
Abstract
The yellow fever vaccine 17D (YF17D) is one of the most effective vaccines. Its wide use and favorable safety profile make it a prime candidate for recombinant vaccines. It is believed that neutralizing antibodies account for a large measure of the protection afforded to YF17D-vaccinated individuals, however cytotoxic T lymphocyte (CTL) responses have been described in the setting of YF17D vaccination. YF17D is an ssRNA flavivirus that is translated as a full-length polyprotein, several domains of which pass into the lumen of the endoplasmic reticulum (ER). The processing and presentation machinery for MHC class I-restricted CTL responses favor cytoplasmic peptides that are transported into the ER by the transporter associated with antigen presentation proteins. In order to inform recombinant vaccine design, we sought to determine if YF17D-induced CTL responses preferentially targeted viral domains that remain within the cytoplasm. We performed whole YF17D proteome mapping of CTL responses in six Indian rhesus macaques vaccinated with YF17D using overlapping YF17D peptides. We found that the ER luminal E protein was the most immunogenic viral protein followed closely by the cytoplasmic NS3 and NS5 proteins. These results suggest that antigen processing and presentation in this model system is not preferentially affected by the subcellular location of the viral proteins that are the source of CTL epitopes. The data also suggest potential immunogenic regions of YF17D that could serve as the focus of recombinant T cell vaccine development.
Similar content being viewed by others


Introduction
More than 95% of YF17D vaccinees mount vaccine-specific immune responses within 10 days of receiving the vaccine (Fields et al. 2001). YF17D has been used in over 500 million individuals with minimal severe side effects (Pulendran et al. 2008; Pugachev et al. 2005). Max Theiler developed YF17D in the 1930s by serial passage of the virulent Asibi strain of yellow fever until an attenuated, avirulent strain was obtained. YF17D, specifically the sub-strain 17D-204, differs from the pathogenic Asibi strain by a mere 32 amino acids (Hahn et al. 1987). Comparisons with other avirulent yellow fever vaccine sub-strains (17DD, 17D-213, and 17D-204) have found commonalities in 22 amino acid changes from wild-type yellow fever virus sequence, indicating that only a few amino acid changes are required for YF17D attenuation (Galler et al. 1998).
The principle mechanism by which YF17D induces immunity is thought to be the generation of potent and long-lived antibody responses. Indeed, patients have been described that still have high neutralizing antibody titers more than 35 years after vaccination (Poland et al. 1981). Cytotoxic T lymphocyte (CTL) responses are also induced by vaccination with YF17D (Querec et al. 2009; Bonaldo et al. 2010; Akondy et al. 2009). While the functional implications of these CTL responses with regards to YF17D clearance and disease prevention are largely unknown, their induction has led to the development of YF17D as a T cell vaccine vector (Bonaldo et al. 2010; Bonaldo et al. 2005; Tao et al. 2005; McAllister et al. 2000).
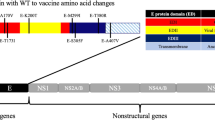
Several insertion sites for exogenous antigens have been developed in YF17D. Two sites allow entire proteins or large domains of proteins to be inserted into YF17D. The first utilizes an artificial domain created between the E and NS1 proteins (Bonaldo et al. 2007). We have introduced a portion of the simian immunodeficiency virus Gag protein into this location and established the immunogenicity of this recombinant YF17D virus in vaccinated rhesus macaques (Bonaldo et al. 2010). More recently, Stoyanov and colleagues have used a second site to insert the circumsporozoite protein from Plasmodium yoelii into the junction between the YF17D C and prM proteins and demonstrated immunogenicity and protection from challenge in a murine malaria model (Stoyanov et al. 2010). Other recombinant YF17D approaches have introduced smaller peptide regions into the C/prM junction (McAllister et al. 2000), into the E protein (Bonaldo et al. 2002), into the NS2b/NS3 junction (Tao et al. 2005; McAllister et al. 2000), or replaced the entire E protein with E proteins from other flaviviruses (Mateu et al. 2007; van Der Most et al. 2000; Lai and Monath 2003). Whether or not any of these insertion sites have an immunologic advantage is currently unknown. It is also not clear if the location of YF17D viral protein domains or recombinant inserts on the cytoplasmic or luminal aspects of the endoplasmic reticulum (ER) membrane (Fig. 1) affects immunogenicity. Therefore, we comprehensively mapped all of the CD8+ T cell responses and some of the CD4+ T cell responses in a group of Indian rhesus macaques vaccinated with YF17D. We found that certain sites appear more immunogenic than others, but location in the cytoplasm or ER lumen did not appear to affect immunogenicity.

Depiction of membrane topology of a generic flavivirus adapted from Fields virology, fourth edition, page 998 (Fields et al. 2001). Membrane topology was predicted from the calculated hydrophobicity of the viral proteins
Materials and methods
Animals
Indian rhesus macaques (Macaca mulatta) from the Wisconsin National Primate Research Center were selected for these studies based upon expression of Mamu-A*02 or Mamu-B*17 as determined by sequence-specific PCR (Knapp et al. 1997). Mamu-A*02+ and Mamu-B*17+ animals were selected because of the extensive characterization of these alleles in Indian rhesus macaques and because of the availability of an online epitope prediction algorithm for both alleles (Peters et al. 2005). All animals were housed and cared for according to the regulations set forth in the Guide for the Care and Use of Laboratory Animals (1996) and all animal experiments were approved by the University of Wisconsin Institutional Animal Care and Use Committee. Six macaques (Table 1) were vaccinated subcutaneously with 100,000 plaque-forming units of YF17D.
Quantitative PCR
Quantification of YF17D virus in plasma from vaccinated animals was determined 7 days after YF17D vaccination, using a previously employed assay (Bonaldo et al. 2010). Viral RNA was isolated from EDTA-anticoagulated plasma by proteinase K digestion of the nuclecapsid in the presence of guanidine hydrochloride. Viral RNA was reverse transcribed and amplified using the SuperScript III Platinum® One-Step Quantitative RT-PCR System (Invitrogen, Carlsbad, CA, USA) in a Roche LightCycler® 480. The final reaction mixtures (100-μL total volume) contained 0.2 mM (each) deoxynucleoside triphosphates, 3.5 mM MgSO4, 750 ng of random hexamer primers (Promega, Madison, WI, USA), 4.0 μL of SuperScript III reverse transcriptase and Platinum Taq DNA Polymerase in a single enzyme mix, 600 nM (each) amplification primers (forward primer YF-17D 10188 [5′- GCGGATCACTGATTGGAATGAC-3′], reverse primer YF-17D 10264 [5′- CGTTCGGATACGATGGATGACTA-3′]), and 100 nM probe (6-carboxyfluorescein [6FAM]-5′- AATAGGGCCACCTGGGCCTCCC-3′-6-carboxytetramethylrhodamine [TamraQ]). The reverse transcription reaction was performed at 37°C for 15 min and then 50°C for 30 min. An activation temperature of 95°C for 2 min was followed by 50 amplification cycles of 95°C for 15 s and 57°C for 1 min with ramp times set to 2.2°C per second. Tenfold serial dilutions of an YF-17D in vitro transcript served as the internal standard curve for each run. The transcript was made using a MEGAscript® (Ambion, Austin, TX, USA) high-yield transcription kit. A plasmid, pCR-Blunt, containing a PCR amplicon with a sequence identical to nucleotides 8,621-10,354 of GenBank accession number U17066, served as the template for transcription.
Peptides
All peptides used in these studies were synthesized to >70% purity by GenScript (Piscataway, NJ, USA). One hundred sixty-three peptides predicted to bind Mamu-A*02 by a published online algorithm (Peters et al. 2005) were pooled into 21 separate groups. The 21 peptides with the highest predicted Mamu-A*02-binding affinity measurements (the lowest predicted IC50 values) were split into each of the pools so that no single pool contained more than one peptide with high predicted Mamu-A*02-binding affinity. For whole proteome mapping, peptides 15-amino acids in length overlapping by 11 amino acids encompassing the YF17D proteome were also synthesized. These peptides were pooled into groups of ten contiguous peptides for initial response screening.
Whole proteome epitope mapping
Fresh peripheral blood mononuclear cells (PBMC) isolated from EDTA-anticoagulated blood were used in interferon (IFN)-gamma enzyme-linked immunosorbent spot (ELISPOT) assays as previously described (Loffredo et al. 2007), however the determination of positive responses was made using a new technique which we have found in control experiments to be more sensitive and reproducible than our previously used method. Test wells were run with two replicates while positive and negative control wells were run in replicates of two, four, or six depending on the assay. Responses containing <50 spot forming units per 1 × 106 cells were considered negative and not tested statistically. Positive responses were determined using a one-tailed t test and alpha = 0.05, where the null hypothesis (H0): background level ≥ treatment level. If determined to be positive statistically, the reported values equal the average of the test wells minus the average of all negative control wells. In the indicated experiments, PBMC were first depleted of CD8+ cells using a non-human primate magnetic bead separation protocol (Miltenyi Biotec, Auburn, CA, USA). Greater than 99% of CD8+ cells were successfully depleted in all experiments and verified by FACS analysis.
Results
To evaluate the relative immunogenicity of various regions of the YF17D proteome in Indian rhesus macaques, we vaccinated four Mamu-A1*00201 + (Mamu-A*02+) animals and two Mamu-B*01701 + (Mamu-B*17+) animals with 100,000 plaque-forming units of YF17D. A list of the animals included in this study along with their MHC class I alleles as defined by sequence-specific PCR is provided in Table 1. We detected viral RNA amplification in four of five animals 7 days after vaccination using a previously described quantitative PCR assay (Bonaldo et al. 2010), indicating that the animals were indeed infected with replicating YF17D (Table 2). The single animal without detectable viremia made anti-YF17D-directed T cell responses post-vaccination, indicating that it was likely infected with replicating YF17D.
To detect Mamu-A*02-restricted epitopes in the YF17D proteome, we used a published, online algorithm (Peters et al. 2005) to generate a list of predicted Mamu-A*02-binding peptides derived from the entire YF17D proteome. We then selected 163 peptides with predicted binding affinities (IC50) < 50 nM for synthesis. We pooled the synthesized peptides into 21 different groups. We then mapped IFN-gamma ELISPOT responses using PBMC in the four Mamu-A*02+ animals on day 17 post-vaccination using the Mamu-A*02-predicted peptide pools. To our surprise, only one of these pools elicited a positive response in two of the four Mamu-A*02+ animals. We broke down the positive pool on day 20 post-vaccination and found that both animals responded to the same E protein peptide, E696–705 (Table 3). In spite of the robust responses directed towards this epitope in two of the Mamu-A*02+ animals, the lack of response in the other two Mamu-A*02+ animals suggests that this response is not recognized by high frequencies of CD8+ T cells in all Mamu-A*02+ animals.
We proceeded to map T cell responses directed against the YF17D proteome in vaccinated macaques using a set of 849 peptides that were 15-amino acids long and overlapped by 11 amino acids. This peptide set covered the entire YF17D polyprotein. Our primary goals were to determine the relative immunogenicity of the various viral proteins, to detect Mamu-B*17-restricted responses, and to see if preferential CD8+ T cell targeting of YF17D protein domains might have occurred based upon their subcellular location in relation to the luminal or cytoplasmic side of the ER. We analyzed responses from all six vaccinated animals to this peptide set on the day of vaccination and found no responses above background (data not shown). On day 17 post-vaccination, we mapped IFN-gamma ELISPOT responses directed against pools of ten sequential overlapping peptides encompassing the entire viral proteome for both total PBMC and CD8-depleted PBMC (representing CD4+ T cell responses) from each animal. All positive responses detected in this initial screen on day 17 were then subsequently verified and broken down to the individual 15-mer peptides on day 20 post-vaccination. These results are summarized in Fig. 2 and Table 3. We reliably detected 21 separate putative CD8+ T cell responses in seven of the ten viral proteins. We did not detect any responses to the C, prM, or NS4b proteins in our vaccinated animals. The two Mamu-B*17+ animals, r00012 and r03031, did not share any responses indicating that we were unable to detect immunodominant Mamu-B*17-restricted YF17D epitopes in our experiment. We also could not detect any immunodominant Mamu-A*02-restricted YF17D epitopes since none of the responses in the four Mamu-A*02+ animals were found in more than two individual animals. However, we did detect the shared predicted Mamu-A*02-restricted E696–705 AF10 epitope in our total proteome scan. The concordance of our overlapping 15-mer scan with our Mamu-A*02 predicted epitope results suggests that YF17D does not contain a large number of broadly immunogenic or immunodominant Mamu-A*02-restricted CTL epitopes. Interestingly, simian immunodeficiency virus contains a total of 17 distinct Mamu-A*02-restricted CTL epitopes, three of which are immunodominant and targeted by CTL in almost all Mamu-A*02+ SIV-infected animals (Loffredo et al. 2004).

Cellular immune responses directed against YF17D peptides in PBMC. Six rhesus macaques (four Mamu-A*02+, left column; two Mamu-B*17+, right column) were vaccinated with 100,000 plaque-forming units of YF17D. Positive IFN-gamma ELISPOT responses to pools of overlapping YF17D 15-mer peptides were broken down to individual 15-mers on day 20 post-vaccination. Locations of positive individual 15-mer peptides and the corresponding magnitude of the indicated responses in IFN-gamma ELISPOT are mapped for each animal. The proportion of each animal's total positive response directed towards individual viral protein domains is diagramed with a pie chart. The total proportions for all six animals and for both groups of MHC-matched animals are also included. Pie charts represent the total magnitude for the individual responses within the indicated viral domain compared to the total magnitude of all responses made
Two high-magnitude CD8+ T cell responses were detected in r04131 and r03031, E681–699 and NS32021–2038, suggesting these animals share one or two MHC class I alleles that restrict these responses (Table 3). Our SSP-PCR MHC class I typing of these two animals (Table 1) did not detect any shared alleles, therefore the restriction of these epitopes remains unknown.
CD4+ T cell epitopes were more difficult to detect in our experiments due to problems with high background on our CD8-depleted IFN-gamma ELISPOT plates. This phenomenon of high background on CD8-depleted IFN-gamma ELISPOT plates, following YF17D vaccination, had been noted in previous experiments and is currently being characterized more in depth (Costa Neves et al., manuscript in preparation). Nevertheless, we were able to detect two low-level CD4+ T cell responses that were shared in r02059 and r03031, E329–343 and NS31837–1851 (Fig. 3). Once again, the shared responses suggest these animals have an MHC class II allele in common that restricts these responses. Due to the high background and other potential confounding issues, we cannot be certain that we were able to map the entire CD4+ T cell repertoire against YF17D. However, these two responses were reproducibly observed in both of these animals.

CD4+ T cell responses reliably detected above background in two YF17D vaccinees. Responses were mapped to individual YF17D 15-mers using magnetically CD8-depleted PBMC. Background responses were higher in CD8-depleted ELISPOT experiments; therefore the statistical test we used to determine positive responses was less sensitive. We could only reliably detect the indicated responses in two of the macaques with the lowest level of background. The fact that these two animals have two responses in common indicates they share one or more MHC class II alleles
Finally, we compiled our comprehensive CD8+ T cell response mapping data to assess the relative immunogenicity of the various viral proteins (Fig. 1). The most immunogenic protein in our six animals was the E protein followed closely by the NS3 and NS5 proteins. Our data may be biased slightly towards E since the Mamu-A*02+ animals, two of which target the E696–705 AF10 epitope, clearly have a preference for the E protein when compared with the Mamu-B*17+ animals. Nevertheless, the most immunogenic regions of YF17D in our cohort of macaques were clearly the E, NS3, and NS5 proteins. All of these proteins were targeted in at least three of the six animals. The NS5 protein was targeted most frequently in six out of six animals followed by E in five out of six animals.
Discussion
YF17D is being developed as a recombinant T cell vaccine vector (Bonaldo et al. 2010; Bonaldo et al. 2005; Tao et al. 2005; McAllister et al. 2000). The relative immunogenicity and efficacy of the various recombinant insertion sites remains unknown. Therefore, we evaluated the immunogenicity of the YF17D viral proteins to determine if certain regions of the viral proteome might be more immunogenic than others. This work was premised on the mechanism of MHC class I antigen processing and presentation which is widely believed to favor the transport of antigenic proteins from the cytoplasm. We postulated that perhaps the location of several viral proteins on the luminal side of the ER membrane (Fig. 1) might diminish their immunogenicity. However, our data do not support this conclusion. In light of work describing the generation of immunogenic MHC class I-restricted peptides from defective ribosomal products (DRiPs) (Lev et al. 2010; Yewdell et al. 1996), it is reasonable to conclude that the irrelevance of the final subcellular location of our antigenic YF17D regions to their immunogenicity is likely due to processing and presentation occurring upstream of this final localization. Because ER translocation occurs in tandem with translation, we presume that antigen processing and presentation of these regions might occur within its own immunologic translation compartment (Lev et al. 2010). Our YF17D immunogenicity data most closely fit the DRiPs model. We did not detect preferential targeting of cytoplasmic polyprotein domains. In fact, the region of YF17D with the highest magnitude of CD8+ T cell responses in our experiments was the ER luminal E protein. Any concerns about using insertion sites in or near the E protein because of its ER luminal location are therefore unjustified.
We also determined that the NS5 protein followed by the E protein and the NS3 protein were the most frequently targeted antigens in YF17D. The NS5 protein was targeted by CD8+ T cell responses of variable magnitudes in all six of our vaccinated animals. NS5 was also a regular target of T cell responses in YF17D-vaccinated humans (Akondy et al. 2009). Our work suggests that a strategy to introduce recombinant YF17D insertion sites in or immediately adjacent to the NS5 protein might be desirable for a recombinant T cell vaccine. Recombinant insertions in this region would likely induce insert-directed responses in most individuals.
Finally, we were surprised by the small number of detected Mamu-A*02- or Mamu-B*17-restricted YF17D epitopes. Mamu-A*02 and Mamu-B*17 are relatively frequent MHC class I alleles in captive populations of rhesus macaques (frequencies in all animals typed to date [N > 8,200] are 20.78% and 10.35% for Mamu-A*02 and Mamu-B*17, respectively). We did not detect any immunodominant Mamu-A*02- or Mamu-B*17-restricted epitopes in our vaccinated cohort. We found a single putative Mamu-A*02-restricted response and were unable to define any Mamu-B*17-restricted responses. It is possible that the transient viremia seen in YF17D-vaccinated humans and macaques is insufficient to induce high-frequency immunodominant responses in all animals, however the description of one such response in HLA-A0201+ YF17D vaccinated humans (Akondy et al. 2009) suggests that this is not always the case. Interestingly, we detected two responses of large magnitude in two of our animals suggesting that they both recognized the same immunodominant epitopes.
Perhaps the discrepancy between Mamu-A*02-restricted responses in acute and chronic SIV infection (Loffredo et al. 2004) and acute YF17D infection is a function both of MHC class I allele specificity for the two viruses and the kinetics of antigen exposure. Notably, the HLA-A0201 allele, which restricts a single immunodominant response in YF17D-vaccinated humans, restricts several immunodominant epitopes in HIV-infected humans (Yusim et al. 2009). It seems reasonable that the detection of only a single immunodominant HLA-A0201-restricted YF17D response in vaccinated humans might indicate a lower antigenic stimulatory capacity of YF17D when compared with HIV infection. In spite of this apparent reduction in YF17D's capacity to induce responses of both high frequency and high magnitude, it is worth noting that the induction of T cell vaccine responses of high frequency and high magnitude in SIV-challenged macaques has not been correlated with improved outcome after infection. Hence, the quality and efficacy of CD8+ T cell responses engendered by a T cell vaccine does not correlate with the induction of such responses. These qualitative aspects of successful antiviral T cell responses are not accurately measured by our current assays. Therefore, we should continue to move forward with several vaccine vectors in efficacy trials in our search for a successful T cell-based vaccine.
References
Akondy RS, Monson ND, Miller JD, Edupuganti S, Teuwen D, Wu H, Quyyumi F, Garg S, Altman JD, Del Rio C, Keyserling HL, Ploss A, Rice CM, Orenstein WA, Mulligan MJ, Ahmed R (2009) The yellow fever virus vaccine induces a broad and polyfunctional human memory CD8+ T cell response. J Immunol 183(12):7919–7930
Bonaldo MC, Garratt RC, Caufour PS, Freire MS, Rodrigues MM, Nussenzweig RS, Galler R (2002) Surface expression of an immunodominant malaria protein B cell epitope by yellow fever virus. J Mol Biol 315(4):873–885
Bonaldo MC, Garratt RC, Marchevsky RS, Coutinho ES, Jabor AV, Almeida LF, Yamamura AM, Duarte AS, Oliveira PJ, Lizeu JO, Camacho LA, Freire MS, Galler R (2005) Attenuation of recombinant yellow fever 17D viruses expressing foreign protein epitopes at the surface. J Virol 79(13):8602–8613
Bonaldo MC, Mello SM, Trindade GF, Rangel AA, Duarte AS, Oliveira PJ, Freire MS, Kubelka CF, Galler R (2007) Construction and characterization of recombinant flaviviruses bearing insertions between E and NS1 genes. Virol J 4:115
Bonaldo MC, Martins MA, Rudersdorf R, Mudd PA, Sacha JB, Piaskowski SM, Costa Neves PC, Veloso de Santana MG, Vojnov L, Sr C, Rakasz EG, Wilson NA, Fulkerson J, Sadoff JC, Watkins DI, Galler R (2010) Recombinant yellow fever vaccine virus 17D expressing simian immunodeficiency virus SIVmac239 gag induces SIV-specific CD8+ T-cell responses in rhesus macaques. J Virol 84(7):3699–3706
Fields BN, Knipe DM, Howley PM, Griffin DE (2001) Fields virology. Lippincott Williams & Wilkins, Philadelphia
Galler R, Post PR, Santos CN, Ferreira II (1998) Genetic variability among yellow fever virus 17D substrains. Vaccine 16(9–10):1024–1028
Hahn CS, Dalrymple JM, Strauss JH, Rice CM (1987) Comparison of the virulent Asibi strain of yellow fever virus with the 17D vaccine strain derived from it. Proc Natl Acad Sci USA 84(7):2019–2023
Knapp LA, Lehmann E, Piekarczyk MS, Urvater JA, Watkins DI (1997) A high frequency of Mamu-A*01 in the rhesus macaque detected by polymerase chain reaction with sequence-specific primers and direct sequencing. Tissue Antigens 50(6):657–661
Lai CJ, Monath TP (2003) Chimeric flaviviruses: novel vaccines against dengue fever, tick-borne encephalitis, and Japanese encephalitis. Adv Virus Res 61:469–509
Lev A, Princiotta MF, Zanker D, Takeda K, Gibbs JS, Kumagai C, Waffarn E, Dolan BP, Burgevin A, Van Endert P, Chen W, Bennink JR, Yewdell JW (2010) Compartmentalized MHC class I antigen processing enhances immunosurveillance by circumventing the law of mass action. Proc Natl Acad Sci USA 107(15):6964–6969
Loffredo JT, Sidney J, Wojewoda C, Dodds E, Reynolds MR, Napoe G, Mothe BR, O'Connor DH, Wilson NA, Watkins DI, Sette A (2004) Identification of seventeen new simian immunodeficiency virus-derived CD8+ T cell epitopes restricted by the high frequency molecule, Mamu-A*02, and potential escape from CTL recognition. J Immunol 173(8):5064–5076
Loffredo JT, Burwitz BJ, Rakasz EG, Spencer SP, Stephany JJ, Vela JP, Martin SR, Reed J, Piaskowski SM, Furlott J, Weisgrau KL, Rodrigues DS, Soma T, Napoe G, Friedrich TC, Wilson NA, Kallas EG, Watkins DI (2007) The antiviral efficacy of simian immunodeficiency virus-specific CD8+ T cells is unrelated to epitope specificity and is abrogated by viral escape. J Virol 81(6):2624–2634
Mateu GP, Marchevsky RS, Liprandi F, Bonaldo MC, Coutinho ES, Dieudonne M, Caride E, Jabor AV, Freire MS, Galler R (2007) Construction and biological properties of yellow fever 17D/dengue type 1 recombinant virus. Trans R Soc Trop Med Hyg 101(3):289–298
McAllister A, Arbetman AE, Mandl S, Pena-Rossi C, Andino R (2000) Recombinant yellow fever viruses are effective therapeutic vaccines for treatment of murine experimental solid tumors and pulmonary metastases. J Virol 74(19):9197–9205
Peters B, Bui HH, Sidney J, Weng Z, Loffredo JT, Watkins DI, Mothe BR, Sette A (2005) A computational resource for the prediction of peptide binding to Indian rhesus macaque MHC class I molecules. Vaccine 23(45):5212–5224
Poland JD, Calisher CH, Monath TP, Downs WG, Murphy K (1981) Persistence of neutralizing antibody 30–35 years after immunization with 17D yellow fever vaccine. Bull World Health Organ 59(6):895–900
Pugachev KV, Guirakhoo F, Monath TP (2005) New developments in flavivirus vaccines with special attention to yellow fever. Curr Opin Infect Dis 18(5):387–394
Pulendran B, Miller J, Querec TD, Akondy R, Moseley N, Laur O, Glidewell J, Monson N, Zhu T, Zhu H, Staprans S, Lee D, Brinton MA, Perelygin AA, Vellozzi C, Brachman PJ, Lalor S, Teuwen D, Eidex RB, Cetron M, Priddy F, del Rio C, Altman J, Ahmed R (2008) Case of yellow fever vaccine-associated viscerotropic disease with prolonged viremia, robust adaptive immune responses, and polymorphisms in CCR5 and RANTES genes. J Infect Dis 198(4):500–507
Querec TD, Akondy RS, Lee EK, Cao W, Nakaya HI, Teuwen D, Pirani A, Gernert K, Deng J, Marzolf B, Kennedy K, Wu H, Bennouna S, Oluoch H, Miller J, Vencio RZ, Mulligan M, Aderem A, Ahmed R, Pulendran B (2009) Systems biology approach predicts immunogenicity of the yellow fever vaccine in humans. Nat Immunol 10(1):116–125
Stoyanov CT, Boscardin SB, Deroubaix S, Barba-Spaeth G, Franco D, Nussenzweig RS, Nussenzweig M, Rice CM (2010) Immunogenicity and protective efficacy of a recombinant yellow fever vaccine against the murine malarial parasite Plasmodium yoelii. Vaccine 28(29):4644–4652
Tao D, Barba-Spaeth G, Rai U, Nussenzweig V, Rice CM, Nussenzweig RS (2005) Yellow fever 17D as a vaccine vector for microbial CTL epitopes: protection in a rodent malaria model. J Exp Med 201(2):201–209
van Der Most RG, Murali-Krishna K, Ahmed R, Strauss JH (2000) Chimeric yellow fever/dengue virus as a candidate dengue vaccine: quantitation of the dengue virus-specific CD8 T-cell response. J Virol 74(17):8094–8101
Yewdell JW, Anton LC, Bennink JR (1996) Defective ribosomal products (DRiPs): a major source of antigenic peptides for MHC class I molecules? J Immunol 157(5):1823–1826
Yusim K, Korber BTM, Brander C, Haynes BF, Koup R, Moore JP, Walker BD, Watkins DI (2009) HIV Molecular Immunology 2009. Theoretical Biology and Biophysics, Los Alamos
Acknowledgements
The authors would like to thank Mauricio Martins for helpful discussions. This work was supported by National Institutes of Health (NIH) grants R37 AI052056, R01 AI049120, R01 AI076114, R24 RR015371, R24 RR016038, and R21 PRJ27JP to DIW. This publication was made possible in part by Grant Number P51 RR000167 from the National Center for Research Resources (NCRR), a component of the NIH, to the Wisconsin National Primate Research Center, University of Wisconsin-Madison. This research was conducted in part at a facility constructed with support from Research Facilities Improvement Program grant numbers RR15459-01 and RR020141-01. This publication's contents are solely the responsibility of the authors and do not necessarily represent the official views of NCRR or NIH.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Mudd, P.A., Piaskowski, S.M., Neves, P.C.C. et al. The live-attenuated yellow fever vaccine 17D induces broad and potent T cell responses against several viral proteins in Indian rhesus macaques—implications for recombinant vaccine design. Immunogenetics 62, 593–600 (2010). https://doi.org/10.1007/s00251-010-0461-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00251-010-0461-0