
Abstract
Streptomyces toxytricini produces lipstatin, a specific inhibitor of pancreatic lipase, which is derived from two fatty acid moieties with eight and 14 carbon atoms. The pccB gene locus in 10.6 kb fragment of S. toxytricini chromosomal DNA contains three genes for acyl-coenzyme A carboxylase (ACCase) complex accA3, pccB, and pccE that are presumed to be involved in secondary metabolism. The pccB gene encoding a β subunit of ACCase [carboxyltransferase (CT)] was identified upstream of pccE gene for a small protein of ε subunit. The accA3 encoding the α subunit of ACCase [biotin carboxylase (BC)] was also identified downstream of pccB gene. When the pccB and pccE genes were inactivated by homologous recombination, the lipstatin production was reduced as much as 80%. In contrast, the accumulation of another compound, tetradeca-5.8-dienoic acid (the major lipstatin precursor), was 4.5-fold increased in disruptant compared with wild-type. It implies that PccB of S. toxytricini is involved in the activation of octanoic acid to hexylmalonic acid for lipstatin biosynthesis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Acyl-coenzyme A(CoA) carboxylase (ACCase) complex is an important metabolic system, which catalyzes the first committed step in the biosynthesis of fatty acids and polyketides in animals, plants, and bacteria (Cronan and Waldrop 2002). This ubiquitous enzyme is responsible for the activation of various organic acids by α-carboxylation of acyl-CoA that can serve as the building blocks for fatty acid biosynthesis.
In Streptomyces and Mycobacterium species, the ACCase consists of α, β, and ε subunits (Diacovich et al. 2002; Gago et al. 2006). In α subunit [biotin carboxylase (BC)], there are biotin binding domain and CO2 fixation domain. A molecule of CO2 is transferred to the biotin moiety at the C terminus of α subunit to form carboxyl–BC. Subsequently, the β subunit [carboxyltransferase (CT)] transfers the carboxyl group from carboxyl–BC to the acyl-CoA (Cronan and Waldrop 2002). Because the acyl-CoA participates only in the second step, the CT determines the substrate specificity of ACCase complex recognizing different acyl-CoAs.
Two types of ACCases, acetyl-CoA carboxylase (ACC) and propionyl-CoA carboxylase (PCC) have been characterized in Streptomyces coelicolor (Diacovich et al. 2002; Rodriguez and Gramajo; 1999; Rodriguez et al. 2001). It has been revealed that those ACCases have different roles in metabolism; ACC plays role in primary metabolism for fatty acid synthesis and PCC in secondary metabolism for polyketide synthesis. A recent study showed that the β subunit encoded by pccB gene and ε subunit by pccE gene in S. coelicolor form a protein complex, which exhibited full activity with longer chain acyl-CoA rather than acetyl-CoA (Diacovich et al. 2002).
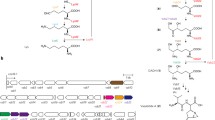
Lipstatin isolated from Streptomyces toxytricini is a potent irreversible inhibitor of pancreatic lipase (Weibel et al. 1987; Hochuli et al. 1987). The data provided from the feeding experiments and tracer studies indicated that the carbon skeleton of lipstatin molecule is biosynthesized via Claisen condensation of two fatty acid precursors, 8-carbon atoms (octanoic acid), and 14-carbon atoms (tetradeca-5,8-dienoic acid; Fig. 1; Eisenreich et al. 1997; Goese et al. 2000, 2001;Schuhr et al. 2002; Eisenreich et al. 2003).

Hypothetical biosynthetic pathway of lipstatin. The lipstatin backbone is formed via Claisen condensation of two fatty acid precursors, octanoic acid, and tetradeca-5,8-dienoic acid
The reaction producing an ultimate 3-oxo or hydroxy intermediate before the β–lactone formation in lipstatin (Goese et al. 2001) resembles the biosynthesis of a mycolic acid (Portevin et al. 2005). A gene knock-out study in Corynebacterium diphtheriae demonstrated that the pks13 gene flanked by two other genes, fadD32 for acyl-AMP ligase and accD4 for ACCase, is responsible for the condensation step to form mycolic acid (Portevin et al. 2004). This Pks13 condensase is a non-iterative type I polyketide synthase that contains four catalytic domains required for the final assembly of mycolic acid from two fatty acid residues. Later the accD4 gene was also confirmed to activate one of the mycolic precursor (Portevin et al. 2005).
Based on the structural similarity between the mycolic acids and 3-hydroxo intermediate of lipstatin, the pks13 gene was screened from S. toxytricini chromosomal DNA but any positive result was not obtained. Instead, pccB gene flanked by accA3, pccE, and bpl genes was identified based on an accD4 gene sequence. The disruption experiment showed that this gene locus is involved in the activation of octanoic acid before generation of lipstatin skeleton by Claisen condensation. Here, we report the biochemical role of pccB gene locus encoding PCC complex in lipstatin biosynthesis.
Materials and methods
Bacterial strains, culture media, and cultivation
The Escherichia coli strains used in this study were listed in Table 1. The E. coli strains were generally grown overnight at 37 °C in Luria-Bertani (LB) medium supplemented with appropriate antibiotics (Sambrook and Russell 2001), but E .coli XL1-Blue MRF′ was cultured in LB broth supplemented with 10 mM MgSO4 and E. coli BW25113/pIJ790 and ET12567/pUZ8002 in SOB–MgSO4 media (Datsenko and Wanner 2000).
Wild-type of S. toxytricini NRRL 15443 was grown on modified mannitol soya flour (MS) or ISP2 agar plates or in tryptic soy broth (TSB) liquid medium at 29 °C. For cultivation of the exoconjugant containing disruption cassette, a normal or modified MS agar plates were supplied with either apramycin/kanamycin or apramycin alone (Datsenko and Wanner 2000). A spore suspension of S. toxytricini strains was inoculated in seed culture medium (per l; soya bean flour 10 g, glycerol 5 ml, Bacto soytone 5 g, soya oil, 10 ml, Triton X-100 2 ml, pH 6.5) and agitated at 29 °C for 48 h under aerobic conditions. This seed culture (3%) was transferred into fermentation medium (per l; soya bean flour 30 g, glycerol 14 ml, Bacto soytone 1 g, Tryton X-100 1 ml, polypropylene glycol 0.2 ml, soya oil 60 ml, pH 7.0) for lipstatin production. Fermentation was carried out at 29 °C for 6.5 days under aerobic condition.
DNA manipulation
Isolation of chromosomal and plasmid DNA, agarose gel electrophoresis, restriction digestion and ligation, preparation of competent cell, and plasmid transformation were performed by conventional methods (Kieser et al. 2000; Sambrook and Russell 2001).
PCR amplification
The gene amplification was done using EF-Taq DNA polymerase or Pfu DNA polymerase (Solgent, Korea) according to the supplier's protocol. For the preparation of pccB probe, the PCR was performed at 58 °C annealing temperature with the combinations of two sets of forward and reverse primers accB–F1 (5′-CCGRTYRT CGGCATYAACGACTC-3′), accB–F2 (5′-AVGAYTCYGGYGGYGCHCGYATCCA-3′), accB–R1 (5′-TGCTTGGABCCCATSACSKCGTA-3′), and accB–R2 (5′-GABCCCATSACSKCRTAVG CDCCGCC-3′).
Construction of S. toxytricini genomic library
The chromosomal DNA was isolated from mycelia of S. toxytricini using a cetyltrimethylammonium bromide procedure (Kieser et al. 2000) and partially digested with Sau3AI. The large-size DNA fragments (30–45 kb) were eluted and ligated with the cosmid vector pOJ446 (Bierman et al. 1992) which was digested with HpaI, dephosphorylated, and further cleaved by BamHI. The ligated DNA products were packaged in vitro by Gigapack III gold packaging extract (Stratagene, USA), and transfected into E. coli XL1-Blue MRF′ cells, to construct the cosmid library of S. toxytricini chromosomal DNA.
Southern hybridization
The probe was labeled with α-32P-dCTP (specific activity; 50 µCi, Amersham, UK) using DecaLabel DNA labeling kit (Fermentas, Lithuania) according to manufacturer's specification and purified through Elutip-D column (Schleicher and Shuell, Germany). The capillary Southern hybridization or in situ colony hybridization was performed by conventional procedure (Sambrook and Russell 2001).
DNA sequencing and analysis
The selected recombinant plasmids were sent to Solgent (Daejeon, Korea) for sequencing. The location of open reading frame (ORF)s in the sequences was determined using FramePlot version 2.3.2. The ORFs were translated into amino acid sequences using translate tool program (http://br.expasy.org/tools/dna.html). The homology search with the obtained DNA or protein sequences was performed with BLAST program (http://blast.ncbi.nlm.nih.gov). The multiple alignments of DNA or protein sequences were performed with EBI–ClustalW2 program (http://www.ebi.ac.uk/Tools/clustalw2/index.html).
Gene disruption by PCR targeting system
A DNA fragment containing the two genes pccB and pccE was amplified from cosmid pSTL24 by Pfu DNA polymerase using primers: BE-up F1 (5′-GCGGATCCGA GGTCGGTGTTGGTGGAGCCCGT–3′) having BamHI site and BE-dn R2 (5′-GCAAGCTTGTCGCCGTGTCGACCACGCAGTG–3′) having HindIII site. The resulting 2.78 kb PCR product was digested with BamHI and HindIII and ligated at the same restriction sites of pHZ1351 plasmid (Sun et al. 2002) to give pHZ-BE24 plasmid (10.8 kb). The confirmed plasmid was transformed again by electroporation into E. coli BW25113/pIJ790 for λ-Red recombination and PCR-targeted gene disruption (Gust et al. 2003). The apramycin resistance cassette having aac(3)IV in plasmid pIJ773 was amplified with two long primers, PCCB Red F1 (5′-GAGTTCGCCCGGCACCGGTCGACCAACTTCGGCTGGAGATTCCGGGGATCCGTCGACC–3′) and PCCB Red R1 (5′–GGGTACTCAGGCGCCGCCTCCGCTTCGCGCCGTCTCTCATGTAGGCTGGAGCTGCTTC–3′) having 19 or 20 nucleotides of target genes at 3′-end flanked FLP recognition targets (FRT). The extended apramycin disruption cassette was introduced by electroporation into E. coli BW25113/(pIJ790 and pHZ-BE24) grown in SOB–MgSO4 containing chloramphenicol and ampicillin with supplementation of 10 mM L-arabinose for the gene replacement by inducing of λ-Red recombinase genes. Immediately after electroporation, 1 ml of ice-cold LB was added to the shocked cells and incubated with shaking for 1.5 h at 30 °C. The revived cells were spread on LB agar plates containing ampicillin and apramycin, followed by incubation at 37 °C to promote the loss of temperature-sensitive plasmid pIJ790 having repA101ts. The newly obtained pHZ-BE-Ap plasmid contained the apramycin disruption cassette instead of the target genes. This apramycin disruption vector was then transformed by electroporation into E. coli ET12567/pUZ8002 to construct a donor strain for its conjugal transfer to S. toxytricini wild strain. The S. toxytricini spores preserved in sterile 20% glycerol and 0.01% Triton X-100 were resuspended in 500 μl of TES buffer (0.05 M N-tris(hydroxymethyl)methyl-2-aminoethanesulfonic acid, pH 8.0), heat-shocked at 50 °C for 10 min for germination, and then mixed with equal volume of E. coli ET12567/(pUZ8002 and pHZ-BE-Ap) cells (Flett et al. 1997). After plating on MS agar and incubation at 30 °C for 16–40 h, 1 ml solution containing 0.5 mg of nalidixic acid and 1.25 mg apramycin was overlaid on the plate and the incubation continued at 29 °C for 3–5 days until potential exoconjugants sensitive to kanamycin but resistant to apramycin grew up.
Metabolite separation and analysis
The lipstatin metabolites were isolated from fermentation broth using standard purification method (Erdei et al. 2005). First 100 ml fermentation broth was extracted with 500 ml of acetone−hexane (1.5:1), dried over sodium sulfate, and concentrated to oil form. The crude oil was further separated by Silica 60 gel (Merck, Germany) column chromatography by eluting with hexane and then with hexane−ethyl acetate mixture in the ordered ratio of 20:1, 10:1, 5:1, and 2.5:1. The fractions were monitored by thin liquid chromatography (TLC) on Silica 60 F254 aluminum sheets (Merck, Germany), which were developed in hexane–ethyl acetate (2.5:1) and visualized with 10% sulfuric acid. The fractions showing spots of the same Rf values were combined and concentrated to dryness. Further metabolites were separated by high performance liquid chromatography (HPLC) (Souri et al. 2007) using SCL-10AVP system (Shimadzu Co., Japan) with YMC-Pack ODS-A reverse-phase column (250 mm × 10 mm internal diameter (l.D.), 12 nm; YMC Co., Ltd., Japan). The isocratic mobile phase of 0.1% orthophosphoric acid–acetonitrile (15:85 v/v) was pumped at a flow rate of 1 ml/min, and the metabolites were detected at 205 nm. The separated pure metabolites by preparative HPLC were subjected to 1H-nuclear magnetic resonance (NMR) for the determination of chemical structures. The samples were dissolved in chloroform-d 1, and the NMR spectra were recorded on Bruker AMX spectrometer (Bruker Biospin, Germany) using Bruker's standard pulse program for 1H NMR (250 MHz, FT).
Determination of lipase inhibitory activity
The inhibitory activity of S. toxytricini metabolites on human pancreatic lipase was assayed with 4-methylumbelliferyl oleate as substrate following the procedure described previously (Imanaka et al. 1983). The fluorescence intensity of 4-methylumbelliferone liberated from the substrate was measured with a fluorescence microplate reader (FLUOstar OPTIMA, BGM Labtechnologies, Germany) using excitation at 340 nm and emission at 460 nm.
Results
Cloning of pccB gene locus from S. toxytricini chromosomal DNA
The two conserved regions were found in the sequence alignment of AccD4 of Mycobacterium species and PccB of Streptomyces species and used to design primers capable of amplifying a DNA fragment with size of 880 bp. The amino acid sequence of the amplified gene fragment showed high level of similarity with PccB of Streptomyces avermetilis MA-4680 (91%) and S. coelicolor A3(2) (89%), as well as AccD4 of Mycobacterium tuberculosis H37Rv (52%), Mycobacterium bovis (52%), and Mycobacterium leprae (51%).
A cosmid library of S. toxytricini genomic DNA constructed in pOJ446 vector was transfected in E. coli XL1-Blue MRF′ and screened by in situ hybridization with 32P-labeled pccB gene fragment as a probe. Southern blot analysis of three positive cosmid clones after digestion with PstI or NcoI revealed the presence of 4.8 kb or 3.5 kb DNA bands containing pccB gene, respectively (Fig. S1). These two bands hybridized with pccB probe were cloned at PstI site of pGEM-3Zf(+) to construct pG3-P4.8 plasmid or NcoI site of pGEM-5Zf(+) to give a pG5-N3.5 plasmid.
The nucleotide sequences of these two DNA fragments were assembled manually in a single contig DNA sequence of 6.2 kb and further extended its upstream and downstream to 10.6 kb. The resulted nucleotide sequence was deposited in GenBank under the accession number of FJ595232.
Organization of S. toxytricini pccB gene locus
This DNA sequence named as the pccB gene locus comprises ten distinct ORFs including bpl, pccB, pccE, and accA3 genes for ACCase complex (Fig. 2a, Table 1). Four genes including bpl is located upstream of pccB gene and expressed in opposite direction. The other six genes including pccB, pccE, and accA3 are with the same orientation (Table 2).

Gene organization of pccB locus (10.6 kb) in the wild-type S. toxytricini (a) and in the disruptant S. toxytricini BE-AP strain having apramycin resistance gene [acc(3)IV] in place of pccB and pccE genes (b). The probes used in Southern hybridization are presented above the respective location, and the primers are denoted below the locations. The restriction sites are shown as abbreviations—N for NcoI; B for BamHI, and P for PstI
Multiple alignment of S. toxytricini PccB with other Streptomyces PccB and Mycobacterium AccD5 revealed the presence of acyl-CoA binding site and biotin binding site (Fig. 3a). The structural analysis of PccB elucidated the involvement of the GG182-183 residues in hydrogen bonding with the carbonyl group of acyl-CoA, whereas GA419-420 residues in hydrogen bonding with carbonyl group of biotin (Diacovich et al. 2004; Lin et al. 2006). The substrate specificity of PCC has known to be dependent upon the residue located outside of substrate pocket; D422 of Streptomyces PccB or C435 of Mycobacterium AccD5. The S. toxytricini PccB has the same amino acids at substrate binding pocket as other Streptomyces PccB proteins.

Amino acid sequence alignment of homologous regions in the active site of the putative PccB (a), AccA3 (b), PccE (c), and Bpl (d) proteins encoded by genes in pccB locus of S. toxytricini. StxPccB, PccB of S. toxytricini NRRL 15443; ScoPccB, PccB of S. coelicolor A3(2) (NP_629079), SavPccB; PccB of S. avermitilis MA-4680 (NP_824507); SprPccB, PccB of S. pristinaespiralis ATCC 25486 (ZP_03173623); SclPccB; PccB of S. clavuligerus ATCC 27064 (ZP_03182649), MtbAccD5, AccD5 of Mycobacterium tuberculosis H37Rv (NP_217797); MboAccD5, AccD5 of M. bovis AF 2122/97 (NP_856953); MleAccD5, AccD5 of M. leprae TN (NP_301571); CdpAccD5, AccD5 of C. diphtheriae NCTC 13129 (NP_939032), StxAccA3, AccA3 of S. toxytricini NRRL 15443; ScoAccA3, S. coelicolor A3(2) (NP_629074); SavAccA3, AccA3 of S. avermitilis MA-4680 (NP_824513); SprJadJ, JadJ of S. pristinaespiralis ATCC 25486 (ZP_05009963); SclJadJ; JadJ of S. clavuligerus ATCC 27064 (ZP_05005117); MtbAccA3; AccA3 of M. tuberculosis H37Rv (NP_217802); MboAccA3; AccA3 of M. bovis AF 2122/97 (NP_856958); MleBccA; BccA of M. leprae TN (NP_301567); CdpAccBC; of C. diphtheriae NCTC 13129 (NP_939023); StxPccE; PccE of S. toxytricini NRRL 15443, ScoPccE; PccE of S. coelicolor A3(2) (NP_629078); SavPccE; PccE of S. avermitilis MA-4680 (NP_824508); SprPccE; PccE of S. pristinaespiralis ATCC 25486 (YP_002201271); SclPccE, S. clavuligerus ATCC 27064 (ZP_03182650); StxBpl, Bpl of S. toxytricini NRRL 15443, SavBirA; BirA of S. avermitilis MA-4680 (NP_824506); ScoBpl; Bpl of S. coelicolor A3(2) (NP_629080); SclBpl; Bpl of S. clavuligerus ATCC 27064 (YP_002193370); SprBpl, Bpl of S. pristinaespiralis ATCC 25486 (YP_002201269), MtbBirA, BirA of M. tuberculosis H37Rv (NP_217796); MboBirA, BirA of M. bovis AF 2122/97 (NP_856952); MleBirA; BirA of M. leprae TN (NP_301572); CdpBirA, BirA of C. diphtheriae NCTC 13129 (NP_939035)
In the translated AccA3, the ATP-binding motif and CO2 fixation site as well as the conserved MKM motif for biotinylation were present (Fig. 3b). The sequence GGGGRGLKVAR161-171 matched the consensus sequence of Gly-rich motif for ATP-binding in all α subunits (BC; Kimura et al. 2000). The Cys residue of RDCS226-229 involved in CO2 fixation and MKM551-553 motif for biotinylation were also found.
It was already demonstrated that the PccB protein in S. coelicolor exhibited higher activity in activation of propionyl-CoA in the presence of PccE subunit (Diacovich et al. 2002). The gene organization in pccB gene locus suggests that those genes might be in a functional relationship and be involved in the activation of acyl-CoAs with longer chain than acetyl-CoA, which can be employed as substrates for biosynthesis of secondary metabolites in S. toxytricini.
The bpl gene encodes a biotin protein ligase transferring biotin molecule to the Lys residue in MKM motif of BC (α subunit; AccA3), which is called a post-translational biotinylation of apo-BC to holo-BC. Residues STN54-56, S99, K154, GIL157-159, and N174 forming strong hydrogen bonds with biotin molecule (Bagautdinov et al. 2008) were found in the sequence (Fig. 3d). The sequence GRGR80-83 for ATP-binding is well-conserved.
Two potential ORFs were identified between pccB and accA3 genes. One resembles Bacillus maf gene involved in arrested septum formation (Butler et al. 1993) with residues S14, S16, R19, E39, K58, D77, and K89 lying on phosphate ion binding pocket of Maf protein (Minasov et al. 2000), but its real biological function is still uncertain. The other gene encodes polypeptide of 147 amino acids, but there was no similar protein found in the BLAST search.
Gene disruption of the pccB locus in S. toxytricini
To confirm the involvement of cloned pccB gene cluster in lipstatin biosynthesis, insertional inactivation of the pccB and pccE genes by homologous recombination was attempted (Gust et al. 2003). The amplified DNA fragment of target genes (pccB and pccE) was inserted in pHZ1351 to construct pHZ-BE24 vector and replaced by homologous recombination with the extended apramycin disruption cassette [acc(3)IV] flanked by FRT sites which was amplified from pIJ773. After the newly obtained disruption plasmid named pHZ-BE-Ap was introduced into E. coli ET12567/pUZ8002, it was transferred conjugally to wild-type S. toxytricini to replace the pccB and pccE genes in chromosomal DNA by homologous recombination (Flett et al. 1997).
Several brownish S. toxytricini exoconjugants were observed on MS agar supplemented with nalidixic acid and apramycin. The double-crossover recombinants were confirmed by a replica plate of the selected clones on nutrient agar supplemented with kanamycin. A single positive clone, resistant to apramycin but sensitive to kanamycin, was selected and designated as S. toxytricini BE-AP.
The double-crossover in disruptant S. toxytricini BE-AP strain was confirmed by Southern hybridization. Wild strain chromosomal DNA digested by BamHI gave strong signal at 4.0 kb DNA fragment after hybridization with pccB probe, while the disruptant did not give any signal (Fig. 2b, Fig. 4a, b). Meanwhile, in another hybridization using acc(3)IV probe, the chromosomal DNA of BE-AP strain gave a strong signal of 2.9 kb DNA, while the wild strain did not.

Confirmation of the double-crossover in S. toxytricini BE-AP strain by Southern hybridization and PCR amplification. a Southern hybridization pattern of BamHI-digested chromosomal DNA with pccB probe. Lane M, 1 kb DNA ladder; lane W, wild-type strain; lane D, BE-AP disruptant. b Southern hybridization pattern with aac(3)IV probe. Lane M, 1 kb DNA ladder; lane W, wild-type strain; lane D, BE-AP disruptant. c PCR amplification of chromosomal DNA from wild-type and BE-AP disruptant. Lane 1, amplification using PCCB-F and PCCB R primers producing a 0.8 kb DNA fragment only in the wild-type strain; lane 2, gene amplification using BE-up F1 and BE-dn R2 primers giving 2.8 kb DNA band in wild-type and 2.5 kb in the disruptant; lane 3, amplification using FRT-F and FRT-R primers giving a 1.4 kb PCR fragment only in disruptant; lane M, 1 kb DNA ladder
PCR amplification of the chromosomal DNAs of the wild-type and BE-AP strains using three different primer set combinations also confirmed the double-crossover (Fig. 2b, Fig. 4c). The combination primer of BE-up-F1 and BE-dn-R2 produced a 2.8 kb DNA band from wild-type chromosomal DNA, but gave a smaller 2.5 kb band from disruptant chromosomal DNA. The 0.8 kb DNA fragment amplified by PCCB-F and PCCB R primers confirmed the presence of pccB gene only in the wild-type, whereas the 1.4 kb PCR fragment obtained with FRT-F and FRT-R primers evidenced the replacement by double-crossover with acc(3)IV gene only in the disruptant chromosomal DNA.
The role of pccB gene locus in lipstatin biosynthesis
The wild-type and BE-AP disruptant were fermented for lipstatin production at 29 °C for 6.5 days under aerobic conditions. The final culture pH of disruptant (pH 6.22 ± 0.09) was lower than that of the wild-type (pH 6.50 ± 0.03).
The fermentation broths were extracted with acetone–hexane. After extraction, the crude oil obtained from 100 ml of fermentation broth was 4.80 ± 0.05 g in case of wild-type and 5.30 g ± 0.21 in case of disruptant. The crude extracts were subjected to the inhibition assay for pancreatic lipase. The 50,000-fold diluted crude extracts from the disruptant exhibited lower inhibitory activity than the wild-type crude extracts (Fig. 5). However, the disruptant extracts still inhibited the pancreatic lipase, which implies that the production of the lipase inhibitor was reduced but not completely hindered in the disruptant strain.

Lipase inhibitory activity of the crude extracts from S. toxytricini wild-type and BE-AP mutant. W/T-F, three independent extracts of wild-type strain; M/T-F, three independent extracts of BE-AP disruptant strain. The bar graphs represent the mean ± SD of three independent experiments. Lipase inhibitory activity of 50,000-fold diluted extracts was determined by measuring the fluorescence intensity of 4-methylumbelliferon liberated in the lipase reaction with 4-methylumbelliferyl oleate. **P < 0.001 compared between the mean values of wild-type and disruptant groups
Both crude extracts of wild-type and BE-AP strain were separated on Silica 60 gel column chromatography, and the metabolites were identified by TLC and further isolated by HPLC (Fig. 6). In HPLC, two major peaks appeared at retention times of 16.6 min (compound 1) and 19.3 min (compound 2). Compound 1 was a major one in the disruptant extract, while compound 2 in the wild-type extract. The peak area calculation showed that the compound 1 increased by four times, whereas, the compound 2 was reduced with 80% in the disruptant extract compared with the wild-type extract.

TLC and HPLC analysis of the crude extracts from S. toxytricini wild-type and disruptant strains. a TLC pattern of crude extracts on Silica 60 F254 sheet. C, orlistat (tetrahydrolipstatin–2 µg/ml); W/T, crude extract of wild-type strain; M/T, crude oil extract from BE-AP disruptant strain; F, fraction of wild-type extract showing the highest lipase inhibition activity which was eluted from Silica 60 column. b HPLC pattern of wild-type extract. c HPLC pattern of disruptant extract. HPLC analysis was performed by isocratic phase of 0.1% orthophosphoric acid and acetonitrile (15:85) and flow rate of 1 ml/min on C18 reverse-phase column. The HPLC chromatograms show two major peaks for compound 1 at 16.6 min and compound 2 at 19.3 min
The two compounds were purified by preparative HPLC from wild-type extracts, and their chemical structures were determined by 1H-NMR (Fig. 7). The 1H-NMR spectrum of the purified compound 2 showed the same spectrum as lipstatin reported by Hochuli et al. (1987). The protons for the unsaturated double bonds were shifted at 5.5 ppm as multiplet and the protons in the lactone ring at 3.2 and 4.3 ppm as multiplets. The characteristic chemical shifts of the protons in the formylleucine residue also appeared at 8.2 ppm for 2′′-amino group and at 5.9 ppm for the aldehyde group. The 1H-NMR of the compound 1 revealed to be tetradeca-5.8-dienoic acid, a precursor of lipstatin, which was confirmed by the earlier reported spectrum (Goese et al. 2000).

1H–NMR spectrum of compound 1 (tetradeca-5,8-dienoic acid) from mutant strain and compound 2 (lipstatin) from wild-type strain isolated by preparative HPLC. Both compounds gave the proton shift at 5.5 ppm as multiplet for the unsaturated double bonds. In lipstatin, the protons in the lactone ring were shifted at 3.2 and 4.3 ppm as multiplets, and the characteristic proton shifts in the formylleucine residue also appeared at 8.2 ppm for 2′′-amino group and at 5.9 ppm for the aldehyde group
Discussion
The formation of lipstatin via Claisen condensation (Goese et al. 2001; Eisenreich et al. 2003; Schuhr et al. 2002) resembles the generation of mycolic acid via condensation of two fatty acids by Pks13 (Portevin et al. 2005). However, any attempts to identify a pks13 (type I pks-like gene) in S. toxytricini did not bring the expected results (data not shown). Only a type II pks gene was previously identified in this strain (Yoo et al. 2006).
Alternatively, the accD gene which is assumed to recognize and activate octanoic acid, one of the main precursors for lipstatin, was selected as the target one. Two consecutive attempts to identify such accD genes in S. toxytricini chromosome lead to identification of two gene clusters in two distinct DNA fragments of 11.2 kb (data not shown) and 10.6 kb from different locus of its genome. Sequence analysis of the first fragment revealed two genes, accA1 and accD1, involved in primary metabolism (fatty acid biosynthesis) (Demirev et al. 2009). In contrast, the ten distinct ORFs identified in the second DNA fragment (10.6 kb) showed high homology with similar gene clusters in other Streptomyces spp. and are thought to be involved in secondary metabolism. The pccE and accA3 genes, encoding ε and α subunits of ACCase, were present downstream of pccB gene encoding β subunit of ACCase.
In S. coelicolor and M. tuberculosis, ACCase consists of three subunits, the α, β, and ε subunit (Diacovich et al. 2002; Gago et al. 2006; Oh et al. 2006). The α subunit (BC) transfers CO2 molecule to form a carboxyl–BC. Subsequently, the β subunit (CT) transfers the carboxyl group from carboxyl–BC to the acyl-CoA and controls the substrate specificity in each ACCase complex. A recent study showed that the gene products of pccB and pccE in S. coelicolor form a protein complex and PccB protein exhibits full activity only in presence of PccE subunit (Gago et al. 2006).
Based on the sequence homology with AccD5 and its putative activity, it was assumed that PccB is related to the secondary metabolism and probably recognizes and activates a fatty acid with longer chain than acetyl-CoA. In addition, the organization of 10.6 kb pccB gene locus showed that they could be functionally related to form a specific ACCase protein complex.
In order to disclose whether the gene products of pccB and pccE are involved in the lipstatin biosynthesis, these two genes were inactivated by homologous recombination in the wild-type S. toxytricini strain. The gene replacement by double-crossover in disruptant S. toxytricini BE-AP was confirmed by DNA hybridization and PCR amplification.
After the fermentative production of active metabolites, the final pH of the fermentation broth of the disruptant strain was lower than the wild-type, probably due to the accumulation of organic acids. There were no visible differences in growth between those strains after 4-day incubation. The metabolites in fermentation broths of wild-type and disruptant strain were extracted by hexane–ethyl acetate mixture, and it was observed that the spot corresponding to lipstatin in wild-type strain was reduced to a great extent in the disruptant in TLC analysis.
In the pancreatic lipase inhibitory assay, the crude extract of the disruptant exhibited considerably lower activity compared with that of the wild-type. However, the disruptant extract still inhibited the pancreatic lipase. Two metabolites purified from wild-type extract by preparative HPLC were subjected to 1H-NMR for determination of their chemical structures. The 1H-NMR spectrum of these compounds showed that they are lipstatin and tetradeca-5.8-dienoic acid, respectively. The HPLC chromatograms of extracts showed that the disruptant did not only accumulate the major precursor tetradeca-5.8-dienoic acid, but also produced a lower level of the active metabolite lipstatin.
Earlier tracer study experiments for elucidations of the specific precursors (Goese et al. 2001; Eisenreich et al. 2003; Schuhr et al. 2002) showed that Claisen condensation of 3-hydroxy-tetradeca-5.8-dienoic acid and octanoic acid leads to formation of the lipstatin backbone. Considering the crystal structure of PccB and its recognition of longer chain fatty acid than acetyl-CoA but shorter than linoleyl-CoA (Diacovich et al. 2004), it can be assumed that PccB of S. toxytricini is involved in the activation of octanoic acid to carboxyl–octanoic acid, which is the second major precursor in the lipstatin biosynthesis.
In addition, it can be speculated that the lipids were actively metabolized by S. toxytricini BE-AP strain to monoglycerides and linoleic acid, which was further metabolized by β–degradation to tetradeca-5.8-dienoic acid. But, the resulting fatty acid could not be utilized by disruptant for lipstatin biosynthesis because octanoic acid was not activated to hexylmalonic acid. However, the lipstatin biosynthesis was not completely turned off in disruptant, probably due to the activation of octanoic acid by another ACC complex system with much lower activity.
The accumulation of tetradeca-5.8-dienoic acid rather than 3-hydroxy-tetradeca-5.8-dienoic acid by disruptant suggests that the hydroxyl group can be introduced at C-5 position of the generated 3-oxo intermediate after the Claisen condensation. The accumulation of this compound also could be the reason for the lower pH after fermentation with the disruptant strain.
References
Bagautdinov B, Matsuura Y, Bagautdinova S, Kunishima N (2008) Protein biotinylation visualized by a complex structure of biotin protein ligase with a substrate. J Biol Chem 283:14739–147550
Bierman M, Logan R, O’Brien K, Seno ET, Rao RN, Schoner BE (1992) Plasmid cloning vectors for the conjugal transfer of DNA from Escherichia coli to Streptomyces spp. Gene 116:43–49
Butler YX, Abhayawardhane Y, Stewart GC (1993) Amplification of the Bacillus subtilis maf gene results in arrested septum formation. J Bacteriol 175:3139–3145
Cronan JE Jr, Waldrop GL (2002) Multi-subunit acetyl-CoA carboxylases. Prog Lipid Res 41:407–435
Datsenko KA, Wanner BL (2000) One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci USA 97:6640–6645
Demirev AV, Lee JS, Sedai BR, Ivanov IG, Nam DH (2009) Identification and characterization of acetyl-CoA carboxylase gene cluster in Streptomyces toxytricini. J Microbiol 47:473–478
Diacovich L, Peiru S, Kurth D, Rodriguez E, Podesta F, Khosla C, Gramajo H (2002) Kinetic and structural analysis of a new group of acyl-CoA carboxylases found in Streptomyces coelicolor A3(2). J Biol Chem 277:31228–31236
Diacovich L, Mitchell DL, Pham H, Gago G, Meglar MM, Khosla C, Gramajo H, Tsai SC (2004) Crystal structure of the β subunit of acyl-CoA carboxylase: structure-based engineering of substrate specificity. Biochemistry 43:14027–14036
Eisenreich W, Kupfer E, Weber W, Bacher A (1997) Tracer studies with crude U-13C-lipid mixtures. Biosynthesis of the lipase inhibitor lipstatin. J Biol Chem 272:867–874
Eisenreich W, Kupfer E, Stohler P, Weber W, Bacher A (2003) Biosynthetic origin of a branched chain analogue of the lipase inhibitor, lipstatin. J Med Chem 46:4209–4212
Erdei J, Gulyas E, Balogh G, Toth L, Keri V, and Csorvasi A (2005) Fermentation process for lipstatin and method of extracting lipstatin from a fermentation broth. U.S. Patent 6,844,174 B2
Flett F, Mersinias V, Smith CP (1997) High efficiency intergeneric conjugal transfer of plasmid DNA from Escherichia coli to methyl DNA-restricting streptomycetes. FEMS Microbiol Lett 155:223–229
Gago G, Kurth D, Diacovich L, Tsai S-C, Gramajo H (2006) Biochemical and structural characterization of an essential acyl coenzyme A carboxylase from Mycobacterium tuberculosis. J Bacteriol 188:477–486
Goese M, Eisenreich W, Kupfer E, Weber W, Bacher A (2000) Biosynthetic origin of hydrogen atoms in the lipase inhibitor lipstatin. J Biol Chem 275:21192–21196
Goese M, Eisenreich W, Kupfer E, Stohler P, Weber W, Leuenberger HG, Bacher A (2001) Biosynthesis of lipstatin. Incorporation of multiply deuterium-labeled (5Z, 8Z)-tetradeca-5, 8-dienoic acid and octanoic acid. J Org Chem 66:4673–4678
Gust B, Challis GL, Fowler K, Kieser T, Chater KF (2003) PCR-targeted Streptomyces gene replacement identifies a protein domain needed for biosynthesis of the sesquiterpene soil odor geosmin. Proc Natl Acad Sci USA 100:1541–1546
Hanahan D (1983) Studies on the transformation of E. coli with plasmids. J Mol Biol 166:557–580
Hochuli E, Kupfer E, Maurer R, Meister W, Mercadal Y, Schmidt K (1987) Lipstatin, an inhibitor of pancreatic lipase, produced by Streptomyces toxytricini. II. Chemistry and structure elucidation. J Antibiot 40:1086–1091
Imanaka T, Moriyama Y, Ecsedi GG, Aoyagi T, Amanuma-Muto K, Ohkuma S, Takano T (1983) Esterastin: a potent inhibitor of lysosomal acid lipase. J Biochem 94:1017–1020
Kieser T, Bibb MJ, Buttner MJ, Chater KF, Hopwood DA (2000) Practical Streptomyces genetics. The John Innes Foundation, Norwich, UK
Kimura Y, Miyake R, Tokumasu Y, Sato M (2000) Molecular cloning and characterization of two genes for the biotin carboxylase and carboxytransferase subunits of acetyl coenzyme A carboxylase in Myxococcus xanthus. J Bacteriol 182:5462–5469
Lin T-W, Melgar MM, Kurth D, Swamidass SJ, Purdon J, Tseng T, Gago G, Baldi P, Gramajo H, Tsai S-C (2006) Structure-based inhibitor design of AccD5, an essential acyl-CoA carboxylase carboxyltransferase domain of Mycobacterium tuberculosis. Proc Natl Acad Sci USA 103:3072–3077
MacNeil DJ, Gewain KM, Ruby CL, Dezeny G, Gibbons PH, MacNeil T (1992) Analysis of Streptomyces avermitilis genes required for avermectin biosynthesis utilizing novel integrating vector. Gene 111:61–68
Minasov G, Teplova M, Stewart GC, Koonin EV, Anderson WF, Egli M (2000) Functional implications from crystal structures of the conserved Bacillus subtilis protein Maf with and without dUTP. Proc Natl Acad Sci USA 97:6328–6333
Oh T-J, Daniel J, Kim H-J, Sirakova TD, Kolattukudy PE (2006) Identification and characterization of Rv3281 as a novel subunit of a biotin-dependent acyl-CoA carboxylase in Mycobacterium tuberculosis H37Rv. J Biol Chem 281:3899–3908
Paget MSB, Chamberlin L, Atrih A, Foster SJ, Buttner MJ (1999) Evidence that the extracytoplasmic function sigma facter σE is required for normal cell wall structure in Streptomyces coelicolor A3(2). J Bacteriol 181:204–211
Portevin D, de Sousa-D'Auria C, Houssin C, Grimaldi C, Chami M, Daffe M, Guilhot C (2004) A polyketide synthase catalyzes the last condensation step of mycolic acid biosynthesis in mycobacteria and related organisms. Proc Natl Acad Sci USA 101:314–319
Portevin D, de Sousa-D'Auria C, Montrozier H, Houssin C, Stella A, Laneelle M-A, Bardou F, Guilhot C, Daffe M (2005) The acyl-AMP ligase FadD32 and AccD4-containing acyl-CoA carboxylase are required for the synthesis of mycolic acids and essential for mycobacterial growth. J Biol Chem 280:8862–8874
Rodriguez E, Banchio C, Diacovich L, Bibb MJ, Gramajo H (2001) Role of an essential acyl coenzyme A carboxylases in the primary and secondary metabolism of Streptomyces coelicolor A3(2). Appl Environ Microbiol 67:4166–4167
Rodriguez E, Gramajo H (1999) Genetic and biochemical characterization of the α and β components of propionyl-CoA carboxylase complex of Streptomyces coelicolor A3(2). Microbiology 145:3109–3119
Sambrook J, Russell DW (2001) Molecular cloning: a laboratory manual, 3rd edn. CSH Press, NY, USA
Schuhr CA, Eisenreich W, Goese M, Stohler P, Weber W, Kupfer E, Bacher A (2002) Biosynthetic precursors of the lipase inhibitor lipstatin. J Org Chem 67:2257–2262
Souri E, Jalalizadeh H, Kebriaee-Zadeh A, Zadehvakili B (2007) HPLC analysis of orlistat and its application to drug quality control studies. Chem Pharm Bull 55:251–254
Sun Y, Zhou X, Liu J, Bao K, Zhang G, Tu G, Kieser T, Deng Z (2002) Streptomyces nanchangensis, a producer of the insecticidal polyether antibiotic nanchangmycin and the antiparasitic macrolide meilingmycin, contains multiple polyketide gene clusters. Microbiology 148:361–371
Weibel EK, Hadvary P, Hochuli E, Kupfer E, Lengsfeld H (1987) Lipstatin, an inhibitor of pancreatic lipase, produced by Streptomyces toxytricini. I. Producing organism, fermentation, isolation and biological activity. J Antibiot 40:1081–1085
Yoo A, Demirev AV, Lee JS, Kim SD, Nam DH (2006) Cloning and analysis of a type II polyketide synthase gene cluster from Streptomyces toxytricini NRRL 15,433. J Microbiol 44:649–654
Acknowledgement
This work was supported by Korea Research Fund (grant number KRF-2009-0071085) in 2009.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary materials
Below is the link to the electronic supplementary material.
Fig. S1
(DOC 95 kb)
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Demirev, A.V., Khanal, A., Sedai, B.R. et al. The role of acyl-coenzyme A carboxylase complex in lipstatin biosynthesis of Streptomyces toxytricini . Appl Microbiol Biotechnol 87, 1129–1139 (2010). https://doi.org/10.1007/s00253-010-2587-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-010-2587-2