
Abstract
Merck Research Laboratories has developed a highly productive Escherichia coli fermentation process to produce plasmid DNA for use as vaccines. The process consists of a fed-batch fermentation in a chemically defined medium. Initiation of the feed stream precedes a growth-limited phase in which plasmid DNA is amplified. The fermentation is only maximally productive for a small fraction of E. coli transformants designated as high-producers, while the predominant low-producer population does not amplify plasmid DNA. In experiments undertaken to probe this phenomenon, transposition of the 768-bp E. coli insertion sequence IS1 into an HIV DNA vaccine vector was observed in several low-producer clones. IS1 was found to insert in or near the neomycin resistance gene in nearly a dozen unique sites from within a single population of plasmid molecules. The fraction of IS1-containing plasmids within several clones was determined by quantitative polymerase chain reaction and was found to increase with increasing cultivation time in the chemically defined medium. Because transposition into an antibiotic-resistance gene is unlikely to affect plasmid amplification, the genomes of high- and low-producers of three different HIV DNA vaccine vectors were subsequently profiled by restriction fragment length polymorphism analysis. In all three cases, IS1 insertional mutations were found in the genomes of the predominant low-producers, while the genomes of the high-producers were indistinguishable from untransformed cells. The insertions reside on similarly sized fragments for two of the low-producer clones, and the fragment size is smaller for the third clone. The third clone also produces much less plasmid DNA than a typical low-producer. The results suggest the presence of an IS1 insertional mutation that affects plasmid replication and amplification, possibly in a position-dependent manner.
Similar content being viewed by others


Introduction
The use of genetic material as a delivery method for immunogens has generated substantial interest in the past several years. The genes that comprise such “DNA vaccines” may be integrated into a viral genome for delivery (Barouch et al. 2000). Alternatively, direct intramuscular injection of plasmid molecules as “naked DNA” can also elicit an immune response (Gurunathan et al. 2000). These novel approaches to vaccination were investigated in numerous laboratory and clinical studies to treat or prevent infectious diseases, including tuberculosis (Lowrie 1999), malaria (Tuteja 2002), influenza (Ulmer 2002), and HIV (Mwau and McMichael 2003). The ability to induce cellular and humoral immune responses has also led to the examination of DNA vaccines for the treatment of noninfectious diseases such as cancer (Leitner and Thalhamer 2003; Schirrmacher 2005). Interest in this vaccination technology further extends to veterinary medicine (Dunham 2002). Indeed, the first licensed DNA vaccines were recently approved in Canada and the United States for use in salmon and horses, respectively (Hensley 2005). The absence of licensed products for humans is due, in part, to low potency (Donnelly et al. 2003). In the case of naked DNA vaccines, milligram quantities of DNA per dose are required to mediate an immune response, and several doses may be required to induce protection. Several current clinical trials utilize both naked and viral-packaged DNA in a prime-boost strategy that may prove to be most effective for some therapeutic areas (Powell 2004).
The supply demands for naked DNA vaccines, either alone or as a primer for a viral boost, are such that a high-yielding production system is required for manufacturing scales. Naked DNA vaccines can be easily propagated as plasmid molecules in the well-studied gram-negative bacterium Escherichia coli (Prather et al. 2003). In particular, vectors based on ColE1/pMB1-derived plasmids such as pUC18/19 are especially attractive due to their high copy numbers per cell. It is well-established that plasmid copy number is inversely related to cell growth rate under a number of conditions for plasmids in the ColE1 incompatibility group (Seo and Bailey 1985, 1986; Lin-Chao and Bremer 1986; Klotsky and Schwartz 1987; Reinikainen et al. 1989; Reinikainen and Virkajärvi 1989; Namdev et al. 1993). Therefore, a reasonable framework for process design would combine a fast-growth stage to accumulate biomass and a slow-growth stage to maximize production of plasmid DNA (see Prather et al. 2003 for review). Two processes that utilize this strategy were patented in the US (Chen 1999; Schmidt et al. 2003). Both employ chemically defined growth media in fed-batch fermentations with nutrient feeding controlled by pH and dissolved oxygen concentration. Chen (1999) reported a tenfold increase in plasmid DNA concentration with a volumetric yield of up 0.10 g/l. Schmidt et al. (2003) reported yields of plasmid DNA up to 0.23 g/l. A two-phase fed-batch that also included a temperature shift resulted in volumetric yields of 1.1 g/l (Carnes 2005).
A fermentation process to produce plasmid DNA in high-yields was also developed within Merck Research Laboratories (MRL, West Point, PA, USA) (Chartrain et al. 2005; Okonkowski et al. 2005). Briefly, the process utilizes a chemically defined growth medium in a fed-batch fermentation to cultivate E. coli DH5 harboring a pUC-derived plasmid. The feed stream is initiated during midexponential growth and is maintained at a constant mass flow rate. Growth is slowed significantly soon after feeding is initiated, and plasmid amplification is subsequently observed. Volumetric yields greater than 1.3 g/l were observed (Okonkowski et al. 2005). This process has reliably provided material in high yields for several HIV DNA vaccine candidates (Lekutis et al. 1997; Casimiro et al. 2002; Caulfield et al. 2002; Liang et al. 2002).
The plasmid-transformed E. coli clones chosen for Master Cell Bank preparation are also selected through a process that involves DME-P5, the chemically defined growth medium used for fermentation. Untransformed DH5 cells that were adapted to DME-P5 are made electrocompetent following standard practices (Dower et al. 1988). These cells are electrotransformed with postshock recovery and nonselective outgrowth also occurring in DME-P5. Single colony isolates are selected from antibiotic-containing DME-P5 agar plates. While this clone selection process has the advantage of maintaining the cultures in a chemically defined growth medium from untransformed state through final fermentation, the transformation produces a heterogeneous population with respect to performance in the fed-batch process (Chartrain et al. 2005). The majority of the transformed clones do not amplify, giving specific yields that are an order of magnitude lower than desired. This heterogeneity necessitates a lengthy procedure to isolate the high-producer clones from a transformed population of predominately low-producers (Chartrain et al. 2005).
We began characterizing the clone selection process in an attempt to both understand the reasons for the appearance of a heterogeneous population and to propose alternatives that would result in increased fractions of high-producers. In the course of the ensuing experiments, we observed the E. coli insertion sequence IS1 in samples of plasmid DNA. The 768-bp IS1 is the smallest known transposable element among the bacterial insertion sequences (Ohtsubo and Sekine 1996). It is found naturally in E. coli genomes at copy numbers up to ten, with six to eight identified in wild-type K-12 strains (Deonier 1987). Upon transposition, a 9-bp duplication of the target sequence is usually generated at the site of insertion (Ohtsubo and Sekine 1996). The IS1s found in E. coli can be grouped into four types; however, only the IS1A/IS1E type was shown to transpose from chromosomal to plasmid DNA (Chen and Yeh 1997). IS1 causes spontaneous insertion mutations with much higher frequency than other insertion sequences (Ohtsubo and Sekine 1996) and was identified as the causative agent for mutations in both plasmid and chromosomal DNA. We therefore speculated that IS1 transposition may be the mechanism by which genetic mutations leading to the differentiation of high- and low-producers arise. In this report, we describe the initial identification and characterization of IS1 transposition in these DNA vaccine-producing E. coli clones and propose a model for the differentiation of clonal variants.
Materials and methods
Strains and growth media
E. coli DH5 [F−deoR recA1 endA1 hsdR17(rk −, mk +) supE44 λ−thi-1 gyrA96 relA1] was the host strain for all DNA vaccine vectors. The strain was originally purchased from Gibco BRL (now Invitrogen; Carlsbad, CA, USA) and was subsequently adapted to the chemically defined medium DME-P5. Preparation of electrocompetent cells and electrotransformation were performed according to standard practices (Dower et al. 1988). Unadapted, untransformed cells purchased from Invitrogen and maintained in Luria–Bertani (LB) medium were also employed. The chemically defined medium, DME-P5, contains the following: 7 g/l KH2PO4, 7 g/l K2HPO4, 6 g/l (NH4)2SO4, 5 g/l l-glutamic acid, 10 g/l glycerol, and 0.5 g/l NaCl, adjusted to pH 7.2 with NaOH. Following heat sterilization, 8.3 ml/l neomycin/thiamine/MgSO4 solution and 1 ml/l trace elements solution were added aseptically. The 120× neomycin/thiamine/MgSO4 solution contains 24 g/l thiamine–HCl, 240 g/l MgSO4·7H2O, and 9.6 g/l neomycin sulfate. The 1,000× trace elements solution contains 27 g/l FeCl3·6H2O, 2 g/l ZnCl2, 2 g/l CoCl2·6H2O, 2 g/l Na2MoO4·2H2O, 1 g/l CaCl2·2H2O, 1.3 g/l CuCl2·2H2O, and 0.5 g/l H3BO3 dissolved in 1.2 N HCl.
DNA vaccine plasmid vectors
Four HIV DNA vaccine vectors were examined: V1Jns-nef (5,540 bp), V1Jns-tpa-pol (7,516 bp), V1Jns-tpa-nef (5,540 bp), and V1Jns-tpa-gag (6,375 bp). All vectors were obtained from the Virus & Cell Biology Department of MRL and consist of the same backbone with various transgenes inserted (Montgomery et al. 1993). Briefly, each DNA vaccine plasmid consists of a pUC19-derived bacterial origin of replication and neomycin/kanamycin resistance gene (nptII) for maintenance and selection in E. coli; and a cytomegalovirus immediate early (CMV-IE) promoter, intron A, and bovine growth hormone terminator/polyadenylation signal for eukaryotic expression of the HIV-derived transgenes (Fig. 1). The three “tpa” vectors also carry a portion of the N-terminal sequence from the tissue plasminogen activator gene in the transgene sequence.

DNA vaccine vector V1Jns-nef with identified sites of IS1 transposon insertion (clone no. 5)
Preparation of DNA vaccine clones for quantitative polymerase chain reaction (Q-PCR) analysis
Cultures were propagated in 25-ml growth medium (LB or DME-P5), supplemented with 80 μg/ml neomycin, in 250-ml baffled shake flasks at 37 °C, 220 rpm in a Kuehner cabinet shaking incubator. DH5 (V1Jns-nef) was grown overnight in LB medium. A small aliquot of the LB culture was used to inoculate DME-P5 and cells were grown to stationary phase. The cells were passaged twice more in DME-P5 medium with harvesting in exponential phase. Each round of cultivation corresponded to approximately ten elapsed generations of growth. Samples from the first round LB medium cultures were also passaged in two additional rounds in LB medium. Plasmid DNA was isolated using the QIAGEN® QIAprep Spin Miniprep Kit (Valencia, CA, USA).
Preparation of samples for restriction fragment length polymorphism (RFLP) analysis
Samples of confirmed high-producers (working seeds) of the three vectors containing the tpa leader sequence were obtained from Bioprocess and Bioanalytical Research of MRL as frozen glycerol stocks. Low-producer clones of V1Jns-tpa-pol and V1Jns-tpa-nef were prepared by first transforming DME-P5-adapted E. coli DH5 with purified plasmid DNA. Transformants were recovered in and plated on DME-P5, and 5–10 single colonies were selected for growth in shake flasks as described previously. The cells were harvested in mid- to late exponential phase and were used to inoculate fresh growth medium. Cultures were passaged in this manner for a total of three rounds. The failure to amplify plasmid DNA in the candidate low-producers was confirmed by fed-batch cultivation in shake flasks. A single clone for each construct was stored as a frozen glycerol stock and designated the representative low-producer. The low-producer clone of V1Jns-tpa-gag, along with a second high-producer culture, was isolated several years before. These laboratory seed cultures had been maintained as frozen glycerol stocks to serve as control cultures for the fed-batch fermentations. Twenty-five-microliter aliquots of each of the frozen glycerol stocks (high- and low-producers) were used to inoculate 25 ml of DME-P5 in 250-ml shake flasks. Cells were grown overnight at 37 °C, 220 rpm in a Kuehner cabinet shaker and harvested for isolation of DNA in mid- to late exponential phase. Total DNA was isolated using the Promega Wizard® Genomic DNA Purification Kit (Madison, WI, USA). Plasmid DNA from each high- and low-producer sample was isolated using the QIAGEN QIAprep Spin Miniprep Kit.
Real time Q-PCR
Primers and probes were designed using Primer Express software version 2.0 from Applied Biosystems (Foster City, CA, USA). Unlabeled primers were purchased from Sigma-Genosys (The Woodlands, TX, USA). TaqMan probes labeled with either 6-carboxyfluorescein (FAM) or the proprietary dye “VIC” at the 5′-end and 6-carboxytetramethylrhodamine (TAMRA) at the 3′-end were purchased from Applied Biosystems. The primers and probe used to quantify the CMV promoter are as follows (working concentrations in the multiplex assay are indicated in parentheses): CMV-Q-F, 5′-GTACGGTGGGAGGTCTATATAAGCA (100 nM); CMV-Q-R, 5′-GGAGGTCAAAACAGCGTGGAT (100 nM); and CMV-Q-P2, 5′-VIC-TCGTTTAGTGAACCGTCAGATCGCCTG-TAMRA (200 nM). The primers and probe used to quantify the transposon IS1 are as follows: IS1-Q-F, 5′-AGGCTCATAAGACGCCCCA (500 nM); IS1-Q-R, 5′-ACGGTTGTTGCGCACGTAT (500 nM); and IS1-Q-P2, 5′-FAM-CGTCGCCATAGTGCGTTCACCG-TAMRA (200 nM). The CMV and IS1 primer-probe sets were run in multiplex mode to quantify transposon (IS1) copies relative to total plasmid (CMV) copies. A plasmid that included copies of IS1 and the CMV promoter in a 1:1 ratio (pnIQ3v2) was constructed and used as an internal standard. Amplification and fluorescence detection of the samples was performed in an ABI 7900HT Sequence Detection System (SDS) (Applied Biosystems) under the following thermal cycler conditions: 50 °C for 2 min, 95 °C for 10 min, followed by 40 cycles of 95 °C for 15 s, and 60 °C for 1 min. Samples were run in a 384-well plate with four to six replicates per experimental sample and six replicates of a tenfold dilution series of the pnIQ3v2 standard. Data analysis was completed using the ABI Prism 7900HT SDS version 2.0 software. Relative quantities of IS1 (IS1/CMV ratios) were calculated using the \(2^{{ - \Delta \Delta C_{T} }}\) method (Livak and Schmittgen 2001).
RFLP analysis
A nonradioactive IS1-specific probe (0.77 kb) was created using the PCR DIG Probe Synthesis Kit (Roche Molecular Biosystems, Mannheim, Germany). Primers were obtained from Sigma-Genosys with sequences as follows: IS1-F2, 5′-GGTAATGACTCCAACTTATTG and IS1-R2, 5′-GGTGATGCTGCCAACTTA. Restriction enzymes were purchased from New England Biolabs (Beverly, MA, USA). Total DNA was digested with AflII and AgeI; plasmid DNA was digested with AflII. (There are no AgeI sites on the plasmid.) Digested samples were run on 0.7% agarose gels overnight (∼16 h) at 34 V, 4 °C. DNA was transferred onto Nytran SuPerCharge nylon membranes (Schleicher and Schuell, Keene, NH, USA) for 1 h using the Turboblotter™ Rapid Downward Transfer System (Schleicher and Schuell). DNA was crosslinked to membranes by UV irradiation at 150 mJ using the BioRad GS Gene Linker® (Hercules, CA, USA). The DIG-labeled IS1 probe was hybridized to the target DNA on Southern blots following the Filter Hybridization Protocol with overnight incubation (Roche Molecular Biosystems). Probe-target hybrids were visualized by an enzyme-linked chemiluminescent assay using an anti-DIG alkaline phosphatase antibody and disodium 3-(4-methoxyspiro {1,2-dioxetane-3,2′-(5′-chloro)tricyclo [3.3.1.13,7]decan)-4-yl)phenyl phosphate (CSPD) (Filter Hybridization Protocol, Roche Molecular Biosystems).
Results
Identification and localization of insertion sequence IS1 in DNA vaccine vectors
The use of a chemically defined (i.e., “minimal”) medium for preparation of electrocompetent cells and subsequent outgrowth results in a suboptimal environment for transformation. This is reflected in typical transformation efficiencies on the order of 105–106 colony-forming units per microgram (cfu/μg) plasmid DNA. To improve the yield at this step, previously adapted cells were reintroduced to LB medium and made electrocompetent. The cells were transformed with the V1Jns-nef HIV DNA vaccine vector. Transformation with LB-grown cells did show a significant improvement, having an efficiency on the order of 109 cfu/μg. Ten well-isolated clones from this transformation were randomly selected for expansion in LB then DME-P5 media. The cells were then tested for the ability to amplify plasmid DNA in fed-batch cultivation, and all ten were found to be low-producers.
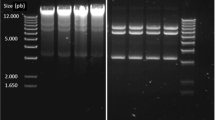
Plasmid DNA was isolated from each of the ten clones after the third round of readaptation to DME-P5 and was visualized by agarose gel electrophoresis. Examination of the plasmid DNA revealed several anomalies (Fig. 2). First, clone no. 8 contained a major, lower molecular weight species in addition to the primary V1Jns-nef plasmid DNA bands. This is most likely the result of an intramolecular rearrangement with exclusion of DNA sequences from the original molecule. Secondly, clone no. 10 appears to exist predominantly as a dimer. Comparison of this sample with plasmid DNA taken from LB-grown cells before the medium shift indicates that the dimer was present after the transformation (data not shown). Lastly, several of the samples contained a minor, higher molecular weight species that was not evident in LB-grown cultures. This suggested the presence of a transposon in a fraction of the plasmid DNA. Restriction digestion and PCR analysis indicated that the insertion was likely IS1. IS1 is present in the genome and would be expected to amplify from most plasmid DNA preparations due to small amounts of residual genomic DNA (Vilalta et al. 2002). To confirm the presence of plasmid-borne IS1 by PCR, one IS1-specific primer was paired with one of the several plasmid-specific primers. Several amplified fragments resulted (data not shown), providing further evidence that the higher molecular weight species observable in these samples is the plasmid V1Jns-nef with an IS1 transposon insertion.

Agarose gel electrophoresis of plasmid DNA samples after ∼30 generations of growth in the defined medium DME-P5. Preparation and transformation of electrocompetent E. coli was accomplished in complex medium. Lanes 1, 7, and 13: 1 kb ladder molecular weight marker (New England Biolabs). Lanes 2–6: clones 1–5. Lanes 8–12: clones 6–10. Higher molecular weight plasmid bands near 4 kb, an indication of transposon insertion, are visible for clones 1, 3, 5, 7, and 8
Confirmation of IS1 insertion and determination of the locations of insertions within the plasmid DNA were accomplished through a series of PCR reactions with one IS1-specific primer and one V1Jns-nef-specific primer, followed by sequencing several of the amplified products. The plasmid DNA of clone no. 5 was selected for characterization because a large fraction of the plasmid DNA appeared to be affected. Both partial and full junctions were resolved, and sequencing results confirmed that the transposon produces a 9-bp target duplication at the site of insertion (Ohtsubo and Sekine 1996). Eleven unique integration sites were identified with insertion of the transposon in either orientation (Fig. 1). One site of insertion was observed in both orientations. All sites were in or within 85 bp of the coding region of the nptII (neoR) gene.
Q-PCR analysis of plasmid DNA extracted from IS1-containing plasmids
The presence of IS1 in these DNA vaccine vector samples provided the first lead toward a mechanism by which high- and low-producers might arise. To further explore the link between transposition and productivity, multiplex Q-PCR was used to determine the fraction of IS1-containing plasmid molecules in each of the ten samples. Total plasmid molecules were quantified by amplifying a portion of the CMV promoter known to be free of insertional mutations. IS1 fractions are calculated as the ratio of IS1/CMV copies.
The first plasmid DNA samples tested were isolated from the aliquots of cells collected at the end of each round of cultivation in the DME-P5 medium. The geometric mean fraction of IS1 in the plasmid DNA increased from an average of 0.06% after the first round to 4% after the third round (Fig. 3, white bars). After the second round (∼20 generations), the samples were very similar to the final round with nine out of ten samples exhibiting IS1/CMV ratios of >1%, and half ≥5%. This result suggested that transposition activity was a function of the cultivation time. To further investigate the time-dependence, the initial LB cultures for all ten samples were grown for an equivalent number of generations in LB medium (i.e., 3 rounds in shake flasks with ∼10 generations per round). Q-PCR analysis of plasmid DNA from these cells indicates that the IS1/CMV ratios varied slightly from a geometric mean of 0.04% after round 1 to 0.06% after round 3 (Fig. 3, dark bars). This suggests that the high transposition activity is specifically associated with prolonged growth in DME-P5.

IS1/CMV copy ratios in V1Jns-nef plasmid DNA samples at the end of each of three rounds of shake flask passaging in either DME-P5 or LB medium (∼10 generations per round). Shaded bars (left): clones previously exposed to DME-P5, transformed and propagated in LB medium. Open bars (center): clones previously exposed to DME-P5, transformed in LB medium, propagated in DME-P5 medium. Striped bars (right): naïve clones transformed in LB medium and propagated in DME-P5 medium. Data is presented in groups of ten, representing clones 1–10, respectively, for each round
Although the samples that displayed a high transposon fraction were transformed and recovered in complex medium, the source of the untransformed cells was a sample of DME-P5-adapted DH5. Therefore, the total exposure time to DME-P5 over the lifetime of the cells was longer than the roughly 30 generations of shake flask cultivation. To determine the impact of preexposure to the defined medium on transposition activity, the medium shift experiment was repeated using E. coli DH5 that had not been previously exposed to DME-P5. Q-PCR assay results indicated that while the preexposed samples had a significant increase in IS1 levels after two rounds in DME-P5, the samples from naïve cells showed almost no increase in IS1 (Fig. 3, striped bars). Only one of the samples contained IS1/CMV copy ratios above 0.1% after any of the three rounds of shake flask passaging. The geometric mean IS1 fraction of these samples was 0.02% in round 3. The Q-PCR assay cannot distinguish between plasmid-based IS1 and that which comes from residual genomic DNA. Plasmid DNA purified with QIAGEN columns may contain up to 3.3% genomic DNA by weight (Vilalta et al. 2002). IS1 is reported to be present six to eight copies in the E. coli genome (Ohtsubo and Sekine 1996). Assuming an average of seven copies of IS1 per genome (see following RFLP analysis), this residual genomic DNA would be reflected as an IS1/CMV ratio of ∼0.03%. Thus, the IS1 present in the nine naïve plasmid samples with fractions <0.1% is likely primarily, if not exclusively, contributed by residual genomic DNA. These results indicate that prior adaptation to the defined medium has a significant impact on the transposition activity in the clones.
IS1 RFLP analysis of high- and low-producer clones
The discovery of an insertion sequence in DNA vaccine vectors suggested that IS1 could be implicated in the differentiation of high- and low-producers. However, it is unlikely that a transposon present in the antibiotic resistance gene of a plasmid, and within 20% or less of the total plasmid population, would result in the observed suppression of plasmid amplification. The genomic DNAs of high- and low-producer clones of three different HIV DNA vaccine vectors were therefore profiled using RFLP analysis to determine whether genome-based mutations were also evident. These three systems were chosen as a representative set of vaccine vectors with different transgenes, for which high-producers had been selected using the same screening protocol as the V1Jns-nef vector profiled by Q-PCR. In addition, high-producing working seed stocks of all three clones, prepared for use in large-scale manufacturing, were available. Restriction enzymes AflII and AgeI were chosen to produce the restriction fragments for analysis, and comparisons were made to genomic DNA from untransformed DH5. Given the Q-PCR results, the untransformed cells were grown in LB medium with no prior adaptation to DME-P5 to isolate potential medium effects at the genomic DNA level. This plasmid-free control was used to establish the baseline number of IS1 sequences in the DH5 genome. The untransformed DH5 profile shows six IS1-containing fragments (Fig. 4a, lane 2). The lowest molecular weight band is approximately twofold more intense than the other bands, suggesting that this fragment may contain more than one IS1 insertion sequence, or that two similarly sized restriction fragments each contain a copy of the transposon. The fragment is less than 2 kb but is large enough to accommodate two 768-bp IS1 insertions. To determine whether the IS1 copy number is 6 or 7, a second blot using restriction enzymes BssHII and AgeI was prepared (data not shown). Seven IS1-containing fragments were evident in this blot; therefore, we believe the IS1 copy number of untransformed DH5 is 7. This is consistent with expectations for a K-12 derived strain of E. coli (Ohtsubo and Sekine 1996), and is equal to the number of IS1 insertions found in the fully sequenced genomes of the K-12 strains MG1655 and W3110 (GenBank accession nos. U00096 and NC_000913, respectively).

IS1 RFLP profiles of a V1Jns-tpa-pol and b V1Jns-tpa-nef clones. Lane 1: IS1-positive control. Lane 2: plasmid-free DH5 control. Lane 3: HP plasmid DNA. Lane 4: HP total DNA. Lane 5: LP plasmid DNA. Lane 6: LP total DNA. The DIG-labeled molecular weight marker is not shown here but is visible with longer exposure times
V1Jns-tpa-pol and V1Jns-tpa-nef clones
AflII-AgeI RFLP profiles were obtained with total DNA from one high-producer and one low-producer clone each of vectors V1Jns-tpa-pol (tpa-pol-HP and tpa-pol-LP, respectively) and V1Jns-tpa-nef (tpa-nef-HP and tpa-nef-LP). AflII-digested, linear plasmid DNA isolated from each of the clones was also included on the blots. All clones were maintained in the defined DME-P5 medium from the transformation of adapted cells through subsequent cultivations and extraction of genomic and/or plasmid DNAs. The tpa-pol-HP total DNA profile sample is consistent with the plasmid-free control sample (Fig. 4a, lanes 2 and 4). No IS1 is evident in the plasmid DNA extracted from tpa-pol-HP (lane 3). The tpa-pol-LP total DNA profile contains two additional fragments not found in the other samples (lane 6). The higher molecular weight band of these two is consistent with the plasmid DNA extracted from tpa-pol-LP (lane 5). The smaller, 3- to 4-kb band is of comparable intensity as the other IS1-positive bands in the tpa-pol-LP profile and is distinct. The profiles of the tpa-nef clones were similar to those obtained with the V1Jns-tpa-pol samples (Fig. 4b). The tpa-nef-HP total DNA profile sample is consistent with the plasmid-free control sample (lanes 2 and 4), and no IS1 is evident in the plasmid DNA extracted from tpa-nef-HP (lane 3). Binding of the IS1 probe to tpa-nef-LP plasmid DNA is also observed in both the plasmid and total DNA samples (lanes 5 and 6), although the plasmid::IS1 band partially overlaps with one of the genomic DNA bands. There remains a band that is not found in either the DH5 control or the tpa-nef-HP sample that is approximately the same size as the unidentified band observed in the tpa-pol-LP sample above. This suggests a common site of IS1 insertional mutation in these low-producer genomes.
V1Jns-tpa-gag clones
In addition to the working seed, a second high-producer clone of V1Jns-tpa-gag was examined. This clone was isolated through the same screening protocol, but had been subcultured repeatedly over several years to serve as an amplification control for the fed-batch fermentation. A low-producer control culture had been similarly maintained to serve as the negative control. The total cultivation time of these clones (designated lab seeds) in DME-P5 is therefore much higher than that of the tpa-pol and tpa-nef clones. Both the working and lab seeds were profiled. The results indicate that the IS1 profiles of total DNA from both high-producer clones are similar (Fig. 5), although there is a faint band in the lab seed total DNA sample (lane 6) that is consistent with plasmid DNA (lane 5). A much lighter band is just visible in the plasmid DNA of the working seed (lane 3). These results indicate that IS1 may be found in high-producer plasmid DNA as well, and the fraction of IS1-containing plasmids may increase with increasing cultivation time in the defined medium. While this dependence of IS1 fraction on cultivation time is consistent with the Q-PCR results obtained with low-producer plasmid DNA, it should be noted that the concentration of IS1 in the high-producer plasmids is much less than that observed in low-producers. The genome profiles of both high-producer samples are also similar to the plasmid-free DH5 control (lane 1).

IS1 RFLP profiles of untransformed DH5 and V1Jns-tpa-gag clones. Lane 1: plasmid-free DH5 control. Lane 2: plasmid-free DH5 adapted to defined medium DME-P5. Lane 3: tpa-gag-HP working seed plasmid DNA. Lane 4: tpa-gag-HP working seed total. Lane 5: tpa-gag-HP laboratory seed plasmid DNA. Lane 6: tpa-gag-HP laboratory seed total DNA. Lane 7: tpa-gag-LP plasmid DNA. Lane 8: tpa-gag-LP total DNA. The DIG-labeled molecular weight marker is not shown here but is visible with longer exposure times
A tpa-gag low-producer equivalent to the tpa-pol and tpa-nef samples described previously was not available, so only the lab seed was evaluated. As with tpa-pol-LP and tpa-nef-LP, an additional band is present in the tpa-gag-LP total DNA IS1 profile that does not correspond to plasmid DNA (lane 8). However, this band is smaller than that observed in the other clones (∼2–3 vs ∼3–4 kb). Unlike tpa-pol and tpa-nef, the IS1 probe did not bind to plasmid DNA isolated from the tpa-gag-LP sample (lane 7). The tpa-gag low-producer lab seed is further distinguished from the tpa-pol and tpa-nef low-producers in both genetic and physiological ways. First, an examination of plasmid DNA on agarose gels reveals an obvious difference between high- and low-producers of tpa-gag (Fig. 6). The low-producer plasmid DNA consists of a mixed population with one group of molecules of the expected size (6.4 kb) and one group that is approximately 1 kb larger. Sequencing indicated that the higher molecular weight species is V1Jns-tpa-gag with the neomycin resistance gene disrupted by the 1,195-bp E. coli insertion sequence IS5 (data not shown). Thus, while tpa-gag-LP plasmid DNA does not contain IS1, it does harbor a different insertion sequence. Secondly, although low-producers are characterized by an inability to amplify plasmid DNA in the fed-batch fermentation, there is usually no significant difference between the DNA content of high- and low-producers before the amplification period. By contrast, the tpa-gag-LP was observed to have a significantly lower plasmid copy number during batch growth and a final plasmid content that is an order of magnitude lower than other low-producers, including the representative clones of tpa-pol and tpa-nef. Therefore, it is possible that the insertional mutation found in tpa-gag-LP is distinct from those found in tpa-pol-LP and tpa-nef-LP, and results in a copy-number suppressed phenotype that has an even greater impact on the ability to amplify plasmid DNA.

Plasmid DNA extracted from high- and low-producer clones of V1Jns-tpa-gag. Lanes 1 and 6: New England Biolabs 1 kb ladder. tpa-gag-HP plasmid DNA: lane 2, uncut and lane 3, linearized with SacI. tpa-gag-LP plasmid DNA: lane 4 uncut and lane 5, linearized with SacI. The higher molecular weight band evident in the linearized sample of the low-producer plasmid DNA was identified as V1Jns-tpa-gag with an IS5 insertion in the neomycin resistance gene by sequencing
E. coli DH5 adapted to DME-P5
The IS1 profiles of high- and low-producers of three different DNA vaccine constructs indicated that all three low-producer clones contain an IS1 insertional mutation, whereas high-producers are similar to plasmid-free DH5. Q-PCR analysis suggested that adaptation of untransformed DH5 to the defined medium DME-P5 resulted in a higher frequency of transposition after transformation with DNA vaccine plasmids. Because Q-PCR cannot distinguish between IS1 localized on a plasmid molecule and that in the genome, it is also possible that the adaptation results in an increase in the copy number or change in location of IS1 in the genome. To examine this, genomic DNA extracted from DH5 that had been adapted to the defined medium DME-P5 was compared to the plasmid-free DH5 control that was propagated in LB medium without prior exposure to DME-P5. The RFLP results indicate that there are no significant differences between the locations of IS1 in these genomes (Fig. 5, lanes 1 and 2). Three faint bands are evident in the naïve, unexposed DH5 genome (lane 1). Given (1) the absence of these bands in either of the previous blots, (2) their faint appearance relative to the other bands, and (3) the increased concentration of this sample relative to the others on this and the previous blots, these bands are most likely a reflection of incomplete digestion of the genomic DNA. We, therefore, conclude that low-producers are correlated with a single IS1 insertional mutation within the host genome and that this insertion occurs after the transformation step.
Discussion
Development of a manufacturing process capable of delivering high volumetric yields of plasmid DNA is critical for the successful commercialization of naked DNA vaccines for human therapeutics. While animal health vaccines were licensed with doses as low as the microgram range, vaccines for human use are projected to require milligram doses, potentially with multiple doses per patient. The demand for vaccine for hundreds of thousands to millions of patients drives the need for a high-yielding process. Merck has developed a fed-batch fermentation process using a chemically defined medium that facilitates amplification of plasmid DNA and results in high specific and volumetric productivities. However, a limitation of this process is the observation that not all transformed clones perform well in the fermentation. The typical fraction of high-producing clones from a single transformation event is ∼0.1%, while the remaining >99% of the population fails to amplify plasmid DNA. In experiments undertaken to understand the reasons for this heterogeneity and potentially improve the yield of high-producers, we identified a link between low-productivity and IS1 insertional mutations.
IS1 was originally found within a 475-bp hotspot in the plasmid DNA that includes the 5′ half of the neomycin resistance coding sequence and ∼100 bp upstream. We observed a preference for insertion sites in which the flanking bases of the 9-bp duplication sequence were G-C pairs (8/11 with two G-C ends, 1/11 with one G-C end) as previously reported (Galas et al. 1980). IS1 insertions were also reported to preferentially occur in adenosine–thymidine (AT)-rich regions (Galas et al. 1980; Zerbib et al. 1985). We found a similar preference; the hotspot has an AT content of 57% compared to 48% for the full plasmid. We did not observe an orientation bias. Loss of antibiotic resistance due to insertional inactivation of the resistance gene by IS1 was reported previously, for example, for tet and rpsL (Chew et al. 1986; Toba-Minowa 1992). Inactivation occurred in the absence of the antibiotic and was presumed to provide an advantage to the host cell by reducing toxicity associated with the expressed genes. In our case, inactivation of the neomycin resistance gene was found when the antibiotic was present.
IS1 insertions in the genomic DNA of three sets of DNA vaccine clones were profiled by examining RFLPs. The RFLP results revealed that the IS1 profiles of high-producer genomic DNA were similar to that of the untransformed host strain. Each of the low-producers contained one additional IS1-positive genomic DNA fragment. This is consistent with the Q-PCR analysis that associated IS1 transposition with low-producers. Although the V1Jns-tpa-pol and V1Jns-tpa-nef plasmid DNA from each of the respective low-producers was positive for IS1, no insertions were evident in the low-producer plasmid DNA on ethidium bromide-stained agarose gels (data not shown). Based on the previous Q-PCR assays, the IS1-containing fraction of this low-producer plasmid DNA is on the order of 1% or less. Plasmid DNAs from two high-producer cultures of V1Jns-tpa-gag were also found to contain a low-level of IS1-positive molecules with a higher fraction among cells with a longer exposure time in the defined medium. In addition, a predecessor to the V1Jns-based vaccine vectors was found to contain a high fraction of IS1 while maintaining high-productivity (P. Duncan, personal communication). Because insertion sequences were found in the plasmid DNAs from both high- and low-producers, we believe that the insertion of a transposon in the plasmid does not result in the differentiation of high- and low-producers. However, the insertional mutations found in the genomic DNA may arise from a plasmid-based intermediate (Itoh et al. 1994).
The presence of IS1 insertional mutations in the genomes of each of these clones provides strong evidence for transposition as a candidate mechanism by which differentiation occurs. IS1 was implicated in a number of mutations of both plasmid and genomic DNA. These mutations were shown to suppress (fully or partially) the expression of toxic or stress-inducing genes (Nakamura and Inouye 1981; Nakahama et al. 1986; Toba-Minowa 1992), activate gene expression by disruption of regulators or distal transcriptional readthrough (Hall 1998; Kobayashi et al. 2001), increase resistance of the host cell to heavy metals (Itoh et al. 1994), and enhance plasmid segregational stability (Chew et al. 1986). The additional presence of IS5 in the V1Jns-tpa-gag low-producer plasmid DNA meant that both IS1 and IS5 had to be considered as the causative agent(s). IS5 is also known to cause insertional mutations, although the frequency of transposition is generally much lower than with IS1 (Rodriguez 1992; Kitamura et al. 1995; Hall 1998). The exception appears to be in resting (stab) cultures of E. coli where the IS5 transposition activity is higher (Naas et al. 1994, 1995). There are also many more IS5 insertions naturally present in E. coli with estimates of up to 20 or more copies per genome, while the IS1 copy number is typically 6 to 8 in K-12 strains (Deonier 1987). IS5 RFLP profiles of all three DNA vaccine constructs were examined, and at least 15 bands of varying intensities were apparent in the total DNA of each sample (data not shown). As with IS1, all IS5 high-producer profiles were similar to untransformed DH5. Among the low-producers, only the total DNA from the V1Jns-tpa-nef clone was distinct, with one band shift among AflII–AgeI digested fragments. This shift could represent a relocation of one of the insertion sequences or a mutation affecting the restriction sites. The total number of IS5 insertions remained the same. Among the plasmid DNAs, only the V1Jns-tpa-gag low-producer carried IS5.
Although differences were observed in the IS5 profile of one low-producer relative to the high-producers and untransformed DH5, the lack of a consistent pattern among several clones makes it unlikely that IS5 mutations cause differentiation. In contrast, the IS1 insertional mutations in V1Jns-tpa-pol and V1Jns-tpa-nef low-producers are contained on similarly sized, 3- to 4-kb fragments. The mutation in the V1Jns-tpa-gag low-producer is found on a smaller fragment (2–3 kb). The smaller fragment size could represent a unique site of insertion, or it could be a reflection of a point mutation nearby that altered the restriction patterns. However, the phenotype of the gag clone is also different, with a lower plasmid copy number both before and after the amplification stage. Therefore, we contend that insertional mutations caused by IS1 result in the differentiation of transformants and that these mutations lead to the creation of a large pool of low-producers, leaving a few high-producers intact.
In an attempt to determine whether the transformant heterogeneity was also observed with a similar plasmid, the control vector pUC19, an ancestor of V1Jns, was used to transform DME-P5-adapted E. coli DH5. It is interesting to note that the transformants were unable to grow on solid defined medium directly after the transformation. Colonies could be obtained on complex solid medium with similar transformation efficiency as the V1Jns-based constructs. DH5(pUC19) clones could be prepared in a complex medium then adapted to the defined medium in liquid culture. In this case, a significant lag time was observed and agarose gel electrophoresis analysis showed that plasmid DNA content was decreased ∼5-fold after the shift from LB to DME-P5 media (data not shown). This is inconsistent with established reports that clearly indicate a correlation between increased copy number and decreased growth rate due to changes in the growth medium (Seo and Bailey 1985; Lin-Chao and Bremer 1986; Klotsky and Schwartz 1987). The pUC19 control clones were also examined for the presence of IS1 insertional mutations and were found to be similar to untransformed DH5 although all were also low-producers (data not shown). The failure of transformants to grow directly on defined medium agar and copy-number suppression were also observed with a modified pUC19 vector containing the same neomycin resistance gene and neighboring sequences found in V1Jns that encompass the hotspot for IS insertion. This suggests that IS1 transposition is affected by the overall plasmid DNA sequence. Boyd et al. (1999) observed the insertion of IS1 in a gene therapy vector with the structural instability leading to enhanced segregational stability. It may be that the presence of eukaryotic elements, perhaps the CMV-IE promoter found in both this gene therapy and our DNA vaccine vectors, induces IS1 transposition in the E. coli clones. IS1 transposition may also be an adaptive mutation for the survival of the majority of the population of transformed low-producers in the DME-P5 medium (Hall 1998). The mutation(s) may result in the ability of clones to maintain plasmid DNA in batch cultivation but an inability to amplify plasmid DNA in fed-batch mode. The absence of the sequences that induce transposition could then result in DH5(pUC) transformants that are not able to grow directly on DME-P5 agar.
Q-PCR analysis indicated that host cells adapted to growth in the defined medium exhibited increased transposition of IS1 into plasmid DNA, while the RFLP analysis revealed that the insertional mutations occur after the transformation step. Our findings suggest that the appearance of the heterogeneous transformant population is in some way directly related to the choice of plasmid used for transformation, and is also influenced by the choice of the defined growth medium. We did not find any link to the method of transformation employed, i.e., electroporation- vs CaCl2-mediated competency. It should be noted that the data presented here point to low-producers as being mutated and high-producers as being “wild-type,” yet high-producers comprise only ∼0.1% of a typical transformant population. A typical transformation is performed with ∼109 cells that were adapted to defined medium DME-P5, plus plasmid DNA present in saturating quantities. The usual yield of clones on defined medium agar is 106 transformants; thus, only 1 in 103 of the initial cells are both transformed and viable. Of the survivors, 1 in 103 is a high-producer. Of the total starting population, 1 in 106 is a high-producer. The latter is more consistent with typical random mutation rates.
We propose the following model for the differentiation of high- and low-producers. First, the adaptation of untransformed E. coli DH5 to the defined medium results in priming of the host cells for IS1 transposition. Second, transformation of the host cells with V1Jns-based plasmids results in IS1 transposition. This transposition enables the survival of transformants in the DME-P5 medium, but the majority (∼99.9%) is unable to amplify plasmid DNA in fed-batch mode. The remaining 0.1% do not contain IS1 insertional mutations and retain the high-producer character that results in amplification of plasmid DNA after fed-batch cultivation with an extended slow-growth phase. Identification of the additional site(s) of IS1 insertion in the low-producers of each of the three DNA vaccine clones would provide valuable information toward confirming and refining this model for the differentiation of high- and low-producers.
The implications of plasmid copy-number suppression may extend beyond the production of DNA vaccines. ColE1-type plasmids are frequently employed for recombinant protein production, as either a product of interest or as a catalyst to mediate transformation of a desired biochemical. The use of well-developed, chemically defined media in fermentations can provide advantages for process development, including increased productivity, lot-to-lot consistency, and simplified analytical descriptions (Zhang and Greasham 1999). Our findings that copy-number suppression (and, perhaps, a concomitant decrease of recombinant protein expression) is linked to the growth medium may have implications for the design of media for high cell-density fermentations to manufacture other biological or biochemical products. Ultimately, understanding of the genetic basis for the high-producer phenomenon and the role of the chemically defined medium therein could result in both the implementation of an improved clone selection process for DNA vaccines and critical knowledge of host–plasmid interactions that may affect the productivity of fermentation processes at large scale.
References
Barouch DH, Santra S, Schmitz JE, Kuroda MJ, Fu TM, Wagner W, Bilska M, Craiu A, Zheng XX, Krivulka GR, Beaudry K, Lifton MA, Nickerson CE, Trigona WL, Punt K, Freed DC, Guan L, Dubey S, Casimiro D, Simon A, Davies ME, Chastain M, Strom TB, Gelman RS, Montefiori DC, Lewis MG, Emini EA, Shiver JW, Letvin NL (2000) Control of viremia and prevention of clinical AIDS in rhesus monkeys by cytokine-augmented DNA vaccination. Science 290:486–492
Boyd AC, Popp F, Michaelis U, Davidson H, Davidson-Smith H, Doherty A, McLachlan G, Porteous DJ, Seeber S (1999) Insertion of natural intron 6a–6b into a human cDNA-derived gene therapy vector for cystic fibrosis improves plasmid stability and permits facile RNA/DNA discrimination. J Gene Med 1:312–321
Carnes AE (2005) Fermentation design for the manufacture of therapeutic plasmid DNA. BioProcess Int 3:36–44
Casimiro DR, Tang A, Perry HC, Long RS, Chen M, Heidecker GJ, Davies M-E, Freed DC, Persaud NV, Dubey S, Smith JG, Havlir D, Richman D, Chastain MA, Simon AJ, Fu T-M, Emini EA, Shiver JW (2002) Vaccine-induced immune responses in rodents and nonhuman primates by use of a humanized Human Immunodeficiency Virus Type 1 pol gene. J Virol 76:185–194
Caulfield MJ, Wang S, Smith JG, Tobery TW, LIu X, Davies M-E, Casimiro DR, Fu T-M, Simon A, Evans RK, Emini EA, Shiver JW (2002) Sustained peptide-specific gamma interferon T-cell response in Rhesus macaques immunized with human immunodeficiency virus gag DNA vaccines. J Virol 76:10038–10043
Chartrain M, Bentley LK, Krulewicz BA, Listner KM, Sun W-J, Lee CB (2005) Process for large scale production of plasmid DNA by E. coli fermentation. WO 2005/078115. Merck, USA
Chen W (1999) Automated high-yield fermentation of plasmid DNA in Escherichia coli. US Patent 5,995,323 (American Home Products)
Chen J-H, Yeh H-T (1997) The seventh copy of IS1 in Escherichia coli W3110 belongs to the IS1A(IS1E) type which is the only IS1 type that transposes from chromosome to plasmids. Proc Natl Sci Counc Repub China B 21:100–105
Chew LCK, Tacon WCA, Cole JA (1986) Increased stability of maintenance of pAT153 in Escherichia coli HB101 due to transposition of IS1 from the chromosome into the tetracycline resistance region of pAT153. FEMS Microbiol Lett 36:275–280
Deonier RC (1987) Locations of native insertion sequence elements. In: Neidhardt F (ed) Escherichia coli and Salmonella typhimurium: cellular and molecular biology. American Society for Microbiology, Washington, DC, pp 982–989
Donnelly J, Berry K, Ulmer JB (2003) Technical and regulatory hurdles for DNA vaccines. Int J Parasitol 33:457–467
Dower WJ, Miller JF, Ragsdale CW (1988) High efficiency transformation of E. coli by high voltage electroporation. Nucleic Acids Res 16:6127–6145
Dunham SP (2002) The application of nucleic acid vaccines in veterinary medicine. Res Vet Sci 73:9–16
Galas D, Calos MP, Miller JH (1980) Sequence analysis of Tn9 insertions in the lacZ gene. J Mol Biol 144:19–41
Gurunathan S, Klinman DM, Seder RA (2000) DNA vaccines: immunology, application, and optimization. Annu Rev Immunol 18:927–974
Hall BG (1998) Activation of the bgl operon by adaptive mutation. Mol Biol Evol 15:1–5
Hensley S (2005) Vaccines that keep salmon safe to eat may help humans. In: Wall St. Journal (Eastern edition). New York, p B.1
Itoh M, Suzuki T, Kimata Y, Kawai K, Horitsu H, Takamizawa K (1994) Cadmium resistance acquirement by IS1 transposition into Escherichia coli C600. J Ferment Bioeng 78:466–468
Kitamura K, Torii Y-i, Matsuoka C, Yamamoto K (1995) DNA sequence changes in mutations in the tonB gene on the chromosome of Escherichia coli K12: insertion elements dominate the spontaneous spectra. Jpn J Genet 70:35–46
Klotsky R-A, Schwartz I (1987) Measurement of cat expression from growth-rate-regulated promoters employing b-lactamase activity as an indicator of plasmid copy number. Gene 55:141–146
Kobayashi K, Tsukagoshi N, Aono R (2001) Suppression of hypersensitivity of Escherichia coliacrB mutant to organic solvents by integrational activation of the acrEF operon with the IS1 or IS2 element. J Bacteriol 183:2646–2653
Leitner WW, Thalhamer J (2003) DNA vaccines for non-infectious diseases: new treatments for tumour and allergy. Expert Opin Biol Ther 3:627–638
Lekutis C, Shiver JW, Liu MA, Letvin NL (1997) HIV-1 env DNA vaccine administered to Rhesus monkeys elicits MHC Class II-restricted CD4+ T helper cells that secrete IFN-g and TNF-a. J Immunol 158:4471–4477
Liang X, Fu T-M, Xie H, Emini EA, Shiver JW (2002) Development of HIV-1 Nef vaccine components: immunogenicity study of Nef mutants lacking myristoylation and dileucine motif in mice. Vaccine 20:3413–3421
Lin-Chao S, Bremer H (1986) Effect of the bacterial growth rate on replication control of plasmid pBR322 in Escherichia coli. Mol Gen Genet 203:143–149
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the \(2^{{ - \Delta \Delta C}} _{T}\) method. Methods 25:402–408
Lowrie DB (1999) DNA vaccines against tuberculosis. Curr Opin Mol Ther 1:30–33
Montgomery DL, Shiver JW, Leander KR, Perry HC, Friedman A, Martinez D, Ulmer JB, Donnelly JJ, Liu MA (1993) Heterologous and homologous protection against Influenza A by DNA vaccination: optimization of DNA vectors. DNA Cell Biol 12:777–783
Mwau M, McMichael AJ (2003) A review of vaccines for HIV prevention. J Gene Med 5:3–10
Naas T, Blot M, Fitch WM, Arber W (1994) Insertion sequence-related genetic variation in resting Escherichia coli K-12. Genetics 136:721–730
Naas T, Blot M, Fitch WM, Arber W (1995) Dynamics of IS-related genetic rearrangements in resting Escherichia coli K-12. Mol Biol Evol 12:198–207
Nakahama K, Itoh Y, Fujisawa Y, Kikuchi M (1986) Increased expression of hepatitis B virus surface antigen gene in Escherichia coli by IS1 insertion. Appl Microbiol Biotechnol 25:262–266
Nakamura K, Inouye M (1981) Inactivation of the Serratia marcescens gene for the lipoprotein in Escherichia coli by insertion sequences, IS1 and IS5; sequence analysis of junction points. Mol Gen Genet 183:107–114
Namdev PK, Irwin N, Thompson BG, Gray MR (1993) Effect of oxygen fluctuations on recombinant Escherichia coli fermentation. Biotechnol Bioeng 41:666–670
Ohtsubo E, Sekine Y (1996) Bacterial insertion sequences. In: Saedler H, Gierl A (eds) Transposable elements. Springer, Berlin Heidelberg New York, pp 1–26
Okonkowski J, Kizer-Bentley L, Listner K, Robinson D, Chartrain M (2005) Development of a robust, versatile, and scalable inoculum train for the production of a DNA vaccine. Biotechnol Prog 21:1038–1047
Powell K (2004) DNA vaccines—back in the saddle again? Nat Biotechnol 22:799–801
Prather KJ, Sagar S, Murphy J, Chartrain M (2003) Industrial scale production of plasmid DNA for vaccine and gene therapy: plasmid design, production, and purification. Enzyme Microb Technol 33:865–883
Reinikainen P, Korpela K, Nissinen V, Olkku J, Soderlund H, Markkanen P (1989) Escherichia coli plasmid production in fermenter. Biotechnol Bioeng 33:386–393
Reinikainen P, Virkajärvi I (1989) Escherichia coli growth and plasmid copy numbers in continuous cultivations. Biotechnol Lett 11:222–230
Rodriguez H (1992) An Escherichia coli plasmid-based, mutational system in which supF mutants are selectable: insertion elements dominate the spontaneous spectra. Mutat Res 270:219–231
Schirrmacher V (2005) T cell-mediated immunotherapy of metastases: state of the art in 2005. Expert Opin Biol Ther 5:1051–1068
Schmidt T, Friehs K, Flaschel E, Schleef M (2003) Method for the isolation of ccc plasmid DNA. US Patent 6,664,078. Qiagen GmbH
Seo J-H, Bailey JE (1985) Effects of recombinant plasmid content on growth properties and cloned gene product formation in Escherichia coli. Biotechnol Bioeng 27:1668–1674
Seo J-H, Bailey JE (1986) Continuous cultivation of recombinant Escherichia coli—existence of an optimum dilution rate for maximum plasmid and gene-product concentration. Biotechnol Bioeng 28:1590–1594
Toba-Minowa M (1992) Characterization of the spontaneous elimination of streptomycin sensitivity (SmS) on high-copy-number plasmids: SmS-enforcement cloning vectors with a synthetic rpsL gene. Gene 121:25–33
Tuteja R (2002) DNA vaccine against malaria: a long way to go. Crit Rev Biochem Mol Biol 37:29–54
Ulmer JB (2002) Influenza DNA vaccines. Vaccine 20:S74–S76
Vilalta A, Whitlow V, Martin T (2002) Real-time PCR determination of Escherichia coli genomic DNA contamination in plasmid preparations. Anal Biochem 301:151–153
Zerbib D, Gamas P, Chandler M, Prentki P, Bass S, Galas D (1985) Specificity of insertion of IS1. J Mol Biol 185:517–524
Zhang J, Greasham R (1999) Chemically defined media for commercial fermentations. Appl Microbiol Biotechnol 51:407–421
Acknowledgments
The authors would like to thank Gwen Heidecker, MRL Virus & Cell Biology for providing plasmid DNA and Paul Duncan, Bioprocess & Bioanalytical Research for providing high-producer working seed samples. We also thank Peter Salmon, Michel Chartrain, Paul Duncan, and Wayne Herber for critical reading of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Prather, K.L.J., Edmonds, M.C. & Herod, J.W. Identification and characterization of IS1 transposition in plasmid amplification mutants of E. coli clones producing DNA vaccines. Appl Microbiol Biotechnol 73, 815–826 (2006). https://doi.org/10.1007/s00253-006-0532-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-006-0532-1