
Abstract
Saturation mutagenesis constitutes a powerful method in the directed evolution of enzymes. Traditional protocols of whole plasmid amplification such as Stratagene’s QuikChange™ sometimes fail when the templates are difficult to amplify. In order to overcome such restrictions, we have devised a simple two-primer, two-stage polymerase chain reaction (PCR) method which constitutes an improvement over existing protocols. In the first stage of the PCR, both the mutagenic primer and the antiprimer that are not complementary anneal to the template. In the second stage, the amplified sequence is used as a megaprimer. Sites composed of one or more residues can be randomized in a single PCR reaction, irrespective of their location in the gene sequence.The method has been applied to several enzymes successfully, including P450-BM3 from Bacillus megaterium, the lipases from Pseudomonas aeruginosa and Candida antarctica and the epoxide hydrolase from Aspergillus niger. Here, we show that megaprimer size as well as the direction and design of the antiprimer are determining factors in the amplification of the plasmid. Comparison of the results with the performances of previous protocols reveals the efficiency of the improved method.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Directed evolution constitutes a powerful method for engineering essentially any property of an enzyme, including thermostability, substrate acceptance, and enantioselectivity, as documented by recent reviews (Arndt and Müller 2007; Arnold and Georgiou 2003a; Brakmann and Schwienhorst 2004; Hibbert et al. 2005; Rubin-Pitel and Zhao 2006; Reetz 2006; Bershtein and Tawfik 2008). It is based on the appropriate combination of gene mutagenesis and expression coupled with high-throughput screening or selection. A variety of mutagenesis techniques have been devised, the most often used methods being error-prone polymerase chain reaction (epPCR; Leung et al. 1989; Cadwell and Joyce 1992), saturation mutagenesis (Georgescu et al. 2003; Hogrefe et al. 2002; Kirsch and Joly 1998; Zheng et al. 2004), and DNA shuffling (Stemmer 1994). Although the success of these studies is indisputable, the quest to make directed evolution more efficient than in the past is the major current challenge (Lutz and Patrick 2004; Fox and Huisman 2008; Reetz 2004; Reetz et al. 2006a). The basic goal is to improve the quality of enzyme libraries to such an extent that the screening effort (Reymond 2005) can be reduced significantly because this is generally the bottleneck of directed evolution.
To this end, we have proposed and implemented iterative saturation mutagenesis (ISM) as a general approach which comes closer to the above goal (Reetz et al. 2006b; Reetz et al. 2006c; Reetz and Carballeira 2007). The concept of ISM can be used to influence very different catalytic properties of an enzyme, such as thermostability in the form of B-FIT (Reetz et al. 2006b; Reetz and Carballeira 2007) or substrate acceptance and enantioselectivity using CASTing (Reetz et al. 2006c; Bartsch et al. 2008; Liang et al. 2007). Based on structural information of the enzyme and by focusing on positions expected to be crucial for a given catalytic property, several sites, composed of one or more residues, can be selected and randomized.
Saturation mutagenesis can be performed by applying a variety of different molecular biological methods developed during the past two decades (Arndt and Müller 2007; Dominy and Andrews 2003; Georgescu et al. 2003; Hogrefe et al. 2002; Kirsch and Joly 1998; Zheng et al. 2004; Reetz 2004). Currently, the most popular approach is the use of the Stratagene QuikChange™ protocol (Hogrefe et al. 2002). The original form of QuikChange™ is limited to the introduction of only one mutation at a given amino acid position. Moreover, problems related to the primer length and design also persist. Different approaches have been reported which overcome the problem of primer design by using partially overlapped (Zheng et al. 2004) or even nonoverlapping oligonucleotides (Kirsch and Joly 1998), where the resulting amplicon is used as a megaprimer (Sarkar and Sommer 1990; Miyazaki and Takenouchi 2002), thereby completing the synthesis of the plasmid in a second PCR. However, we encountered difficulties in the application of these methods to certain recalcitrant targets such as plasmids containing P450-bm3 from Bacillus megaterium and Pseudomonas aeruginosa lipase A genes. In such cases, the amplification and the subsequent introduction of mutations proved to be unsuccessful.
Given the importance of saturation mutagenesis in enzyme evolution (Reetz 2004; Reetz 2006), any substantial simplification and/or extension of existing protocols would be of great interest. Here, we present an improved method to perform saturation mutagenesis in the gene of difficult-to-amplify templates using a single two-stage whole-plasmid PCR.
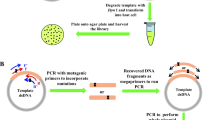
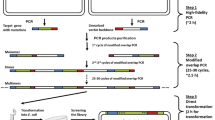
In the first stage of the PCR, two mutagenic primers or a mutagenic primer and an antiprimer (a non-mutagenic (silent) primer used to complete the complementary extension as well as to help in opening and uncoiling the DNA) are used for only a few cycles which are needed to generate the megaprimer. Once the megaprimer is generated, the second stage begins in which the annealing temperature is increased to eliminate priming by the oligonucleotide primers, and a further 20 cycles are carried out to amplify the mutated plasmid (Fig. 1).

Reaction scheme with variation of the antiprimer position. The gene is represented in blue, the vector backbone in gray, and the formed megaprimer in black. In the first stage of the PCR, both the mutagenic primer (positions randomized represented by a red square) and the antiprimer (or another mutagenic primer, shown to the right) anneal to the template and the amplified sequence is used as a megaprimer in the second stage. Finally, the template plasmids are digested using DpnI, and the resulting library is transformed in bacteria. The scheme to the left of the figure illustrates the three possible options in the choice of the megaprimer size for a single site randomization experiment. The scheme to the right represents an experiment with two sites simultaneously randomized
This procedure is an extension of the method proposed by Kirsch and Joly, modified to work not only on the complicated templates mentioned above but also on any template as an alternative to standard techniques and especially in cases where QuikChange™ fails. Recently, Tseng et al. (2008) published a note on related work, also based on the work of Kirsch and Joly, while we were finalizing our experiments. Our findings broadly agree with their results, but our method probes further into the possible uses of this approach, as in saturation mutagenesis at multiple sites and more importantly into the quality of the resulting libraries. We demonstrate that optimal primer localization and orientation are essential to increase the yield in the amplification of these difficult sequences. Additionally, this method intrinsically avoids problems arising from palindromes, hairpins or self-pairing in oligonucleotides that plague other methods based on overlapping primers.
Materials and methods
General information
Plasmids pETM11-P450-BM3 (8474 bp), pUCPCL6AN (6994 bp; Jaeger et al. 1997), pETM11-CALB (6282 bp), and pQE-ANEH (4631 bp; Cedrone et al. 2003) were used as templates to create saturation mutagenesis libraries. The P450-bm3 gene from B. megaterium (Narhi and Fulco 1982) was PCR amplified from genomic DNA (ATCC 14581/strain No. 32 at DSMZ GmbH, Germany) and cloned into the expression vector pETM11 (EMBL vector collection, Germany) using the NcoI and SacI restriction sites. The calB gene from Candida antarctica was PCR amplified from genomic DNA (ATCC 32657 at DSMZ GmbH, Germany) and cloned into the expression vector pETM11 (EMBL vector collection, Germany) using the NcoI and NotI restriction sites.
All degenerate oligonucleotides (see Table 1 for more details) were synthesized by Invitrogen (Germany). PCR amplifications were carried out with KOD Hot Start DNA polymerase (Novagen, USA) and digested with DpnI (New England Biolabs, UK). The sequencing was performed on plasmid DNA extracted from pooled colonies using QIAprep Miniprep Kit (Qiagen, Germany). All transformations were carried out with the same batch of homemade Escherichia coli DH5α chemocompetent cells according to a standard protocol. 1 kb DNA ladder was obtained from Fermentas.
Method of QuikChange™
The reactions were performed in a total volume of 50 μl, and the reaction mixtures were prepared according to the QuikChange™ protocol, using 50 ng of template. PCR conditions were: initial denaturation 3 min at 95 °C followed by 16 cycles of 1 min at 95 °C, 1 min of annealing at the required temperature (T m–5 °C), 1 min/kb of extension at 72 °C; and 2 min/kb of final extension (Table 1). To remove template plasmid, PCR amplified mixtures were digested with DpnI (20 U, 10× DpnI buffer) for 1 h at 37 °C, after which another aliquot of DpnI (20 U) was added and digestion continued for 1 h more. A 2-μl aliquot was used to transform E. coli DH5α. As P. aeruginosa lacks the Dam methylation system (Stover et al. 2000), the PCR template used in this protocol had to be obtained from E. coli in order to be recognized by DpnI.
Method of Kirsch and Joly
The method reported by Kirsch and Joly (1998) was applied as described by the authors. This called for 50 ng of template to be mixed with 12.5 pmol of each oligonucleotide in a 50-μl reaction. The amplification program was as follows: initial denaturation 3 min at 95 °C followed by 9 cycles of 30 s at 95 °C, annealing 1 min at 55 °C, extension 1 min at 68 °C, with a further 9 cycles of 30 s at 95 °C, extension 6 min at 68 °C; and final extension 16 min at 68 °C. PCR-amplified reaction mixtures were digested with DpnI and transformed in E. coli as described above.
Method of Zheng
The method reported by Zheng et al. (2004) was carried out using 50 ng of template and 12.5 pmol of each oligonucleotide in a 50 μl reaction. PCR cycle conditions were: initial denaturation 3 min at 94 °C followed by 16 cycles of 1 min at 94 °C, annealing 1 min at 52 °C, extension 1 min/kb at 68 °C; and final extension 1 h at 68 °C. DpnI digestion and transformation were performed as described above.
Improved method of the present study
The sequence of the antiprimers was designed to amplify megaprimers of different lengths: small, with the antiprimer annealing site close to the open reading frame; medium, with the annealing site that gives a megaprimer encompassing half length of the plasmid; and large, with the annealing site giving the whole plasmid minus a small part of the gene. The sequence of each antiprimer was adjusted to have an identical or similar T m value as the mutagenic oligonucleotide used to produce the library. The reactions were performed in a final volume of 50 μl containing 50–100 ng of template and 5 pmol of each primer. The amplification program was as follows: initial denaturation 3 min at 95 °C (for P. aeruginosa lipA template, 5 min at 98 °C) followed by 5 cycles of 30 s at 95 °C, annealing 1 min at 55 °C (this parameter depend on the T m of the oligonucleotides used, and in all cases a gradient PCR was performed), extension 1 min/kb according to the megaprimer size at 72 °C. The second stage consisted of 20 cycles of 30 s at 95 °C and extension at 1 min/kb of template at 68 °C; and a final extension of 2 min/kb of template at 68 °C. For each reaction, DpnI digestion and transformation were performed as described above.
Results
Experiments with the P450-bm3 gene
Here, we use our method for the introduction of 1- and 2-codon mutations in the P450-bm3 gene cloned in pETM11. We first applied it to the site saturation mutagenesis of position F87 (one codon, NNK randomization; Table 2). The yields of the amplification were analyzed by agarose gel electrophoresis (Fig. 2). The QuikChange™ protocol gave an average of 80 colonies (see experimental procedures in “Materials and methods” for details). This result was 3.5-fold better than both the Kirsch and Joly method and the Zheng et al. method (28 and 23 colonies, respectively). The application of our method gave significantly different results with respect to the size of the generated megaprimer. In the above case, the medium-sized megaprimer gave results comparable to QuikChange™ with 71 colonies. The small-sized megaprimer yielded 168 colonies, which are 2.5-fold better. Ultimately, the generation of the large megaprimer gave the best results with more than 200 colonies. This is almost one order of magnitude more than the Kirsch and Joly and Zheng et al. methods and over threefold greater compared to QuikChange™.

Agarose gel analysis of the PCR amplification of pETM11-P450-BM3 (8474 bp) after DpnI digestion using the different protocols. Lanes 1 to 6 are F87 randomization experiments. Lanes 7 to 10 are M185/L188 simultaneous randomization experiments. 1 QuikChange™ protocol; 2 Kirsch and Joly protocol; 3 and 7 Zheng et al. protocol; 4 and 8 our protocol with small megaprimer; 5 and 9 our protocol with medium megaprimer; 6 and 10 our protocol with large megaprimer; M 1 kb DNA ladder standard (Fermentas)
The randomization frequencies at position F87 were assessed for each protocol. The inserts harbored the desired NNK randomization in that position. The statistical distribution of the different nucleotides, assuming complete randomization, should have been 25% of each nucleotide for the first two positions and 50% (only T and G are expected) for the third nucleotide in the codon. However, the observed distribution was different with respect to the protocol used (Table 3). When QuikChange™ was used for F87 saturation, the wild-type TTT codon was only partially randomized since it turned out that all three deoxythymidines were highly conserved (from 67% to 83% conservation for individual nucleotides). When the Kirsch and Joly protocol was used, the native codon was less conserved (from 60% to 72% conservation for individual nucleotides). With our method, the randomization was more efficient (large megaprimer), reaching around 50% for the whole codon.
We further extended our method to the simultaneous randomization of two codons corresponding to positions M185 and L188 in P450-BM3. In this part of the study, we compared our improved method with the Zheng et al. method (Table 2). Around 40 colonies were obtained on average with the Zheng et al. protocol. Our method yielded 15 colonies with the small- and 37 colonies with the medium-sized megaprimers. Furthermore, the generation of the large megaprimer gave 245 colonies after transformation. This result demonstrates that even for randomization at two positions, our method is 6.5-fold more efficient than the previously published protocol by Zheng et al.
Sequencing performed on the plasmid DNA confirmed that the wild-type codons at positions M185 and L188 were substituted by two NNK randomized codons. As with position F87, similar randomization efficiency was obtained for the simultaneous saturation of M185 and L188 where the Zheng et al. protocol generated conservation rates from 39% to 79% for individual nucleotides, while our method (large megaprimer) gave rates ranging from 39% to 53% for individual nucleotides (Table 4). The latter results demonstrate that this method never underperformed the existing mutagenesis strategies and is more efficient in almost every case.
Experiments with P. aeruginosa lipase A gene
Site-saturation mutagenesis was also performed at two adjacent positions in the same oligonucleotide in the lipA gene of P. aeruginosa PAO1 (accession number X63390). The yields of amplification were analyzed by agarose gel electrophoresis (Fig. 3). One oligonucleotide for the library and different antiprimers annealing outside the cloning site were used (Fig. 4).

Gel electrophoresis of saturation mutagenesis reactions using pUCPCL6AN (6994 bp) after DpnI digestion containing the lipA gene from P. aeruginosa as template. Lane 1 small-sized megaprimer, clockwise; lane 2 medium-sized megaprimer, clockwise; lane 3 large-sized megaprimer, clockwise; lane M 1 kb DNA ladder standard (Fermentas); lane 4 small-sized megaprimer, anticlockwise; lane 5 medium-sized megaprimer, anticlockwise; lane 6 large-sized megaprimer, anticlockwise

Our method applied to a plasmid template containing P. aeruginosa lipA gene (pUCPCL6AN). Different positions and orientations of the antiprimers were tried for the amplification of megaprimers both in the clockwise (a) and anticlockwise (b) directions. These different positions and directions encompass regions of the plasmid with differing GC content. Note that the mutagenic primer and only one of the antiprimers are used in each experiment
The three amplification reactions were each carried out with the two possible directionalities. Firstly, three PCR reactions were performed using the forward mutagenic primer and a different reverse antiprimer in each one to produce megaprimers, (small, medium, and large size, respectively) clockwise (CW) with respect to lipA gene (Fig. 4a). Secondly, we used the reverse complementary primers: reverse mutagenic primer and forward antiprimers to synthesize the anticlockwise (ACW) megaprimers (small, medium, and large) with respect to lipA gene (Fig. 4b). The results from these experiments were compared with other protocols described for site-directed and saturation mutagenesis (Table 5).
The standard QuikChange™ and the Kirsch and Joly methods failed to amplify the plasmid, and no colonies were found after transformations. Implementation of the Zheng et al. method to this template yielded only five colonies. On the other hand, the yields from the application of our method were extremely variable depending on the size and position of the megaprimers. The small megaprimers were successfully amplified in both directions in spite of the differences in GC content between these two fragments (Table 5, Fig. 3: lanes 1 and 4). Nevertheless, no colonies were obtained after transformation with the small ACW megaprimer and only seven in the case of small CW megaprimer, showing that the whole plasmid PCR was not completely accomplished. The yield was slightly increased when the medium and large megaprimers were produced in the anticlockwise direction, giving almost the same number of colonies as the Zheng et al. method. However, it was with the medium-sized clockwise megaprimer that we achieved the best result with 277 colonies: an improvement of more than 55-fold with respect to the Zheng et al. method. The quality of these libraries was determined by sequencing the PCR products before transformation: they showed the expected degeneracy at the mutated positions.
Experiments with the Candida antarctica lipase B and Aspergillus niger epoxide hydrolase genes
In order to further demonstrate the broad applicability of this method, we introduced site-directed mutations and saturation mutagenesis in two different templates. The first example is the application of our method to C. antarctica lipase B, cloned in pETM11 plasmid. Here, we attempted to perform site-saturation mutagenesis at two distant positions simultaneously in a single PCR. These two positions, separated by 171 bp, were successfully randomized giving a high quality saturation mutagenesis library (data not shown) and yielding an average of 400 colonies after transformation of DpnI digested reactions.
The second example utilizes the gene of A. niger epoxide hydrolase cloned in the pQE vector (Cedrone et al. 2003). Our improved method was successfully applied in the case of site-directed mutagenesis or saturation mutagenesis in regions where the restricted possibilities of primer design given by QuikChange™ force the inclusion of sequences that contain palindromes or overlapping regions (oligonucleotides BWT-F, BWT-R, PRI B2-R, PRI B3-R; Table 6). The problem was easily solved by the selection of a reverse antiprimer outside of the gene (SP1-F), in an area free of those inconveniences, with a similar T m as the forward mutagenic primer. The application of our method resulted in a high percentage of the expected mutants and insertions were never observed (Table 6).
Discussion
The previously reported method of Kirsch and Joly (1998) is essentially a combination of QuikChange™ (Stratagene, La Jolla, CA, USA) and megaprimer (Sarkar and Sommer 1990) leading to one-step PCR mutagenesis. In our extension of this protocol, we applied saturation mutagenesis at specific residues using modified oligonucleotide concentrations and annealing cycles in order to increase the yield of the final product. Furthermore, we compared different annealing positions of the helper oligonucleotide—called the antiprimer—which can be silent or mutagenic, obtaining different megaprimer sizes.
During the first stage of PCR, the megaprimer is generated by amplifying the region between the two oligonucleotide primers. Contrary to what was reported by Kirsch and Joly (same number of cycles per stage), we suggest a drastic reduction in the number of cycles during the first stage. When we have tried the Kirsch and Joly method in our systems, overproduction of megaprimer was observed in the exponential stage. As a result, in the second or linear stage, megaprimers self anneal leading to unproductive complexes. A reduced amount of oligonucleotides (5 pmol) in the PCR reaction, an increased amount of template and only 3 to 5 cycles, enhance the yield of the final plasmid library (Fig. 5).

Recommended working conditions. The amount of template and oligonucleotides are given for 50 μl of reaction mixture. The figure also depicts the three possible choices in the election of the antiprimer position
We applied our method in several practical examples where existing protocols (namely QuikChange™, Kirsch and Joly as well as Zheng et al.) failed to generate saturation mutagenesis libraries. We discuss them here separately.
Experiments with the P450-bm3 gene
The optimization of P450-BM3 is an intensive field of research in protein engineering as recently reviewed by Tee and Schwaneberg (2007). To the best of our knowledge, two methods are currently used for the creation of BM3 saturation mutagenesis libraries, namely overlap extension PCR and QuikChange™ (Peters et al. 2003; Arnold and Georgiou 2003b; Wong et al. 2008; Li et al. 2008). Overlap extension PCR is a time-consuming protocol that often yields a low number of colonies which is not well suited for the creation of libraries. Moreover, in our hands, applying the standard QuikChange™ protocol to P450-BM3 gene failed in almost all cases (no amplification, insertions or deletions were observed, data not shown). A major problem in applying saturation mutagenesis concerns those cases in which large plasmids are involved. Indeed, P450-bm3 was cloned in the vector pETM11, resulting in a large construct of 8474 bp.
For the construction of the F87 library of P450-BM3, our method gave equal or better results than QuikChange™, the Kirsch and Joly method, and the Zheng et al. protocol with respect to both quality (randomization) and quantity (number of colonies). The size of the megaprimer has a considerable impact on the results. In this particular case, the large megaprimer was the most successful for library creation.
The ideal methodology for performing site saturation mutagenesis is the one yielding (1) a uniform statistical distribution of the desired mutations (quality criteria) and (2) a number of colonies sufficient to get 95% coverage of the given library in one step (quantity criteria). Accordingly, our method is one step closer to the ideal site saturation mutagenesis protocol which allowed us to construct two different saturation libraries for P450-BM3, in less time, with the greatest number of transformants and with the highest level of randomization.
Experiments with P. aeruginosa lipase A gene
Different techniques such as overlap extension PCR and/or subcloning of a mutated DNA fragment have been applied to mutate certain residues by saturation mutagenesis or site-directed mutagenesis in P. aeruginosa lipase A gene (Liebeton et al. 2000; Reetz et al. 2001; Liebeton et al. 2001; Fuji et al. 2005; Reetz et al. 2006a; Reetz et al. 2007). Whole-plasmid PCR, QuikChange™, and other methods have proven to be rapid and highly efficient methods to introduce these mutations directly in the plasmid in only one step. However, these protocols failed when applied to P. aeruginosa lipase A constructs, giving very poor yields or no amplification.
It is known that high GC containing templates cause particular problems such as inconsistent amplification or none at all even with the addition of enhancers or the use of different polymerases. Thus, we analyzed the GC content of this template revealing that the region encompassing the lipA gene (coding for lipase A) and the lipH gene (coding for the foldase) has a GC content of 68% compared to the overall value of 54%. To address this problem, we again applied our method, placing the antiprimer at three positions and with two directionalities, resulting in six megaprimers as described above. In this case, we hypothesize that the megaprimer might aid in the amplification of the GC rich regions by wedging the DNA strands open, thereby facilitating extension.
From the results of our experiments (Fig. 3, Table 5), it appears that the combination of size and possibly GC content in the megaprimer are responsible for the efficient amplification of the whole plasmid. Although the medium sized megaprimers (clockwise and anticlockwise) amplified fragments of almost identical length, those fragment contained different parts of the template having different GC content. The anticlockwise megaprimer mainly encompasses vector DNA with lower GC content and leads to unsuccessful amplification probably for the same reason as in the other methods, i.e., the polymerase cannot replicate the remaining region (with high GC content). On the other hand, the clockwise megaprimer, which includes the lipA and lipH genes (with high GC content) as well as part of the vector, yields a successful amplification. This seems to confirm the hypothesis that opening the GC rich region with a large DNA fragment (megaprimer) aids the polymerase in accomplishing the whole plasmid PCR.
The oligonucleotides giving the small sized clockwise megaprimer were positioned in the lipA and lipH genes. Although the GC content in this region of the plasmid is very high (68%), the megaprimer was successfully amplified; however, it is possible that the size of the megaprimer was insufficient to keep the plasmid open during the second stage of the PCR. The large anticlockwise megaprimer was not properly amplified during the first stage of the PCR (Fig. 4) where the main product was an indistinct band. Due to the large size of this megaprimer and its orientation, the polymerase is probably facing the same problems that plague the standard methods: a long fragment to amplify, primed from within a region of high GC content.
To summarize, the orientation and the annealing positions of the primers used to amplify the medium size clockwise megaprimer created conditions favorable to the polymerase for amplification of the plasmid. This megaprimer which encompasses a large portion of the high GC area efficiently disrupted this region of the template, facilitating whole plasmid amplification in the second stage of the PCR.
Experiments with C. antarctica lipase B and A. niger epoxide hydrolase genes
Our protocol was also applied to templates that could be effectively mutated by the QuikChange™ protocol but where the use of the improved method represents an advantage or is able to overcome some problems derived from the sequence of the template. We illustrate this with two examples: C. antarctica lipase B cloned in pETM11 and A. niger epoxide hydrolase cloned in pQE60.
The advantage gained in the case of C. antarctica lipase B was the achievement of simultaneous randomization at two distant sites in one PCR. With the QuikChange™ method, the same result would have required two PCRs and four mutagenic primers. Therefore, using accessible laboratory materials, our method saves time and costs, representing an alternative to high-priced kits as, e.g., QuikChange™ (Stratagene) Multi-Site Directed Mutagenesis and QuikChange™ XL that depend on special enzyme mixtures and tailor-made solutions.
In the case of A. niger epoxide hydrolase, we wanted to perform site-directed mutagenesis in a problematic region of the gene. One of the most limiting features of QuikChange™ relates to the primer design when performing site-directed mutagenesis or saturation mutagenesis in regions containing repetitions or palindromes. In these regions, the formation of primer-dimers is facilitated, frequently leading to multiple insertions of the primer sequence or less often to deletions of one or more nucleotides. Due to the small difference in the size of these erroneously mutated plasmids with respect to the original template, it is very difficult to distinguish and isolate them from the desired mutant by gel electrophoresis, thereby contaminating and decreasing the quality of the library and consequently increasing the screening effort in directed evolution. The fact that QuikChange™ requires the use of complementary forward and reverse primers can make it impossible to overcome the problem of primer-dimers. Because our method uses an antiprimer located outside of the target (palindrome containing) region, we were able to circumvent the problem of primer dimers. Additionally, the flexibility in the choice of the antiprimer allows a sequence matching the T m of the mutagenic primer to be designed, all of which leads to successful amplification of palindromic targets as illustrated by the experiments with A. niger epoxide hydrolase (Table 6).
In conclusion, the modified and extended protocol we present offers high applicability and rapidity in performing saturation mutagenesis. It avoids traditional subcloning steps and requires only one randomized oligonucleotide per library plus an antiprimer (non-mutagenic oligonucleotide) which can be used repeatedly in different saturation mutagenesis reactions—in contrast to QuikChange™, which requires two primers per mutation. This antiprimer can be designed in an area that avoids palindromes, hairpins, or overlapping regions with the mutagenic primer. Additionally, as HPLC-purified oligonucleotide primers are highly recommended for generating saturation mutagenesis libraries, the repeated use of one specific antiprimer for each template is very cost effective.
We engaged in this research in order to generate site-saturation mutagenesis libraries in difficult templates that could not be randomized with standard methods. The result of our work is that we have produced a generally applicable protocol where we have methodically defined parameters for number of cycles per stage, antiprimer position, and primer concentration, each of which contribute to the successful outcome of the reaction. Primer location and orientation as enhancers of amplification reaction are a new concept in optimizing this type of PCR, which provide a new option in cases where standard protocols and optimization methods have failed. We anticipate that the method we present here will be of considerable interest to molecular biologists and protein engineers.
References
Arndt KM, Müller KM (2007) Protein engineering protocols (Methods in molecular biology), vol. 352. Humana, Totowa
Arnold FH, Georgiou G (2003a) Directed enzyme evolution: Screening and selection methods (Methods in molecular biology), vol. 230. Humana, Totowa
Arnold FH, Georgiou G (2003b) Directed evolution library creation: methods and protocols. In: Methods in molecular biology, vol. 231. Humana, Totowa, p 75-84
Bartsch S, Kourist R, Bornscheuer UT (2008) Complete inversion of enantioselectivity towards acetylated tertiary alcohols by a double mutant of a Bacillus subtilis esterase. Angew Chem Int Ed 47:1508–1511 Angew Chem 120:1531−1534
Bershtein S, Tawfik DS (2008) Advances in laboratory evolution of enzymes. Curr Opin Chem Biol 12:151–158
Brakmann S, Schwienhorst A (2004) Evolutionary methods in biotechnology: clever tricks for directed evolution. Wiley, Weinheim
Cadwell RC, Joyce GF (1992) Randomization of genes by PCR mutagenesis. PCR Methods Appl 2:28–33
Cedrone F, Niel S, Roca S, Bhatnagar T, Ait-Abdelkader N, Torre C, Krumm H, Maichele A, Reetz MT, Baratti JC (2003) Directed evolution of the epoxide hydrolase from Aspergillus niger. Biocatal Biotransform 21:357–364
Dominy CN, Andrews DW (2003) Site-directed mutagenesis by inverse PCR. In: Casali N, Preston A (eds) Methods in molecular biology, vol. 235. Humana, Totowa, pp 209–223
Fox RJ, Huisman GW (2008) Enzyme optimization: moving from blind evolution to statistical exploration of sequence-function space. Trends Biotechnol 26:132–138
Fujii R, Nakagawa Y, Hiratake J, Sogabe A, Sakata K (2005) Directed evolution of Pseudomonas aeruginosa lipase for improved amide-hydrolyzing activity. Prot Eng Des Sel 18:93–101
Georgescu R, Bandara G, Sun L (2003) Saturation mutagenesis. In: Arnold FH, Georgiou G (eds) Directed evolution library creation. vol. 231. Humana, Totowa, pp 75–83
Hibbert EG, Baganz F, Hailes HC, Ward JM, Lye GJ, Woodley JM, Dalby PA (2005) Directed evolution of biocatalytic processes. Biomol Eng 22:11–19
Hogrefe HH, Cline J, Youngblood GL, Allen RM (2002) Creating randomized amino acid libraries with the QuikChange® multi site-directed mutagenesis kit. BioTechniques 33:1158–1165
Jaeger K-E, Schneidinger B, Rosenau F, Werner M, Lang D, Dijkstra BW, Schimossek K, Zonta A, Reetz MT (1997) Bacterial lipases for biotechnological applications. J Mol Catal B: Enzym 3:3–12
Kirsch RD, Joly E (1998) An improved PCR-mutagenesis strategy for two-site mutagenesis or sequence swapping between related genes. Nucleic Acids Res 26:1848–1850
Leung DW, Chen E, Goeddel DV (1989) A method for random mutagenesis of a defined DNA segment using a modified polymerase chain reaction. Technique (Philadelphia) 1:11–15
Li H-M, Mei L-H, Urlacher VB, Schmid RD (2008) Cytochrome P450 BM-3 evolved by random and saturation mutagenesis as an effective indole-hydroxylating catalyst. Appl Biochem Biotechnol 144:27–36
Liang L, Zhang J, Lin Z (2007) Altering coenzyme specificity of Pichia stipitis xylose reductase by the semi-rational approach CASTing. Microb Cell Fact 6:36
Liebeton K, Zonta A, Schimossek K, Nardini M, Lang D, Dijkstra BW, Reetz MT, Jaeger K-E (2000) Directed evolution of an enantioselective lipase. Chem Biol 7:709–718
Liebeton K, Zacharias A, Jaeger K-E (2001) Disulfide bond in Pseudomonas aeruginosa lipase stabilizes the structure but is not required for interaction with its foldase. J Bacteriol 183:597–603
Lutz S, Patrick WM (2004) Novel methods for directed evolution of enzymes: quality, not quantity. Curr Opin Biotechnol 15:291–297
Miyazaki K, Takenouchi M (2002) Creating random mutagenesis libraries using megaprimer PCR of whole plasmid. BioTechniques 33:1033–1038
Narhi LO, Fulco AJ (1982) Phenobarbital induction of a soluble cytochrome P-450-dependent fatty acid mono-oxygenase in Bacillus megaterium. J Biol Chem 257:2147–2150
Peters MW, Meinhold P, Glieder A, Arnold FH (2003) Regio- and enantioselective alkane hydroxylation with engineered cytochromes P450 BM-3. J Am Chem Soc 125:13442–13450
Reetz MT (2004) Controlling the enantioselectivity of enzymes by directed evolution: Practical and theoretical ramifications. Proc Natl Acad Sci U S A 101:5716–5722
Reetz MT (2006) Directed evolution of enantioselective enzymes as catalysts for organic synthesis. In: Gates BC, Knözinger H (eds) Advances in catalysis. vol. 49. Elsevier, San Diego, pp 1–69
Reetz MT, Carballeira JD (2007) Iterative Saturation Mutagenesis (ISM) for rapid directed evolution of functional enzymes. Nat Protoc 2:891–903
Reetz MT, Wilensek S, Zha D, Jaeger K-E (2001) Directed evolution of an enantioselective enzyme through combinatorial multiple cassette mutagenesis. Angew Chem Int Ed 40:3589–3591 Angew Chem 113:3701–3703
Reetz MT, Carballeira JD, Peyralans JJ-P, Höbenreich H, Maichele A, Vogel A (2006a) Expanding the substrate scope of enzymes: Combining mutations obtained by CASTing. Chem-Eur J 12:6031–6038
Reetz MT, Carballeira JD, Vogel A (2006b) Iterative saturation mutagenesis on the basis of B Factors as a strategy for increasing protein thermostability. Angew Chem Int Ed 45:7745–7751 Angew Chem 118:7909–7915
Reetz MT, Wang L-W, Bocola M (2006c) Directed evolution of enantioselective enzymes: Iterative cycles of CASTing for probing protein-sequence space. Angew Chem Int Ed 45:1236–1241 Erratum 2494, Angew Chem 118:1258–1263; Erratum 2556
Reetz MT, Puls M, Carballeira JD, Vogel A, Jaeger K-E, Eggert T, Thiel W, Bocola M, Otte N (2007) Learning from directed evolution: Further lessons from theoretical investigations into cooperative mutations in lipase enantioselectivity. ChemBioChem 8:106–112
Reymond J-L (2005) Enzyme assays—high-throughput screening, genetic selection and fingerprinting. Wiley, Weinheim
Rubin-Pitel SB, Zhao H (2006) Recent advances in biocatalysis by directed enzyme evolution. Comb Chem High Throughput Screening 9:247–257
Sarkar G, Sommer SS (1990) The “megaprimer” method of site-directed mutagenesis. BioTechniques 8:404–407
Stemmer WPC (1994) Rapid evolution of a protein in vitro by DNA shuffling. Nature (London, UK) 370:389–391
Stover CK, Pham XQ, Erwin AL, Mizoguchi SD, Warrener P, Hickey MJ, Brinkmann FSL, Hufnagle WO, Kowalik DJ, Lagrou M, Garber RL, Goltry L, Tolentino E, Westbrock-Wadman S, Yuan Y, Brody LL, Coulter SN, Folger KR, Kas A, Larbig K, Lim R, Smith K, Spencer D, Wong GK-S, Wu Z, Paulsen IT, Reizer J, Saier MH, Hancock REW, Lory S, Olson MV (2000) Complete genome sequence of Pseudomonas aeruginosa PAO1, an opportunistic pathogen. Nature (London, UK) 406:959–964
Tee KL, Schwaneberg U (2007) Directed evolution of oxygenases: Screening system, success stories and challenges. Comb Chem High Throughput Screening 10:197–217
Tseng W-C, Lin J-W, Wei T-Y, Fang T-Y (2008) A novel megaprimed and ligase-free, PCR-based, site-directed mutagenesis method. Anal Biochem 375:376–378
Wong TS, Arnold FH, Schwaneberg U (2008) Laboratory evolution of cytochrome P450 BM-3 monooxygenase for organic cosolvents. Biotechnol Bioeng 85:351–358
Zheng L, Baumann U, Reymond J-L (2004) An efficient one-step site-directed and site-saturation mutagenesis protocol. Nucleic Acids Res 32:e115
Acknowledgement
This research was supported by the German–Israeli Project Cooperation (DIP), the Deutsche Forschungsgemeinschaft (Schwerpunkt 1170; “Directed Evolution to Optimize and Understand Molecular Biocatalysis”; Project RE 359/13-1) and the Fonds der Chemischen Industrie.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
Joaquin Sanchis, Layla Fernández, and J. Daniel Carballeira contributed equally.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Sanchis, J., Fernández, L., Carballeira, J.D. et al. Improved PCR method for the creation of saturation mutagenesis libraries in directed evolution: application to difficult-to-amplify templates. Appl Microbiol Biotechnol 81, 387–397 (2008). https://doi.org/10.1007/s00253-008-1678-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-008-1678-9