
Abstract
Cytochrome P450 monooxygenases are valuable biocatalysts due to their ability to hydroxylate unactivated carbon atoms using molecular oxygen. We have cloned the gene for a new cytochrome P450 monooxygenase, named CYP154H1, from the moderately thermophilic soil bacterium Thermobifida fusca. The enzyme was overexpressed in Escherichia coli at up to 14% of total soluble protein and purified to homogeneity in three steps. CYP154H1 activity was reconstituted using putidaredoxin reductase and putidaredoxin from Pseudomonas putida DSM 50198 as surrogate electron transfer partners. In biocatalytic reactions with different aliphatic and aromatic substrates of varying size, the enzyme converted small aromatic and arylaliphatic compounds like ethylbenzene, styrene, and indole. Furthermore, CYP154H1 also accepted different arylaliphatic sulfides as substrates chemoselectively forming the corresponding sulfoxides and sulfones. The enzyme is moderately thermostable with an apparent melting temperature of 67°C and exhibited still 90% of initial activity after incubation at 50°C.
Similar content being viewed by others


Introduction
Cytochrome P450 monooxygenases (E.C. 1.14) form a large and diverse class of enzymes with currently >9,000 members (Nelson 2009). They are widely distributed in nature where they play important roles in the assimilation of carbon sources, the detoxification of xenobiotics, or the synthesis of secondary metabolites (Ortiz de Montanello 2005). Due to their ability to catalyze highly diverse chemical reactions using molecular oxygen, e.g., hydroxylation of non-activated carbon atoms, epoxidation of double bonds, oxidation of heteroatoms, dealkylations, etc. (Isin and Guengerich 2007), P450s are highly interesting and valuable biocatalysts (Bernhardt 2006). Cytochrome P450 monooxygenases contain a heme prosthetic group and depend on electron transfer components which deliver electrons from the cofactor to the heme iron during catalysis. Depending on the kind of electron transfer partners and their organization, P450s are divided into different classes (Hannemann et al. 2007). Class 1 comprises bacterial (soluble) and mitochondrial (membrane-bound) three-component P450 monooxygenases that besides the P450 monooxygenase consist of a ferredoxin reductase and a ferredoxin which shuttle electrons from the cofactor NAD(P)H to the monooxygenase component.
The best studied bacterial three-component P450 monooxygenase is CYP101A1 (P450cam) from Pseudomonas putida DSM 50198. Together with its electron transfer partners putidaredoxin reductase (PdR) and putidaredoxin (Pdx), it catalyzes the first step in the degradation of camphor by stereospecific hydroxylation of the substrate giving 5-exo-hydroxycamphor (Hedegaard and Gunsalus 1965). One special feature of the P450cam system is that all three components necessary for camphor hydroxylation in P. putida are encoded together in one operon. In contrast, for many other bacterial three-component P450 monooxygenases, the natural electron transfer partners are not known. Since it was found that putidaredoxin is also able to deliver electrons to a range of other bacterial P450s, PdR and Pdx have been used as substitute electron transfer partners (Agematu et al. 2006; Furuya and Kino 2009). However, the electron transfer efficiency varies significantly which may lead to rather high uncoupling of NADH consumption and substrate oxidation.
So far, only few thermostable P450 monooxygenases are described in the literature, most of them of archaeal origin. The best studied archaeal P450 is CYP119, a hyperthermostable enzyme from Sulfolobus solfataricus. It was shown to epoxidize styrene and cis-stilbene and to hydroxylate fatty acids (Koo et al. 2000, 2002; McLean et al. 1998). The crystal structure was solved and the natural electron donors of this system were discovered to consist of a ferredoxin and a 2-oxoacid-ferredoxin reductase (Puchkaev and Ortiz de Montellano 2005; Yano et al. 2000). The only characterized thermostable P450 monooxygenase of bacterial origin is CYP175A1 from Thermus thermophilus, an enzyme displaying β-carotene hydroxylase activity (Blasco et al. 2004). Its crystal structure was also solved and recently the natural electron donor partners, a ferredoxin and a NADPH-dependent ferredoxin reductase, were identified (Mandai et al. 2009; Yano et al. 2003).
The moderately thermophilic soil bacterium Thermobifida fusca with an optimum growth temperature of 50–55°C belongs to the phylum of Actinobacteria and is a major degrader of plant cell walls in heated organic materials (Bachmann and McCarthy 1991). This bacterium is of high interest due to the secretion of different thermostable cellulolytic enzymes that display high activity and a broad pH range (Wilson 2004). Recently, the genome sequence of this actinomycete was published (Lykidis et al. 2007). The single chromosome contains ten different genes encoding putative cytochrome P450 monooxygenases of which four belong to family CYP154. To the same family belong currently 18 members, all P450s originating from actinomycetes (Nelson 2009). However, only few of them have been studied so far. CYP154C1 from Streptomyces coelicolor A3(2) was shown to convert the macrolides narbomycin and YC-17 to pikromycin and neomethymycin, respectively (Podust et al. 2003), whereas CYP154 from Nocardia farcinica IFM 10152 hydroxylates testosterone (Agematu et al. 2006). Also the crystal structures of two members of this family, CYP154A1 and CYP154C1 from S. coelicolor A3(2), were determined revealing an unusual 180° flip of the heme in the active site of CYP154A1 (Podust et al. 2003, 2004). Besides that, none of the P450 monooxygenases belonging to family CYP154 has been characterized in detail yet.
We became interested in CYP154H1 from T. fusca during our work on steroid hydroxylation, since the enzyme exhibits 46% sequence identity at the protein level with CYP154 from N. farcinica IFM 10152, an enzyme that regio- and stereoselectively hydroxylates testosterone (Agematu et al. 2006). Furthermore, CYP154H1 shares 52% sequence identity at the protein level with CYP154C1 from S. coelicolor A3(2), of which the three-dimensional structure was solved, allowing us to generate a structural model of CYP154H1. Additionally, the anticipated higher thermal stability makes the enzyme an attractive candidate for biocatalytic applications. In this study, we report on the recombinant expression, purification, and characterization of CYP154H1, a new moderately thermostable bacterial P450 monooxygenase from T. fusca.
Materials and methods
Materials
All chemicals were purchased from Sigma-Aldrich (Zwijndrecht, The Netherlands), unless otherwise specified. Isopropyl-1-thio-β-d-galactopyranoside (IPTG) was obtained from Roth GmbH (Karlsruhe, Germany).
DNA primers were obtained from Sigma Genosys (Germany). DNA-modifying enzymes were purchased from New England Biolabs (Beverly, MA, USA).
Bacterial strains and plasmids
The Escherichia coli TOP10 strain (Invitrogen, Carlsbad, CA, USA) was used for genetic manipulations, while E. coli C43(DE3) was used for expressions. Strain P. putida DSM 50198 was obtained from the DSMZ (Braunschweig, Germany), and strain T. fusca was kindly provided by Dr. Diana Irwin (Cornell University, New York, NY, USA).
Plasmids pACYC-Duet1 and pET28a(+) were purchased from Novagen (EMD Biosciences, San Diego, CA, USA). The broad host-range expression vector pIT2-MCS was derived from vector pBBR1 (Kovach et al. 1994; kindly provided by Kenneth Peterson, Louisiana State University Medical Center, Shreveport, LA, USA) by introduction of the trc promotor from pKK233-2 (Takara Bio Europe/Clontech, Saint-Germain-en-Laye, France), exchanging the bla ampicillin resistance gene by the tet gene conferring tetracycline resistance and introduction of the multiple cloning site of vector pBAD/Myc-His (Invitrogen).
Cloning of cyp154H1, camA, and camB
The gene of CYP154H1 was amplified by PCR from genomic DNA of T. fusca using primers TF-P-NdeI-FW: 5′-GGAGGTCACATATGGCTTCGCCTACCGACAATCCG-3′ and TF-P-HindIII-RV: 5′-GTTTTGCGGGAAGCTTACGGGCGCAGGATGAC-3′. Thereby restriction sites NdeI and HindIII were introduced for subsequent cloning of cyp154H1 into vector pIT2-MCS. The resulting vector was named pIT2cyp154H1.
Genes camA and camB coding for PdR and Pdx, respectively, were amplified by PCR from genomic DNA of P. putida DSM 50198 using primers PP-R-BspHI-FW: 5′-AAAAAAAATCATGAACGCAAACGACAACGTGG-3′ and PP-R-SacI-RV: 5′-ATTTAGAGCTCAGGCACTACTCAGTTCAGCTTTGG-3′ for camA as well as PP-X-NdeI-FW: 5′-AGGATAATCATATGTCTAAAGTAGTGTATGTGTC-3′ and PP-X-XhoI-RV: 5′-TTTTTTTTCTCGAGGTTTACCATTGCCTATCG-3′ for camB. The PCR product carrying the camA gene was digested with BspHI and SacI and ligated into vector pACYC-Duet1 cut with NcoI and SacI. The resulting vector, named pACYCcamA, was then used as template in a QuikChange site-directed mutagenesis PCR to remove the XhoI site present in camA. The PCR was performed according to the manufacturer’s instructions using primers QC-R-XhoI-FW: 5′-ATACCTGCGCACACTGGAGGACGCCGAGTGCATTC-3′ and QC-R-XhoI-RV: 5′-AATGCACTCGGCGTCCTCCAGTGTGCGCAGGTATC-3′. The resulting vector was digested with NdeI and XhoI and ligated with the camB gene, previously cut with the same pair of enzymes. The final plasmid was named pACYCcamAB. Similarly, camB digested with NdeI and XhoI was also ligated into pACYC-Duet1 to give vector pACYCcamB.
Expression of CYP154H1 together with Pdx and PdR in E. coli
E. coli C43(DE3)(pACYCcamAB)(pIT2cyp154H1) cells were grown in 500 ml TB medium containing 25 μg ml−1 chloramphenicol and 10 μg ml−1 tetracycline (TBCm, Tc) at 37°C to an optical density at 600 nm of 1. Then, 0.8 mM IPTG and 0.5 mM δ-amino levulinic acid (δ-ala) were added to start protein expression, and the culture was incubated for another 48 h at 28°C. The cells were harvested by centrifugation at 5,000×g for 20 min and washed once with 50 mM potassium phosphate buffer, pH 7.5. The cell pellet was resuspended in 20 ml of the same buffer and sonicated for 15 min (15 s on, 30 s off, 70% amplitude) on ice. Cell debris was removed by centrifugation at 31,000×g for 45 min at 4°C. The resulting cell-free extract was stored at −20°C after addition of 10% glycerol.
Purification of CYP154H1
For easy purification of CYP154H1 via an N-terminal his6-tag, the corresponding gene was subcloned from plasmid pIT2cyp154H1 into pET28a(+) using restriction enzymes NdeI and HindIII. The resulting plasmid was named pET28cyp154H1 and transformed into E. coli C43(DE3) for expression. E. coli C43(DE3) cells containing pET28cyp154H1 were grown in 500 ml TB medium supplemented with 50 μg ml−1 kanamycin at 37°C to OD600 = 1.0. At this point, 0.8 mM IPTG and 0.5 mM δ-ala were added to start protein expression, and the culture was incubated for another 48 h at 30°C. The cells were harvested by centrifugation at 5,000×g for 20 min and washed once with 50 mM potassium phosphate buffer, pH 7.5. The cell pellet was resuspended in 20 ml of the same buffer and sonicated for 15 min (15 s on, 30 s off, 70% amplitude) on ice. Cell debris was removed by centrifugation at 31,000×g for 45 min at 4°C. The resulting cell-free extract was heated for 30 min at 50°C and centrifuged afterwards for 15 min at 17,000×g and 4°C to remove denatured protein. The supernatant carrying active CYP154H1 was afterwards loaded on a 60-ml Q Sepharose FF column (GE Healthcare, Hoevelaken, The Netherlands) equilibrated with 50 mM potassium phosphate buffer, pH 7.5. The column was washed with three column volumes of the same buffer before elution of CYP154H1 with a linear gradient of 0–500 mM KCl in 50 mM potassium phosphate buffer, pH 7.5. Fractions with the highest A416/A280 ratio were combined and concentrated by ultrafiltration using a 30-kDa cutoff membrane. This protein concentrate was then loaded on a 5-ml HisTrap HP column (GE Healthcare) equilibrated with 50 mM potassium phosphate buffer, pH 7.5. The column was washed with the same buffer containing 20 mM imidazole until all unbound protein was removed. Finally, CYP154H1 was eluted using 50 mM potassium phosphate buffer containing 200 mM imidazole, pH 7.5, and desalted using Econo-Pac 10DG columns (Bio-Rad Laboratories B.V., Veenendaal, The Netherlands) and 50 mM potassium phosphate buffer, pH 7.5. Purified CYP154H1 was concentrated by ultrafiltration using a 30-kDa cutoff membrane and stored at −20°C after addition of 10% glycerol.
Purification of PdR and Pdx
PdR expression was performed at 30°C for 48 h using E. coli C43(DE3)(pACYCcamA). The protein was purified from CFE according to the protocol of Sevrioukova et al. (2001).
Expression of Pdx was also performed at 30°C for 48 h using E. coli C43(DE3)(pACYCcamB). Purification of Pdx from CFE was carried out according to a published protocol (Sevrioukova et al. 2003).
Enzyme assays
CYP154H1 concentration was determined by measuring CO-difference spectra (Omura and Sato 1964). The amount of P450 was calculated based on the maximum absorbance of CO-bound P450 at 450 nm (ε 450 = 91 mM−1 cm−1). Putidaredoxin reductase activity was determined by monitoring the decrease in ferricyanide concentration at 420 nm (ε 420 = 1.02 mM−1 cm−1; Roome et al. 1983).
Putidaredoxin activity was determined by monitoring the reduction of cytochrome c at 550 nm (ε 550 = 19.1 mM−1 cm−1) with PdR as electron-supplying reductase (Lacour et al. 1998). Similarly, activity of PdR and Pdx together in CFE of E. coli C43(DE3) (pACYCcamAB)(pIT2cyp154H1) was also measured using the cytochrome c reduction assay.
Bioconversions
All enzyme reactions for determining the substrate scope of CYP154H1 were first carried out using CFE of E. coli C43(DE3)(pACYCcamAB)(pIT2cyp154H1) and later confirmed in reactions using purified CYP154H1, Pdx, and PdR. Bioconversions using CFE were performed in 10 ml closed glass tubes at 30°C with shaking. Small-scale reactions of 1 ml total volume consisted of 0.5 ml CFE (containing CYP154H1, Pdx, and PdR), 50 μM NADH, and 5 mM substrate (as 1 M stock in ethanol) as well as 150 mM sodium formate and 0.5 U formate dehydrogenase (FDH; Sigma-Aldrich) for cofactor regeneration in 50 mM potassium phosphate buffer. Reactions were extracted with each 500 μl ethyl acetate containing 0.5% dodecane as internal standard. Organic phases were dried over anhydrous sodium sulfate and analyzed by GC, GC-MS, and HPLC. Conversions were usually calculated from relative peak areas of products after GC analysis using a standard curve. In case of complex product mixtures, conversions were calculated from respective relative peak areas of substrates.
Bioconversions using purified CYP154H1, Pdx, and PdR were performed accordingly containing 10 μM CYP154H1, 10 μM PdR, and 30 μM Pdx instead of CFE. Furthermore, sodium formate concentration was lowered to 50 mM since no other NADH-consuming enzymes besides PdR were present and 300 U/ml catalase was added to efficiently remove H2O2. Catalase addition was not necessary in the case of conversions using CFE due to E. coli-born catalase activity.
Kinetic measurements and coupling efficiency
NADH oxidation rates were measured spectrophotometrically at 30°C using purified P450, PdR, and Pdx by following NADH absorbance at 340 nm over time. The reaction mixture typically contained 1 μM PdR, 5 μM Pdx, 1 μM CYP154H1, and 5 mM substrate (1 M stock in DMSO) in 50 mM potassium phosphate buffer, pH 7.5. Reactions were started by adding 100 μM NADH (ε 340 = 6.22 × 103 M−1 cm−1). All measurements were done in triplicate.
After complete oxidation of cofactor NADH, each 200-μl reaction mix was used to measure the amount of H2O2 that was formed during the reaction. This was done according to a protocol by Heuts et al. (2007).
Turnover numbers for the conversion of ethylbenzene (1) and styrene (8) by CYP154H1 were determined by following product formation in time using GC. Reactions of 5 ml total volume consisted of 2 μM P450, 4 μM PdR, 20 μM Pdx, 5 mM substrate (1 M stock in ethanol), 150 mM sodium formate, 0.5 U FDH, 10 U catalase, and 100 μM NADH in 50 mM potassium phosphate buffer, pH 7.5. Reactions were incubated at 30°C with shaking, and samples of each 1 ml volume were taken over a period of 60 min. Samples were extracted and analyzed by GC as described under “Bioconversions”.
Determination of thermostability
The apparent melting temperature of CYP154H1 was determined by measuring ellipticity at 220 nm for increasing temperatures on a Jasco J-815 CD spectrometer (Jasco Inc, Easton, MD, USA). A 1-ml sample of 35 μg/ml purified P450 in 50 mM potassium phosphate buffer, pH 7.5, was heated from 35°C to 100°C with a rate of 0.5°C/min. Every 30 s, the ellipticity was measured at 220 nm. The obtained data were plotted against the temperature and the transition point of the resulting graph corresponds to the apparent melting temperature.
Additionally, thermal stability of purified CYP154H1 was determined by measuring the residual activity of the enzyme at 25°C after incubation at higher temperatures. Thus, a 1-μM solution of CYP154H1 in 50 mM potassium phosphate buffer, pH 7.5, was heated with 2°C/min to various temperatures between 40°C and 100°C and incubated for 10 min at the respective temperature before cooling down again to 25°C with 2°C/min. The residual activity of the enzyme was determined by measuring the decrease in NADH absorbance at 340 nm. The reaction mixture contained 0.1 μM PdR, 1 μM Pdx, 0.1 μM CYP154H1, and 2 mM ethylbenzene in 50 mM potassium phosphate buffer, pH 7.5. Reactions were started by adding 100 μM NADH.
GC, GC-MS, and HPLC analyses
Achiral GC analysis was performed on a Shimadzu GC14 gas chromatograph equipped with an AT-5 column (Grace, Deerfield, IL, USA). The temperature profile for the column was as follows: linear gradient starting at 40°C and heating with 10°C/min to 300°C. Injector and detector temperature were set to 300°C. Retention times were as follows: 1, 4.8 min; 2, 7.9 min; 3, 8.0 min; 4, 6.2 min; 5, 9.3 min; 6, 9.0 min; 7, 9.5 min; 8, 5.2 min; 9, 8.0 min; 10, 8.7 min; 11, 11.4 min; 16, 14.2 min; 17, 17.0 min; 18, 14.7 min; 20a, 8.3 min; 20b, 9.2 min; 20c, 9.9 min; 20d, 9.6 min; 21a, 11.9 min; 21b, 13.1 min; 21c, 13.5 min; 21d, 13.9 min; 22a, 12.8 min; 22b, 13.9 min; 22c, 14.3 min; 22d, 14.4 min; 23, 18.3 min; and 24, 20.3 min. GC-MS measurements were performed on an Agilent HP 6890 gas chromatograph with a HP5973 MS detector equipped with a HP1 column (Agilent, Santa Clara, CA, USA) using the same temperature profile.
Chiral GC analysis of sulfoxides 21a–d was performed on an Agilent HP 6890 gas chromatograph equipped with a Chiraldex G-TA column (Astech, Whippany, NJ, USA). The column was heated at 160°C; injector and detector temperature were set to 200°C. Retention times were as follows: (R)-21a, 5.8 min; (S)-21a, 7.3 min; (R)-21b, 6.9 min; (S)-21b, 8.3 min; (R)-21c, 7.7 min; (S)-21c, 8.1 min; (R)-21d, 13.5 min; and (S)-21d, 9.4 min.
Chiral HPLC measurements were performed with a Shimadzu LC instrument consisting of a CTO-10A column oven, a SIL-10AD autoinjector and a SPD-M10A diode array detector using different chiral columns (Chiral Technologies Europe, Illkirch-Cedex, France) and varying heptane/isopropanol mixtures as running solvent (Table 1). The flow rate was set to 1 ml/min. Absolute configurations were determined by comparison to enantiomerically pure standards.
Results
Cytochrome P450 monooxygenases from T. fusca
In the genome sequence of the moderately thermophilic soil bacterium T. fusca, a total of ten open reading frames coding for putative cytochrome P450 monooxygenases were identified (Nelson 2009). A high number of P450-encoding genes are also present in the genome sequences of other actinomycetes such as Streptomyces sp. or Nocardia sp. which produce P450s that are mainly involved in secondary metabolism (Lamb et al. 2003).
According to the P450 classification, four of the P450s from T. fusca belong to family CYP154 (namely CYP154E1, CYP154F1, CYP154G1, and CYP154H1) and one to family CYP157 (CYP157A3), whereas the other five (namely CYP215A1, CYP216A1, CYP217A1, CYP218A1, and CYP222A1) constitute new families (Nelson 2009). A BLAST search revealed that there are no homologs of CYP216A1, CYP218A1, and CYP222A1 with >40% sequence identity at the protein level present in the non-redundant protein (nr) and Swissprot databases and that would fall into the same CYP families. Thus, P450 monooxygenases CYP216A1, CYP218A1, and CYP222A1 are the only members of their respective family so far, and no close homologs of identified functionality are known. For CYP157A3 and CYP215A1, other members of the same families can be found in the nr and Swissprot databases, but none of these proteins has been studied until now. The CYP217A1 gene likely is part of a polyketide synthesis (PKS) gene cluster. The closest homologs of CYP217A1 that are present in Streptomyces albaduncus and Streptomyces griseoflavus are part of PKS clusters involved in the synthesis of chrysomycin and gilvocarcin V, respectively (Fischer et al. 2003; Kharel et al. 2010).
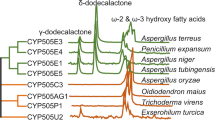
According to the phylogenetic tree in Fig. 1, none of the P450s from T. fusca is closely related to known thermostable P450s originating from various archaea or the bacterium T. thermophilus. Cytochrome P450s CYP154E1, CYP154F1, CYP154G1, and CYP154H1 belong to one family of P450 monooxygenases and thus also fall into one clade. However, CYP154H1 is more distantly related to the other three P450s as it lies in another branch of the same subtree. The closest homologs of CYP154H1 with known functionality are CYP154C1 from S. coelicolor and CYP154 from N. farcinica sharing 52% and 46% sequence identity at the protein level with CYP154H1. Since these enzymes were shown before to catalyze the conversion of several macrolides and the hydroxylation of testosterone, respectively (Agematu et al. 2006; Podust et al. 2003), we decided to characterize CYP154H1 in more detail.

Phylogenetic tree of the cytochrome P450 monooxygenases from T. fusca and their closest homologs together with known hyperthermostable P450s. The multiple sequence alignment was made using ClustalX version 2.0.12. The phylogenetic tree was constructed by the neighbor-joining method and was schematically presented using the ClustalX output and MEGA version 4 (Tamura et al. 2007). Prefixes in front of the CYP number abbreviate the genus and species of the respective bacterial organism where the enzyme originates from (Tf T. fusca, Nd Nocardiopsis dassonvillei, Nf Nocardia farcinica, Sal Streptomyces albaduncus, Sco Streptomyces coelicolor, Sgr Streptomyces griseoflavus, Shy Streptomyces hygroscopicus, Ssp Streptomyces sp., Sso Sulfolobus solfataricus, Sto Sulfolobus tokodaii, Pt Picrophilus torridus, Tt Thermus thermophilus). P450s from T. fusca are shown in red; hyperthermostable P450s are given in blue. The bar in the lower left corner represents 0.1 amino acid substitutions per amino acid for the branch length
Cloning and recombinant expression of CYP154H1
The gene of CYP154H1 was amplified from genomic DNA using PCR and cloned into the broad host-range vector pIT2-MCS, which contains an IPTG-inducible trc promoter and a tetracycline resistance gene. Putidaredoxin and putidaredoxin reductase from P. putida DSM50198 served as electron transfer components in bioconversions (Peterson et al. 1990). The corresponding genes were also amplified by PCR from genomic DNA of this strain and cloned into the vector pACYCDuet-1 that contains two T7 promoters and a chloramphenicol resistance gene. Thereby, each gene was put behind a separate promoter. For efficient coexpression of all three proteins in one host, E. coli C43(DE3) was used, which is an E. coli BL21(DE3) mutant specially suited for recombinant expression of difficult proteins (Miroux and Walker 1996). Thus, much higher levels of soluble PdR and less inclusion bodies could be obtained for expression in E. coli C43(DE3) at 30°C compared to E. coli BL21(DE3). Protein expression was induced by IPTG addition and δ-aminolevulinic acid was added for enhanced heme synthesis. Using this expression system, CYP154H1 was produced at up to 4% of the total soluble protein, and a specific activity of the electron transfer components of ≥1 U/mg total protein was obtained under optimized conditions as determined by the cytochrome c reduction assay.
Purification of CYP154H1
For easy purification of CYP154H1, the corresponding gene was subcloned into vector pET28a(+) to generate an N-terminal his6-tagged protein. Recombinant expression was also performed in E. coli C43(DE3) at 28°C yielding up to 14% P450 of total soluble protein with a molecular weight of 48 kDa. The enzyme was purified to homogeneity in three steps (Table S1, Figure S1, Online Resource): a heating step (30 min at 50°C), anion exchange chromatography using a Q Sepharose column, and immobilized metal affinity chromatography taking advantage of the N-terminal his6-tag. Thus, 140 mg of pure CYP154H1 could be obtained from a 500-ml culture as determined by CO-difference spectra.
Substrate spectrum of CYP154H1
To explore the range of substrates that can be converted by CYP154H1, different aliphatic and aromatic compounds of varying size were tested in bioconversions with CFE containing PdR, Pdx, and P450. It was found that the enzyme was active with small arylaliphatic substrates like ethylbenzene (1) and styrene (8; Scheme 1; Table 2) which were exclusively hydroxylated or epoxidized at the aliphatic side chain. In contrast, toluene, anisole, and dimethoxybenzene were hydroxylated at the aromatic ring, but conversions were only below 5%. Phenol and naphthalene were not substrates of CYP154H1. Of the tested aliphatic compounds, cyclohexane, cyclohexene, cyclohexanone, testosterone, and lauric acid methyl ester were not converted whereas 1-octene was transformed into the corresponding epoxide and 3-hydroxy-1-octene in a 2:1 ratio.

Conversion of ethylbenzene (1), propylbenzene (4), and styrene (8) by CYP154H1 from T. fusca. CamA (PdR) and CamB (Pdx) served as electron transfer components
In the case of the arylaliphatic hydrocarbons ethylbenzene (1), propylbenzene (4), and styrene (8), chiral products (R)-1-phenylethanol ((R)-2), (R)-1-phenyl-1-propanol ((R)-5) and (S)-styrene oxide ((S)-9), respectively, were formed by CYP154H1 with low to moderate enantiomeric excess. The enantioselectivity seems to increase with increasing length of the aliphatic side chain giving 5 with 66% ee. Interestingly, CYP101A1 (P450cam) gives also preferably (S)-styrene oxide as product in the conversion of 8, while CYP102A1 (P450BM3) and CYP108A1 (P450terp) preferentially produce the (R)-epoxide (Fruetel et al. 1994). In all three cases, the enantiomeric excess of the formed products are also only low to moderate. In the conversion of propylbenzene (4) by CYP154H1, small amounts of 1-phenyl-2-propanol (6) were also detected, but the enantiomeric excess was below 2%. To rule out that some of the identified products are not formed by CYP154H1 but by other enzymes present in the CFE, all bioconversions were also tested using CFE of E. coli C43(DE3)(pACYCcamAB) not expressing CYP154H1 (negative control). For none of the substrates, product formation was detected in these negative controls. Furthermore, all bioconversions were also carried out using purified PdR, Pdx, and P450 in order to investigate if some of the initially formed products were further converted by E. coli enzymes (e.g., oxidation of formed alcohols to the respective ketones). However, in all cases, products were the same as in reactions with CFE indicating that the formation of these products was due to P450 activity.
CYP154H1 also converted indole (11), forming a mixture of indigo (14) and indirubin (15) already during coexpression of PdR, Pdx, and the P450 in E. coli. This became evident by a blue to violet coloration of the cell pellet and the presence of a blue and a pink band on TLC after extraction. Thus, indole (11), N-acetylindole (16), and 3-acetylindole (19) were also tested in bioconversions using CFE. It appeared that only 11 and 16 were transformed with 44% and 70% conversion, respectively, whereas 19 was not converted. This could be explained by a preferential hydroxylation of the indole ring at the 3-position leading to autocatalytic dimerization with formation of products 14 and 15 in the case of indole (Scheme 2). Accordingly, for substrate 16, the main product was found by GC-MS to be 3-oxo-N-acetylindole (17) and in much smaller amounts also the corresponding 2-oxo product 18 was formed (ratio 4:1, respectively).

Conversion of indole derivatives 11, 16, and 19 by CYP154H1 from T. fusca. CamA (PdR) and CamB (Pdx) served as electron transfer components
To investigate, if CYP154H1 is also able to chemoselectively oxidize organic sulfides, reactions with CFE containing PdR, Pdx as well as P450 and different arylaliphatic sulfides (20a–d) were carried out. The enzyme converted 20a–d to the corresponding sulfoxides 21a–d and further to the sulfones 22a–d (Scheme 3, Table 3), with the sulfoxides as the main products. Product ratios of 21:22 varied significantly depending on the used substrate. The ee values of the different chiral sulfoxides were low to moderate. Interestingly, the stereopreference of the enzyme changed from R to S with increasing size of the aromatic substituent. While 20a was converted to the (R)-sulfoxide with 56% ee, addition of a methyl group in para-position at the phenyl ring (20c) or replacement of the phenyl by a benzyl moiety (20d) led to the formation of (S)-sulfoxide with 20% and 46% ee, respectively. Such a change in stereopreference in the oxidation of thioanisole (20a) and p-methyl thioanisole (20c) was also found for CYP101A1 (P450cam) yielding (R)-21a and (S)-21c with 44% and 4% ee, respectively (Fruetel et al. 1994). In the conversion of 20b by CYP154H1, two more products were detected, which were identified by GC-MS to be diphenyldisulfide (23) and a corresponding sulfone (24; Figure S2, Online Resource). The latter are probably formed through S-dealkylation of the substrate instead of S-oxidation (Watanabe et al. 1981). Both products were also found in incubations with purified CYP154H1 but not in control reactions with CFE of E. coli C43(DE3)(pACYCcamAB), confirming that their formation depends on the presence of P450.

Oxidation of sulfides 20a–d by CYP154H1 from T. fusca. CamA (PdR) and CamB (Pdx) served as electron transfer components
Kinetics and coupling efficiency
Usually a significant rate of uncoupling between electron transfer to the heme and substrate hydroxylation is observed for conversions of non-natural substrates or when artificial electron transfer partners are applied in bioconversions with P450 monooxygenases. Thus, turnover numbers (TN) for different substrates should be determined based on product formation rates rather than measuring the rates of cofactor (NADH) oxidation. For CYP154H1, TN for ethylbenzene (1) and styrene (8) conversion at 30°C were determined using purified PdR, Pdx, and P450 by following product formation in time. Using 5 mM substrate, TN for 1 and 8 were calculated to be 0.28 and 0.31 nmol min−1 nmol−1 P450, respectively. In contrast, corresponding NADH oxidation rates for 1 and 8 with 5.2 and 4.6 nmol min−1 nmol−1 P450, respectively, were ≥15-fold higher, indicating rather low coupling efficiencies. This was also confirmed by the detection of high amounts of H2O2 that were produced during measurements of NADH oxidation (data not shown). In comparison, in controls using only purified PdR and Pdx without CYP154H1, NADH oxidation rates of 1.6 nmol min−1 nmol−1 PdR were obtained under the same assay conditions. Thus, PdR already oxidizes NADH without the presence of a P450 monooxygenase, but the rate is significantly lower compared to reactions with CYP154H1.
Thermostability
In order to determine the thermal stability of the enzyme, residual activity of purified CYP154H1 towards 1 was measured at 25°C after incubation for 10 min at elevated temperatures (Figure S3, Online Resource). It was found that the enzyme hardly lost activity when incubated at 40°C, 50°C, and 60°C. For higher temperatures, the decrease in activity was more dramatic, which is in agreement with the optimum growth temperature of T. fusca of 50–55°C (Lykidis et al. 2007). Nevertheless, even after incubation at 80°C, still 10% of initial activity could be measured. Furthermore, the apparent melting temperature of CYP154H1 was determined by CD spectrometry to be 67°C.
Discussion
The moderately thermophilic soil bacterium T. fusca is a rich source of valuable biocatalysts with high stability that are still active at ambient temperature (Wilson 2004). The genome sequence of this bacterium contains also ten open reading frames coding for putative cytochrome P450 monooxygenases. Although it can be anticipated that the P450s from T. fusca exhibit increased stability compared to P450s from mesophilic hosts—a characteristic that is highly desired for biocatalytic applications—none of them had been studied in detail so far. In fact, the usually low stability of P450 monooxygenases is one major limitation for their use on industrial scale. Thus, there is a clear need for the discovery of thermostable P450s with desired activities and substrate specificities. Nevertheless, up to now, only few naturally occurring, thermostable P450 monooxygenases have been reported in the literature and the substrate specificities of only two of them are known (Blasco et al. 2004; Koo et al. 2000, 2002; McLean et al. 1998).
In the present paper, we described the recombinant expression and characterization of CYP154H1, the first P450 monooxygenase from T. fusca that has been cloned and studied regarding substrate specificity and stability. The enzyme can be highly overexpressed in E. coli in active form and purified with the help of an N-terminal his6-tag (Table S1). Since the natural electron transfer partners of this three-component P450 monooxygenase are unknown, putidaredoxin reductase and putidaredoxin from P. putida were employed to supply CYP154H1 with electrons. Using this surrogate three-component system, the substrate specificity of CYP154H1 was investigated revealing a preference for small (aromatic) compounds. Thus, it was shown that the enzyme is able to enantioselectively convert different arylaliphatic substrates (1, 4, and 8) to the corresponding chiral products (2, 5, and 9, respectively, Table 2), which represent key chiral building blocks in organic synthesis. Of the different aliphatic compounds that were tested, only 1-octene was transformed. Furthermore, despite the 46% sequence identity with CYP154 from N. farcinica IFM 10152—an enzyme that hydroxylates testosterone regio- and stereoselectively—CYP154H1 is not able to convert steroids or even larger aromatic compounds like naphthalene derivatives. This might be due to a smaller active site compared to that of CYP154 from N. farcinica or a restricted access for these compounds to the active site. According to the homology models of CYP154H1 and CYP154 from N. farcinica—both are based on the crystal structure of CYP154C1 which shares 52% sequence identity at the protein level with both P450s (Podust et al. 2003)—the active site shapes are rather similar. Furthermore, the expected substrate entrance channel of CYP154 from N. farcinica is even narrower than the one from CYP154H1 (data not shown). Thus, reliable conclusions about why larger compounds are not converted by this P450 cannot be drawn based on the homology models. For that, it will be necessary to solve the crystal structure of CYP154H1 with a bound substrate in the active site, since it is known for P450s that they undergo structural changes upon substrate binding in order to adopt a structure that better fits the substrate molecule into the active site (Li and Poulos 1997; Raag et al. 1993).
Besides compounds 1, 4, and 8, different organic sulfides (20a–d) are oxidized by CYP154H1 to the corresponding sulfoxides (21a–d) in an enantioselective fashion (Table 3). Such sulfoxides are also important due to their use as chiral auxillaries in organic synthesis and their use as building blocks in the pharmaceutical industry. Another important feature of CYP154H1 is the selective synthesis of 3-oxo-N-acetylindole (17) from N-acetylindole (16) since the product is used as an important intermediate in the synthesis of different phosphodiesterase inhibitors (Weinbrenner et al. 2008), calcium channel blockers (Shcherbakova and Nikoyukin 2008), and HIV inhibitors (Kesteleyn et al. 2005).
Since CYP154H1 comes from a thermophilic host, the enzyme was expected to display also a higher stability compared to its mesophilic counterparts. Indeed, an apparent melting temperature (T m) of 67°C was determined by circular dichroism. This apparent T m is significantly higher than the one found for CYP101A1 (P450cam, T m = 54°C, measured by differential scanning calorimetry (DSC)) but also much lower than the apparent T m reported for CYP119 (T m = 91°C, measured by DSC), a hyperthermostable cytochrome P450 monooxygenase from S. solfataricus (McLean et al. 1998). This was also in agreement with activity measurements showing that the enzyme still displayed 54% of initial activity after heating for 10 min at 65°C (Figure S3, Online Resource). Thus, the enzyme exhibits improved stability compared to cytochrome P450s from mesophilic hosts. Due to the low stability of the applied electron transfer components, PdR and Pdx, at elevated temperatures (Sevrioukova et al. 2003), it was not possible to determine the temperature optimum of CYP154H1. Furthermore, the overall activity of the surrogate three-component system seems to be rather low. There is a big discrepancy between the rates of product formation and NADH oxidation rates that were measured for 1 and 8. This could be due to an insufficient transfer of electrons from Pdx to the P450 as CYP154H1 and CYP101A1 (the natural monooxygenase partner of PdR and Pdx) share only 26% sequence identity on protein level. For example, slow transfer of the second electron (after binding of oxygen to the heme iron) during catalysis can lead to uncoupling and formation of superoxide (Karuzina and Archakov 1994). Thus, identification of the natural electron transfer partners of CYP154H1 is of major importance in order to optimize the performance of the enzyme system. On the other hand, the used substrates themselves might cause uncoupling in the P450 catalytic cycle due to non-productive binding in the active site. As an example, coupling efficiencies in the conversion of styrene and thioanisole by the P450cam three-component system were ≤10% in contrast to the tight coupling of NADH consumption and substrate oxidation in the case of camphor, the natural substrate of P450cam (Fruetel et al. 1992, 1994). Thus, we cannot exclude that much better substrates with higher coupling efficiencies exist for CYP154H1 since the natural substrate of this enzyme is not known.
In summary, CYP154H1 from T. fusca constitutes a new moderately thermostable P450 monooxygenase that can be easily expressed in E. coli in high yield. The enzyme represents a promising new biocatalyst for oxidative reactions since it is able to produce a range of industrially relevant compounds. To further improve the stereoselectivity of the enzyme, protein engineering and directed evolution methods can be employed (Bell et al. 2001; Jaeger and Reetz 2000; Seifert et al. 2009). Currently we are working on the crystallization of CYP154H1 to elucidate its three-dimensional structure. Furthermore, we are trying to identify the natural electron transfer partners from T. fusca to improve catalytic efficiency of the three-component system in bioconversions.
References
Agematu H, Matsumoto N, Fujii Y, Kabumoto H, Doi S, Machida K, Ishikawa J, Arisawa A (2006) Hydroxylation of testosterone by bacterial cytochromes P450 using the Escherichia coli expression system. Biosci Biotechnol Biochem 70:307–311
Bachmann SL, McCarthy AJ (1991) Purification and cooperative activity of enzymes constituting the xylan-degrading system of Thermomonospora fusca. Appl Environ Microbiol 57:2121–2130
Bell SG, Sowden RJ, Wong L-L (2001) Engineering the haem monooxygenase cytochrome P450cam for monoterpene oxidation. Chem Comm 7:635–636
Bernhardt R (2006) Cytochromes P450 as versatile biocatalysts. J Biotechnol 124:128–145
Blasco F, Kauffmann I, Schmid RD (2004) CYP175A1 from Thermus thermophilus HB27, the first β-carotene hydroxylase of the P450 superfamily. Appl Microbiol Biotechnol 64:671–674
Fischer C, Lipata F, Rohr J (2003) The complete gene cluster or the antitumor agent gilvocarcin V and its implication for the biosynthesis of the gilvocarcins. J Am Chem Soc 125:7818–7819
Fruetel J, Collins JR, Camper DL, Loew GH, Ortiz de Montellano PR (1992) Calculated and experimental absolute stereochemistries of the styrene and β-methyl styrene epoxides formed by cytochrome P450 cam . J Am Chem Soc 114:6987–6993
Fruetel J, Chang Y-T, Collins J, Loew G, Ortiz de Montellano PR (1994) Thioanisole sulfoxidation by cytochrome P450 cam (CYP101): experimental and calculated absolute stereochemistries. J Am Chem Soc 116:11643–11648
Furuya T, Kino K (2009) Discovery of 2-naphthoic acid monooxygenases by genome mining and their use as biocatalysts. ChemSusChem 2:645–649
Hannemann F, Bichet A, Ewen KM, Bernhardt R (2007) Cytochrome P450 systems—biological variations of electron transport chains. Biochim Biophys Acta 1770:330–344
Hedegaard J, Gunsalus IC (1965) Mixed function oxidation. J Biol Chem 240:4038–4043
Heuts DP, van Hellemond EW, Janssen DB, Fraaije MW (2007) Discovery, characterization and kinetic analysis of an alditol oxidase from Streptomyces coelicolor. J Biol Chem 282:20283–20291
Isin EM, Guengerich FP (2007) Complex reactions catalyzed by cytochrome P450 enzymes. Biochim Biophys Acta 1770:314–329
Jaeger K-E, Reetz MT (2000) Directed evolution of enantioselective enzymes for organic chemistry. Curr Op Chem Biol 4:68–73
Karuzina II, Archakov AI (1994) The oxidative inactivation of cytochrome P450 in monooxygenase reactions. Free Radical Bio Med 16:73–97
Kesteleyn BRR, van de Vreken W, Kindermans NMF, Canard MFJMG, Hertogs K, Bettens E, de Vroey VCP, Jochmans DED, Wigerinck PTBP, Wang J, Tahri A, Surleraux DLNG (2005) Combinations of substituted 1-phenyl-1,5-dehydro-pyrido-[3,2-B] indol-2-ones and other HIV inhibitors. WO 2005110411 A1
Kharel MK, Nybo SE, Shepherd MD, Rohr J (2010) Cloning and characterization of the ravidomycin and chrysomycin biosynthetic gene clusters. Chembiochem 11:523–532
Koo LS, Tschirret-Guth RA, Straub WE, Moënne-Loccoz P, Loehr TM, Ortiz de Montellano PR (2000) The active site of the thermophilic CYP119 from Sulfolobus solfataricus. J Biol Chem 275:14112–14123
Koo LS, Immoos CE, Cohen MS, Farmer PJ, Ortiz de Montellano PR (2002) Enhanced electron transfer and lauric acid hydroxylation by site-directed mutagenesis of CYP119. J Am Chem Soc 124:5684–5691
Kovach ME, Phillips RW, Elzer PH, Roop RM, Peterson KM (1994) pBBR1MCS: a broad-host-range cloning vector. Biotechniques 16:800–802
Lacour T, Achstetter T, Dumas B (1998) Characterization of recombinant adrenodoxin reductase homologue (Arh1p) from yeast. J Biol Chem 273:23984–23992
Lamb DC, Ikeda H, Nelson DR, Ishikawa J, Skaug T, Jackson C, Omura S, Waterman MR, Kelly SL (2003) Cytochrome P450 complement (CYPome) of the avermectin-producer Streptomyces avermitilis and comparison to that of Streptomyces coelicolor A3(2). Biochem Biophys Res Comm 307:610–619
Li H, Poulos TL (1997) The structure of the cytochrome P450BM-3 haem domain complexed with the fatty acid substrate, palmitoleic acid. Nat Struct Biol 4:140–146
Lykidis A, Mavromatis K, Ivanova N, Anderson I, Land M, DiBartolo G, Martinez M, Lapidus A, Lucas S, Copeland A, Richardson P, Wilson DB, Kyrpides N (2007) Genome sequence and analysis of the soil cellulolytic actinomycete Thermobifida fusca YX. J Bacteriol 189:2477–2486
Mandai T, Fujiwara S, Imaoka S (2009) A novel electron transport system for thermostable CYP175A1 from Thermus thermophilus HB27. FEBS J 276:2416–2429
McLean MA, Maves SA, Weiss KE, Krepich S, Sligar SG (1998) Characterization of a cytochrome P450 from the acidothermophilic archaea Sulfolobus solfataricus. Biochem Biophys Res Comm 252:166–172
Miroux B, Walker JE (1996) Over-production of proteins in Escherichia coli: mutant hosts that allow synthesis of some membrane proteins and globular proteins at high levels. J Mol Biol 260:289–298
Nelson DR (2009) The cytochrome P450 homepage. Hum Genomics 4:59–65
Omura T, Sato R (1964) The carbon monoxide-binding pigment of liver microsomes. Evidence for its hemoprotein nature. J Biol Chem 239:2370–2378
Ortiz de Montanello P (2005) Cytochrome P450: structure, mechanism and biochemistry. Springer, The Netherlands
Peterson JA, Lorence MC, Amarneh B (1990) Putidaredoxin reductase and putidaredoxin. Cloning, sequence determination, and heterologous expression of the proteins. J Biol Chem 265:6066–6073
Podust LA, Kim Y, Arase M, Neely BA, Beck BJ, Bach H, Sherman DH, Lamb DC, Kelly SL, Waterman MR (2003) The 1.92-Å structure of Streptomyces coelicolor A3(2) CYP154C1. J Biol Chem 278:12214–12221
Podust LA, Bach H, Kim Y, Lamb DC, Arase M, Sherman DH, Kelly SL, Waterman MR (2004) Comparison of the 1.85 Å structure of CYP154A1 from Streptomyces coelicolor A3(2) with the closely related CYP154C1 and CYPs from antibiotic biosynthetic pathways. Protein Sci 13:255–268
Puchkaev AV, Ortiz de Montellano PR (2005) The Sulfolobus solfataricus electron donor partners of thermophilic CYP119: an unusual non-NAD(P)H-dependent cytochrome P450 system. Arch Biochem Biophys 434:169–177
Raag R, Li H, Jones BC, Poulos TL (1993) Inhibitor-induced conformational change in cytochrome P-450 cam . Biochemistry 32:4571–4578
Roome PW, Philley JC, Peterson JA (1983) Purification and properties of putidaredoxin reductase. J Biol Chem 258:2593–2598
Shcherbakova I, Nikoyukin Y (2008) Indoloquinoline compounds as calcium channel blockers. WO 2008027182 A2
Seifert A, Vomund S, Grohmann K, Kriening S, Urlacher VB, Laschat S, Pleiss J (2009) Rational design of a minimal and highly enriched CYP102A1 mutant library with improved regio-, stereo- and chemoselectivity. Chembiochem 10:853–861
Sevrioukova IF, Hazzard JT, Tollin G, Poulos TL (2001) Laser flash induced electron transfer in P450cam monooxygenase: putidaredoxin reductase–putidaredoxin interaction. Biochemistry 40:10592–10600
Sevrioukova IF, Garcia C, Li H, Bhaskar B, Poulos TL (2003) Crystal structure of putidaredoxin, the [2Fe–2S] component of the P450cam monooxygenase system from Pseudomonas putida. J Mol Biol 333:377–392
Tamura K, Dudley J, Nei M, Kumar S (2007) MEGA4: molecular evolutionary genetics analysis (MEGA) software version 4.0. Mol Biol Evol 24:1596–1599
Watanabe Y, Numata T, Iyanagi T, Oae S (1981) Enzymatic oxidation of alkyl sulfides by cytochrome P450 and hydroxyl radical. Bull Chem Soc Jpn 54:1163–1170
Weinbrenner S, Dunkern T, Marx D, Schmidt B, Stengel T, Flockerzi D, Kautz U, Hauser D, Diefenbach J, Christiaans J, Menge W (2008) 6-Benzyl-2,3,4,7-tetrahydro-indolo-[2,3-C] quinoline compounds useful as PDE5 inhibitors. WO 2008095835 A1
Wilson DB (2004) Studies of Thermobifida fusca plant cell wall degrading enzymes. Chem Rec 4:72–82
Yano JK, Koo LS, Schuller DJ, Li H, Ortiz de Montellano PR, Poulos TL (2000) Crystal structure of a thermophilic cytochrome P450 from the archaeon Sulfolobus solfataricus. J Biol Chem 275:31086–31092
Yano JK, Blasco F, Li H, Schmid RD, Henne A, Poulos TL (2003) Preliminary characterization and crystal structure of a thermostable cytochrome P450 from Thermus thermophilus. J Biol Chem 278:608–616
Acknowledgment
This project was financially supported by the Netherlands Ministry of Economic Affairs and the B-Basic partner organizations through B-Basic, a public-private NWO-ACTS program.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 118 kb)
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Schallmey, A., den Besten, G., Teune, I.G.P. et al. Characterization of cytochrome P450 monooxygenase CYP154H1 from the thermophilic soil bacterium Thermobifida fusca . Appl Microbiol Biotechnol 89, 1475–1485 (2011). https://doi.org/10.1007/s00253-010-2965-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-010-2965-9