B (Fig. 4a, b). Trees obtained both in Phyml and PhyloBayes gave identical or almost identical topologies for each of these proteins. In agreement with clustering analysis, the Klebsiella phage KP34 proteins clustered in all trees significantly with its homologues found in Vibrio phage VP93 and belonged to a major well-supported clade containing sequences from φKMV-like viruses infecting γ-Proteobacteria. All the analysed proteins exhibit homologues in members of different bacterial groups, which indicate that these phage genes were subjected to intensive transduction process. Of particular interest was the finding of tail tubular proteins A and B homologues in a number of bacterial genomes including α-, β- and γ-Proteobacteria and Synergistetes. The latter are a phylum of obligately anaerobic Gram-negative bacteria (Vartoukian et al. 2007). While a T7-like prophage had been identified in the genome of Pseudomonas putida strain KT2440 (Canchaya et al. 2003, 2004), this is the first time that similar prophages have been reported for the strains indicated here.


Phyml tree of tail tubular proteins A (a) and tail tubular proteins B (b). Sequence from Klebsiella phage KP34 indicated in bold font. Sequences from phages are placed in the grey rectangle. Numbers at nodes, in the shown order, correspond to the minimum of support values calculated in Phyml by χ2 and Shimodaira–Hasegawa-like procedure (χ 2-SH); support values resulted from bootstrap analysis in Phyml (PH) and posterior probabilities estimated in Phylobayes (PB). Values of the posterior probabilities and bootstrap percentages lower or equal to 0.50 and 50%, respectively, were omitted or indicated by a dash. GenBank identifiers (gi) of sequences are shown in parenthesis
Discussion
K. pneumoniae is one of the most common etiological factors of nosocomial infections particularly of Gram-negative bacteraemias and urinary tract infections (Matsen 1973; Center for Disease Control 1977; Jones et al. 2000; Coque et al. 2008). Widespread use of antibiotic treatment has been held responsible for the occurrence of Klebsiella strains producing ESBLs resulting in multidrug resistance (Gniadkowski 2001; Nijssen et al. 2004; Coque et al. 2008; Miriagou et al. 2010). At present, the prevalence of ESBL-producing Klebsiella strains in Europe has reached 10–30% among invasive isolates (European Antibiotic Resistance Surveillance System, http://www.ecdc.europa.eu/en/activities/surveillance/EARS-Net). Since multidrug-resistant bacteria appeared, therapeutic options have become limited. One possible method of bacteria eradication could be the application of bacteriophages. The idea of using phages as natural bacterial parasites is well-known, but most of available virus collections are ineffective against currently isolated clinical strains. There is a need to look for novel phages with lytic potential to multidrug resistance K. pneumoniae isolates. Recently isolated Klebsiella phages belonged to Podoviridae family (Kumari et al. 2009; 2010a; b; Verma et al. 2009) or pseudo-T-even family (Wu et al. 2007). During our team’s experiments, five different viruses from the Podoviridae, Siphoviridae and Myoviridae were characterised regarding their biology and genomics (data not shown) and genomes of three phages have already been deposited in GenBank. In the present study, one of these phages (KP34) was described. The KP34 virus was propagated on K. pneumoniae subsp. pneumoniae isolate carrying ESBL plasmid. The host specificity of KP34 among most common Enterobacteriaceae rods was limited to one species, but the lytic activity was found to be in the high range (41.6%) regardless of whether they were ESBL(+) or ESBL(−) strains. Klebsiella KP34 phage, with its narrow host specificity, could be applied in phage cocktail as alternative or as supportive treatment simultaneously with antibiotics, for K. pneumoniae subsp. pneumoniae strains eradication with no harmful action on normal bacterial flora.
With respect to its physicochemical stability, KP34 is similar in its sensitivity to chemical agents such pH and chloroform to the previously described T7-like Klebsiella phage, KPO1K2 (Verma et al. 2009), but is much more susceptible to high temperature. The average burst size determined from the one-step growth experiment is three times lower for KP34 (∼50 virions per cell) comparing to KPO1K2 (∼140 virions per cell). We identified ten KP34 structural proteins by SDS-PAGE, but only nine were predicted on the basis of our bioinformatic analysis of the sequence data. The presence of an additional 42-kDa protein in the gel could be the result of a frameshifting event in the major capsid gene as was observed previously with coliphage T7 phage (Condron et al. 1991). Similar results were also obtained with the T7-like Pseudomonas phage φIBB-PF7A (Sillankorva et al. 2008). Other investigators analysing structural proteins of K. pneumoniae T7-like phages by SDS-PAGE technique obtained wide range of band numbers, from 12 (29 to 120 kDa) with K. pneumoniae phage B5055 (Verma et al. 2009) to two to five bands representing proteins from 20 to 55 kDa in five phages (Kumari et al. 2010b). This would suggest that considerable variation occurs in this group of bacteriophages with respect to protein number and size. It is also significant that KP34 phage genome encoded only one type of tail fiber protein (307 amino acid; 32.7 kDa) as opposed to the closely related φKMV described by Lavigne et al. (2006) which produces two fiber proteins (gp38 and gp40).
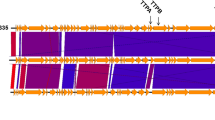
This bacteriophage was also characterised genomically. Genetic and proteomic analysis helped us to discover new types of viruses and determine the phylogenetic relationships within phage groups. The whole-genome comparisons and phylogenetic studies showed a clear relationship between K. pneumoniae phage KP34 and the φKMV-like bacterial viruses. Among phages with completely sequenced genomes, Vibrio phage VP93 seems to be the closest relative of KP34. However, detailed comparison of genomes from KP34 and other φKMV-like viruses revealed some differences in gene organisation at the 3′-end genomic region in these phages and lysozyme domain distribution (Fig. 5). Lavigne et al. (2006) described that one of the features distinguishing Pseudomonas phage φKMV from “T7-like viruses” is that the gp36 protein containing a functional C-terminal lysozyme domain. They reported that the corresponding gp35 proteins in two “SP6-like viruses” also have a proper lysozyme domain at their C-terminal end. However, the “T7-like viruses” have a functional transglycosylase domain at the N-terminal end of gp16 proteins. We found that proteins with the C-terminal lysozyme domain are also coded by genomes of other “phiKMV-like viruses” (listed in Table S2 in electronic supplementary material) with exception of Ralstonia phage RSB1, Klebsiella phage KP34 and Vibrio phage VP93. Interestingly, Ralstonia phage RSB1, similar to “T7-like viruses”, have a lysozyme domain at the N-terminal end of gp37 protein which is annotated as putative transglycosylase (Fig. 5). Although gene 46 (locus tag: KP-KP34p47) from Klebsiella phage KP34 and ORF33 (locus tag: VPP93_gp33) from Vibrio phage VP93 are in the same gene context as gp36 (locus tag: phiKMVp36) from Pseudomonas phage φKMV, their products show neither significant similarity to the gp36 nor to the lysozyme domain. We found only marginal similarity in pairwise BLAST comparison the C-terminal lysozyme domain proteins from “phiKMV-like” viruses with KP-KP34p47 and VPP93_gp33 proteins (identity ∼20%; similarity ∼40%; E-value = 0.001 and 0.014, respectively). Probably, the genes in Klebsiella phage KP34 and Vibrio phage VP93 have undergone a high rate of divergence rate which minimized the sequence similarity. The most striking differences in the analysed genome region are visible just at its 3′-end (Fig. 5). Among the compared genomes only Vibrio phage VP93 does not code for a lysozyme but instead has a putative glycosyl hydrolase. Pseudomonas phage φKMV genome encodes additional putative tail fiber proteins and two minor structural proteins while Ralstonia phage RSB1 has a gene coding a putative type II and III secretion system protein precursor. The different gene contents may be responsible for differences in host recognition and infection mechanisms between “typical” φKMV viruses and Klebsiella phage KP34, Ralstonia phage RSB1 and Vibrio phage VP93. This is in agreement with whole-genome clustering analysis which showed outstanding position of three latter phages from the “typical” φKMV viruses (Fig. 3).

Comparison of 3′-end genomic sequence in four phages classified to “φKMV-like” viruses. ORFs whose products show significant sequence similarity are presented in the same colour. ORFs in grey have their products annotated as hypothetical protein
References
Abascal F, Zardoya R, Posada D (2005) ProtTest: selection of best-fit models of protein evolution. Bioinformatics 21:2104–2105
Ackermann HW, DuBow MS (1987) Viruses of prokaryotes. CRC, Boca Raton, pp 143–172
Adams M (1959) Bacteriophages. Interscience, New York, pp 137–159
Bastías R, Higuera G, Sierralta W, Espejo RT (2010) A new group of cosmopolitan bacteriophages induce a carrier state in the pandemic strain of Vibrio parahaemolyticus. Environ Microbiol 12:990–1000
Bogovazova GG, Voroshilova NN, Bondarenko VM (1991) The efficacy of Klebsiella pneumoniae bacteriophage in the therapy of experimental Klebsiella infection. Zh Mikrobiol Epidemiol Immunobiol 4:5–8
Canchaya C, Proux C, Fournous G, Bruttin A, Brüssow H (2003) Prophage genomics. Microbiol Mol Biol Rev 67:238–276
Canchaya C, Fournous G, Brüssow H (2004) The impact of prophages on bacterial chromosomes. Mol Microbiol 53:9–18
Castresana J (2000) Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol 17:540–552
Center for Disease Control (1977) National nosocomial infections study report, annual summary 1975. Center for Disease Control, Atlanta
Chopin A, Deveau H, Ehrlich SD, Moineau S, Chopin MC (2007) KSY1, a lactococcal phage with a T7-like transcription. Virology 365:1–9
Clokie MRJ, Kropinski AM (2009) Bacteriophages: methods and protocols, volume 1: isolation, characterization, and interactions. Humana, New York
Condron BG, Atkins JF, Gestland RF (1991) Frameshifting in gene 10 of bacteriophage T7. J Bacteriol 173:6998–7003
Coque TM, Baquero F, Canton R (2008) Increasing prevalence of ESBL-producing Enterobacteriaceae in Europe. Euro Surveill 13:19044
Dąbrowski M, Weber-Dąbrowska B, Doroszkiewicz W (1988) The estimation of usefulness of the Pseudomonas bacteriophages in epidemiological investigations of Pseudomonas clinical strains. Acta Microbiol Pol 37:295–307
Frickey T, Lupas AN (2004) CLANS: a Java application for visualizing protein families based on pairwise similarity. Bioinformatics 20:3702–3704
Gallet R, Shao Y, Wang I-N (2009) High adsorption rate is detrimental to bacteriophage fitness in a biofilm-like environment. BMC Evol Biol 9:241–253
Gniadkowski M (2001) Evolution and epidemiology of extended-spectrum beta-lactamases (ESBLs) and ESBL-producing microorganisms. Clin Microbiol Infect 7:597–608
Guindon S, Gascuel O (2003) A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol 52:696–704
Hughes KA, Sutherland IW, Clark J, Jones MV (1998) Bacteriophage and associated polysaccharide depolymerases—novel tools for study of bacterial biofilms. J Appl Microbiol 85:583–590
Jones RN, Croco MTA, Kugler KC, Pfaller MA, Beach ML, The SENTRY participants group (North America) (2000) Respiratory tract pathogens isolated from patients hospitalized with suspected pneumonia: frequency of occurrence and antimicrobial susceptibility patterns from SENTRY antimicrobial surveillance program (United States and Canada, 1997). Diagn Microbiol Infect Dis 37:115–125
Katoh K, Kuma K, Toh H, Miyata T (2005) MAFFT version 5: improvement in accuracy of multiple sequence alignment. Nucleic Acids Res 33:511–518
Kotsonis SE, Powell IB, Pillidge CJ, Limsowtin GK, Hillier AJ, Davidson BE (2008) Characterization and genomic analysis of phage asccφ28, a phage of the family Podoviridae infecting Lactococcus lactis. Appl Environ Microbiol 74:3453–3460
Kumari S, Harjai K, Chhibber S (2009) Efficacy of bacteriophage treatment in murine burn wound infection induced by Klebsiella pneumoniae. J Microbiol Biotechnol 19:622–628
Kumari S, Harjai K, Chhibber S (2010a) Evidence to support the therapeutic potential of bacteriophage Kpn5 in burn wound infection caused by Klebsiella pneumoniae in BALB/c mice. J Microbiol Biotechnol 20:935–941
Kumari S, Harjai K, Chhibber S (2010b) Isolation and characterization of Klebsiella pneumoniae specific bacteriophages from sewage samples. Folia Microbiol Praha 55:221–227
Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–686
Lartillot N, Philippe H (2004) A Bayesian mixture model for across site heterogeneities in the amino-acid replacement process. Mol Biol Evol 21:1095–1109
Lavigne R, Noben JP, Hertveldt K, Ceyssens PJ, Briers Y, Dumont D, Roucourt B, Krylov VN, Mesyanzhinov VV, Robben J, Volckaert G (2006) The structural proteome of Pseudomonas aeruginosa bacteriophage φKMV. Microbiol 152:529–534
Lavigne R, Seto D, Mahadevan P, Ackermann HW, Kropinski AM (2008) Unifying classical and molecular taxonomic classification: analysis of the Podoviridae using BLASTP-based tools. Res Microbiol 159:406–414
Lima-Mendez G, Van Helden J, Toussaint A, Leplae R (2008) Reticulate representation of evolutionary and functional relationships between phage genomes. Mol Biol Evol 25:762–777
Marchler-Bauer A, Anderson JB, Cherukuri PF, DeWeese-Scott C, Geer LY, Gwadz M, He S, Hurwitz DI, Jackson JD, Ke Z, Lanczycki CJ, Liebert CA, Liu C, Lu F, Marchler GH, Mullokandov M, Shoemaker BA, Simonyan V, Song JS, Thiessen PA, Yamashita RA, Yin JJ, Zhang D, Bryant SH (2005) CDD: a conserved domain database for protein classification. Nucleic Acids Res 33:D192–D196
Matsen JM (1973) The sources of hospital infection. Medicine 52:271–277
Miriagou V, Cornaglia G, Edelstein M, Galani I, Giske CG, Gniadkowski M, Malamou-Lada E, Martinez-Martinez L, Navarro F, Nordmann P, Peixe L, Pournaras S, Rossolini GM, Tsakris A, Vatopoulos A, Cantón R (2010) Acquired carbapenemases in gram-negative bacterial pathogens: detection and surveillance issues. Clin Microbiol Infect 16:112–122
Nijssen S, Florijn A, Bonten MJM, Schmitz FJ, Verhoef J, Fluit AC (2004) Beta-lactam susceptibilities and prevalence of ESBL-producing isolates among more than 5000 European Enterobacteriaceae isolates. Int J Antimicrob Agents 24:585–591
Pajunen M, Kiljunen S, Skurnik M (2000) Bacteriophage φYeO3-12, specific for Yersinia enterocolitica serotype O:3, is related to coliphages T3 and T7. J Bacteriol 182:5114–5120
Richardson JF, Rosdahl VT, van Leeuwen WJ, Vickery AM, Vindel A, Witte W (1999) Phages for methicillin-resistant Staphylococcus aureus: an international trial. Epidemiol Infect 122:227–233
Roncero C, Darzins A, Casadaban MJ (1990) Pseudomonas aeruginosa transposable bacteriophages D3112 and B3 require pili and surface growth for adsorption. J Bacteriol 172:1899–1904
Sakandelidze VM, Meipariani AN (1974) Use of combined phages in suppurative-inflammatory diseases. Zh Mikrobiol Epidemiol Immunobiol 6:135–136
Sambrook J, Russell DW (2001) Molecular cloning, 2nd edn. Cold Spring Harbor Laboratory, New York
Scholl D, Kieleczawa J, Kemp P, Rush J, Richardson CC, Merril C, Adhya S, Molineux IJ (2004) Genomic analysis of bacteriophages SP6 and K1-5, an estranged subgroup of the T7 supergroup. J Mol Biol 335:1151–1171
Sillankorva S, Neubauer P, Azeredo J (2008) Isolation and characterization of a T7-like lytic phage for Pseudomonas fluorescens. BMC Biotechnol 8:80
Ślopek S, Durlakowa I, Kucharewicz-Krukowska A, Krzywy T, Ślopek A, Weber B (1972) Phage typing of Shigella flexneri. Arch Immunol Ther Exp 20:1–60
Ślopek S, Durlakova I, Weber-Dąbrowska B, Kucharewicz-Krukowska A, Dąbrowski M, Bisikiewicz R (1981a) Results of bacteriophage treatment of suppurative bacterial infections I. General evaluation of the results. Arch Immunol Ther Exp 31:267–291
Ślopek S, Durlakova I, Weber-Dąbrowska B, Kucharewicz-Krukowska A, Dąbrowski M, Bisikiewicz R (1981b) Results of bacteriophage treatment of suppurative bacterial infections II. Detailed evaluation of the results. Arch Immunol Ther Exp 31:293–327
Ślopek S, Durlakova I, Weber-Dąbrowska B, Dąbrowski M, Kucharewicz-Krukowska A (1984) Results of bacteriophage treatment of suppurative bacterial infections III. Detailed evaluation of the results obtained in further 150 cases. Arch Immunol Ther Exp 32:317–335
Sulakvelidze A, Alavidze Z, Morris JG Jr (2001) Bacteriophage therapy. Antimicrob Agents Chemother 45:649–659
Vartoukian SR, Palmer RM, Wade WG (2007) The division “Synergistes”. Anaerobe 13:99–106
Verma V, Harjai K, Chhibber S (2009) Characterization of a T7-like lytic bacteriophage of Klebsiella pneumoniae B5055: a potential therapeutic agent. Curr Microbiol 59:274–281
Weber-Dąbrowska B, Mulczyk M, Górski A (2000) Bacteriophage therapy of bacterial infections: an update of our institute’s experience. Arch Immunol Ther Exp 48:547–551
Weber-Dąbrowska B, Mulczyk M, Górski A (2001) Bacteriophage therapy for infections in cancer patients. Clin Appl Immunol Rev 1:131–134
Weber-Dąbrowska B, Mulczyk M, Górski A (2003) Bacteriophages as an efficient therapy for antibiotic-resistant septicemia in man. Transp Proc 35:1385–1386
Wu LT, Chang SY, Yen MR, Yang TC, Tseng YH (2007) Characterization of extended-host-range pseudo-T-even bacteriophage Kpp95 isolated on Klebsiella pneumoniae. Appl Environ Microbiol 73:2532–2540
Zhukov-Verezhnikov NN, Peremitina LD, Berillo EA, Komissarov VP, Bardymov VM, Khvoles AG, Ugryumov LB (1978) A study of the therapeutic effect of bacteriophages agents in a complex treatment of suppurative surgical diseases. Sov Med 12:64–66
Acknowledgements
We are grateful to Professor Marek Gniadkowski for making the NMI collection accessible. This study was supported by Polish Ministry of Science and Higher Education research grant no. N N401 3550 33. A.M.K. was supported by a Discovery Grant from the Natural Sciences and Engineering Research Council of Canada
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Drulis-Kawa, Z., Mackiewicz, P., Kęsik-Szeloch, A. et al. Isolation and characterisation of KP34—a novel φKMV-like bacteriophage for Klebsiella pneumoniae . Appl Microbiol Biotechnol 90, 1333–1345 (2011). https://doi.org/10.1007/s00253-011-3149-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-011-3149-y