
Abstract
Enveloped viruses infect host cells by fusion of viral and target membranes. This fusion event is triggered by specific glycoproteins in the viral envelope. Fusion glycoproteins belong to either class I, class II or the newly described third class, depending upon their arrangement at the surface of the virion, their tri-dimensional structure and the location within the protein of a short stretch of hydrophobic amino acids called the fusion peptide, which is able to induce the initial lipid destabilization at the onset of fusion. Viral fusion occurs either with the plasma membrane for pH-independent viruses, or with the endosomal membranes for pH-dependent viruses. Although, viral fusion proteins are parted in three classes and the subcellular localization of fusion might vary, these proteins have to act, in common, on lipid assemblies. Lipids contribute to fusion through their physical, mechanical and/or chemical properties. Lipids can thus play a role as chemically defined entities, or through their preferential partitioning into membrane microdomains called “rafts”, or by modulating the curvature of the membranes involved in the fusion process. The purpose of this review is to make a state of the art on recent findings on the contribution of cholesterol, sphingolipids and glycolipids in cell entry and membrane fusion of a number of viral families, whose members bear either class I or class II fusion proteins, or fusion proteins of the recently discovered third class.
Similar content being viewed by others

Two classes of viral fusion proteins... and more ?
Enveloped animal viruses are classified into several families, depending on genome structure, virion morphology and replication cycle. However, common features are emerging, in particular, at the level of the envelope glycoproteins these viruses use to enter in host cells. These transmembrane proteins are meant to destabilize lipid bilayers in a controlled fashion, known as viral membrane fusion. This fusion step can occur either at the plasma membrane for viruses that do not depend on pH for their cell entry (e.g., Paramyxoviruses, some Herpesviruses and Retroviruses) (Fackler and Krausslich 2006; Rey 2006; Russell and Luque 2006), or inside the endosome after activation by low pH for endocytosis-dependent virions, such as Orthomyxoviruses, Filoviruses, Coronaviruses, Arenaviruses, Flaviviruses, Alphaviruses and Rhabdoviruses (Alazard-Dany et al. 2006; Chu et al. 2006; Gaudin 2000b; Kobayashi et al. 2006; Kunz et al. 2002; Lavillette et al. 2006; Monath and Heinz 1996; Schlesinger and Schlesinger 1996; Yonezawa et al. 2005). Table 1 presents an overview of the viral protein families that will be discussed in this review.
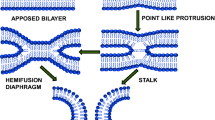
Fusion proteins undergo a number of conformational changes to become fusion-competent; these changes are triggered either by receptor binding onto the plasma membrane for viruses that fuse at neutral pH, or by protonation of specific residues in the endosome for viruses that penetrate their host cells via endocytosis. Fusion consists in a succession of subtly controlled steps, beginning with close apposition of viral and cellular membranes and local dehydration at the contact points. It is commonly thought that the energy released during the protein structural changes to reach the most stable conformation is used to drive the apposition and subsequent merging of the bilayers. This could apply to viral and intracellular fusion processes (Blumenthal et al. 2003; Chernomordik et al. 2006). The initial apposition step is followed by fusion of the outer leaflets of membranes (the hemifusion step), leading to the formation of a transient fusion intermediate called stalk (Chernomordik and Kozlov 2005). This evolves into the fusion of inner leaflets and the formation of a pore. This allows the internal compartments of both fusion partners to get mixed. Ultimately, the viral genome is transferred to the cytoplasm of the host cell and viral replication can start. This is illustrated in Fig. 1.

Schematic pathway of membrane deformation during fusion (fusion proteins are not represented). Intact viral and cellular membranes first come into close apposition, which leads to the first step of the fusion process, the fusion of the outer leaflets of membranes (hemifusion). This step can be blocked by the presence of lysolipids in that leaflet, or promoted by unsaturated PE, cholesterol or monoolein. The mixing of the inner leaflets of membranes evolves into an early fusion pore, which enlarges to give rise to the late fusion pore. At that stage viral genetic material is delivered to the cell cytoplasm
For viral fusion to occur, two minimal partners are required: a fusion machinery and lipids. These two partners must act in concert and in a cooperative fashion so that the fusion process can be completed. Fusion machineries have therefore in common: (i) to interact with lipids, so they possess hydrophobic segments (fusion peptides) or are able after rearrangements to enter into hydrophobic interactions with membranes; (ii) to adopt specific conformations related to the fusogenic and non-fusogenic states, since fusion is limited in space and time. Dissimilar viruses would therefore infect their host cells by very similar mechanisms at the molecular level of proteins and lipids. However, similar mechanisms can be brought about by structurally different fusion machineries. Indeed, recent studies on Flaviviruses such as the tick-borne encephalitis virus (TBEV) and the Dengue virus, and on Alphaviruses such as the Semliki forest virus (SFV) have revealed the characteristics of another class of viral fusion proteins, unrelated to the well-studied influenza virus hemagglutinin, the prototype for fusion proteins of Orthomyxoviruses, Paramyxoviruses, Retroviruses and Filoviruses. This class is known as class II fusion proteins and regroup proteins of the Flaviviridae and Togaviridae families of viruses, whereas class I proteins closely resemble the influenza hemagglutinin.
Class I viral fusion proteins form spiky projections at the surface of the virion, and do not give any scaffold to the virion envelope (e.g., Fig. 2 for the influenza hemagglutinin). Glycoprotein spikes might even be very few on the envelope surface, as exemplified recently by cryoelectron microscopy tomography with HIV-1 (Zhu et al. 2006). Viruses bearing such proteins are therefore called irregular. Class I protein maturation is achieved by a proteolytic cleavage which releases at their amino-terminus the fusion peptide. In Filoviruses such as the Ebola virus, the fusion peptide is not located at the N-terminus of the fusion glycoprotein, but a few amino acids downstream (Ito et al. 1999). These proteins undergo irreversible and major conformational changes leading to a hairpin post-fusion structure formed by a three-stranded coiled coil of α helices [reviewed in Colman and Lawrence ( 2003)].

3D-structures of a prototype of each class of fusion proteins; for class I, trimer of the influenza hemagglutinin at low pH [1HTM PDB accession number, (Bullough et al. 1994)], displaying a three-stranded coiled coil of alpha-helices (note that the fusion peptides are absent from this structure); for class II, monomer of the E protein of TBEV at neutral pH, mainly composed of beta-strands (the fusion peptide is at the top) [1SVB PDB accession number, (Rey et al. 1995)]; for the newly described class of proteins, trimer of VSV-G at low pH [2CMZ PDB accession number, (Roche et al. 2006)], displaying three-stranded coiled coils (bottom), a beta-strand-rich region (top), and a pleckstrin homology domain absent in class I and II fusion proteins (middle). The fusion peptides are at the top
In contrast to class I fusion proteins, class II proteins lie almost flat on the virion surface, and do not exhibit coiled coils (Fig. 2). They are predominantly arranged as β-strands forming three distinct domains (DI to DIII) (see also Fig. 4), and form head-to-tail orientated dimers [reviewed in Kielian and Rey (2006)]. Flaviviruses such as the TBE, the Dengue and West Nile viruses possess two major envelope glycoproteins prM and E. E, the fusion glycoprotein, is arranged as a homodimer in the mature state of the virus (Kuhn et al. 2002; Modis et al. 2003; Mukhopadhyay et al. 2003; Rey et al. 1995; Zhang et al. 2003). In the Semliki forest and Sindbis Alphaviruses, the fusion activity is contained into E1 which forms a heterodimer with E2 (Fuller et al. 1995; Smit et al. 1999; Zhang et al. 2002). In both E and E1, a crucial region for the fusion activity is called the hinge region, which lies between DI and DII and whose flexibility is essential for its biological function. The flat orientation of class II fusion proteins on the viral surface plays a major role in maintaining the envelope structure and curvature of the virion, in contrast to spiky class I fusion proteins. Through a network of lateral interactions, one dimer makes contact with the neighboring dimers, which creates a highly ordered shell with icosahedral symmetry (Ferlenghi et al. 2001; Kuhn et al. 2002; Lescar et al. 2001; Mukhopadhyay et al. 2003). These viruses are therefore called regular. The fusion peptide is internal, located in a loop at the tip of domain II, buried at the dimer interface for Flaviviruses and exposed for Alphaviruses (Lescar et al. 2001; Rey et al. 1995). A proteolytic cleavage does not occur within the fusion protein itself, but in the accompanying protein. Conformational changes are reported to be irreversible, but recent data indicate that low pH-induced inactivation of SFV E1 could be reverted (Waarts et al. 2005). These conformational changes include trimerization of E or E1, and the projection of the β barrels of domain II toward the cellular membrane, facilitating the interaction of the fusion peptide with the lipid bilayer (Bressanelli et al. 2004; Gibbons et al. 2004).
The envelope of the hepatitis C virus (HCV), an Hepacivirus of the Flaviviridae family, is constituted by two glycoproteins assembled as a heterodimer, E1 and E2. Whether E1 or E2 is the fusion protein remains controversial since no structural data are available at present. Using the 3D-structure of the TBEV E as a template, tentative molecular 3D-modelization of the E1 and E2 molecules has been proposed, which would rank them among class II fusion proteins (Garry and Dash 2003; Yagnik et al. 2000). However, due to poor amino acid sequence conservation and in the absence of any experimental evidence, these molecular models must be taken with caution.
Fusion proteins of viruses from the Rhabdoviridae and Herpesviridae families seem to constitute a separate class of fusion proteins, both structurally and biochemically (Fig. 2). The recently published 3D-structures of the G protein of the vesicular stomatitis virus (VSV), and of the gB protein of herpes simplex virus type 1 (HSV-1) revealed features common to both class I and class II proteins, i.e., three-stranded coiled coils at the trimer axis (reminiscent of class I) and a long three-stranded β-sheet with a structure similar to that of a class II motif but with different strand topology (Heldwein et al. 2006; Roche et al. 2006). Both contain a pleckstrin homology (PH) domain, a domain present in several cellular proteins and serving as a scaffold for phosphatidylinositol phosphates (PIP) and protein binding (Lemmon 2004). Both carry an internal “fusion peptide” [see Durrer et al. (1995) for VSV], organized in two fusion loops (Heldwein et al. 2006; Roche et al. 2006), exposed in the prefusion protein conformation for VSV-G (Roche et al. 2007). The “fusion peptide” of VSV-G is exposed in a reversible manner after low pH application (Gaudin 2000b). This reversibility implies that, in contrast with most proteins from other classes, the native state of the protein can be recovered by shifting to neutral pH in the absence of target membranes, although exposure to the low pH-trigger had led to protein inactivation (Roche and Gaudin 2002). Overall, these proteins little resemble the canonical class I and class II molecules, which suggests that they form a third class (Da Poian et al. 2005; Steven and Spear 2006).
However, it appears that all fusion proteins induce membrane fusion very much alike, and would end up in similar hairpin structures after fusion activation. In all cases, the fusion peptide would be a major element of the fusion machinery, being the trigger for controlled membrane destabilization. Another element essential to the completion of the fusion reaction is the transmembrane domain (TMD) of the fusion proteins. Indeed, once the peptide has been inserted into the target membrane, a series of conformational changes take place, eventually leading to the observed hairpin. This implies that the bulky trimer of class I or II fusion proteins, the minimal fusion subunit when the fusion trigger has been applied, “lies down” between viral and cellular membranes (Colman and Lawrence 2003; Kielian 2006). Fusion then proceeds through trimer oligomerization. Such an architecture would allow for pore formation and enlargement, and release of the viral genetic material into the host cell. TMD were shown to be essential to this final step, since fusion proteins devoid of their TMD can only induce hemifusion (Kemble et al. 1994; Tong and Compans 2000; Weiss et al. 1996).
We will now examine the second partner for fusion: lipids.
Lipids in membrane fusion
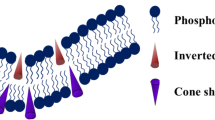
The lipid composition of animal membranes is complex, but three main categories of lipids can be distinguished (van Meer 2005): glycerophospholipids, sphingolipids (SPL) and sterols. Glycerophospholipids regroup phosphatidylcholine (PC), phosphatidylethanolamine (PE), phosphatidylserine (PS) and phosphatidylinositol (PI). PC represents 50% of cellular membranes, and forms stable bilayers. PE (20% of most membranes) has a cone-shaped molecular structure and the ability to promote the bilayer-to-hexagonal transition that may facilitate fusion, by increasing the negative curvature of the membrane (Chernomordik and Kozlov 2005; Siegel and Epand 1997). PS is mostly located in the inner leaflet of the plasma membrane, and appears on its exoplasmic face during apoptosis and blood coagulation. PI is the basis for the biosynthesis of PIP. Phospholipids can be metabolized by phospholipases A into lysophospholipids (lysoPL), which fulfill signaling roles upon cell activation. These lysoPL display an inverted-cone molecular shape, that would inhibit the formation of the hemifusion stalk when present in the outer leaflet of the bilayer, by increasing the positive curvature of the membrane (Chernomordik and Kozlov 2005). Sphingomyelin (SM) is the prototype for sphingolipids (SPL), with a phosphocholine head like PC but a hydrophobic ceramide backbone. In glycoSPL, ceramide carries carbohydrates, the simplest ones being glucosyl- (GlcCer) and galactosyl-ceramide (GalCer). By themselves, SPL do not form membranes, but display a liquid–crystalline phase. Membranes are fluidized by cholesterol (chol), the mammalian sterol. The SPL- and chol-to-phospholipid ratios decrease from the plasma membrane to the Golgi apparatus and to the endoplasmic reticulum (van Meer 1998). Mixtures of PC, SM and chol spontaneously tend to segregate into a liquid-ordered (Lo) phase enriched in SM and chol, and a disordered phase. The Lo phase would form lateral heterogeneities in the membrane, known as the (small, dynamic and transient) “rafts” [for recent reviews, see for e.g., Kahya (2006); McIntosh and Simon (2006) and references therein].
Lipids could play a role in membrane fusion by themselves as chemically-defined molecules, thereby critically affecting peptide and protein binding, or by the physico-chemical and mechanical properties they induce on/in target membranes (“raft” formation, membrane curvature). This will now be further discussed in the context of viral fusion, and the reader can refer Table 1 for details concerning the relation between the three protein classes and lipids.
“Rafts” in viral entry: a complex interplay of lipids, viral proteins and cellular receptors
Retroviruses and chol/SPL
The physiological function of lipid microdomains in HIV entry is still a matter of great controversy, as is the location of HIV-1 receptors and coreceptors into lipid microdomains. Since the purpose of this review is not to go into the mechanistic details of HIV entry into cells, the reader is directed to recent reviews and articles in the field (Gaibelet et al. 2006; Rawat et al. 2005; Viard et al. 2004; Yi et al. 2006).
The HIV fusion glycoprotein is composed of two subunits, the surface (SU) gp120 and the transmembrane (TM) gp41 that contains the fusion peptide (Roux and Taylor 2007). Both subunits are arranged as trimers. HIV infects permissive cells by binding to its receptor CD4 through gp120, and this initial interaction with CD4 promotes a conformational change in gp120 that enables viral glycoprotein interaction with the co-receptors CXCR4 or CCR5. This binding occurs through a variable loop called V3, and variations in the amino acid sequence of this loop determine HIV isolates which recognize either CXCR4, or CCR5, or both. Essentially, two lines of controversy emerge, one concerning the arrangement of receptors and co-receptors in rafts, the other concerning the involvement of lipid rafts in HIV entry and fusion. This is illustrated in Fig. 3. Very schematically, in the first line of controversy, reports argue that CD4 is present in rafts, but not the co-receptors [see for e.g., Chazal and Gerlier (2003; Viard et al. (2004)] (Hypothesis 1, Fig. 3), whereas other reports argue that CD4 and the co-receptors are in rafts [see for e.g., Becher and McIlhinney (2005; Manes et al. (2001)] (Hypothesis 2, Fig. 3). In the second line of controversy, results report either that rafts are involved in HIV entry, or that rafts are not involved. Bachelerie et al. argued that the presence of receptor and coreceptors in lipid microdomains was not required for fusion (Percherancier et al. 2003). They recently demonstrated a constitutive interaction between CD4 and CCR5 at the plasma membrane; however, the ternary interaction between HIV gp120/CD4/CCR5 would take place outside the “raft” domains of the plasma membrane (Gaibelet et al. 2006) (Hypothesis 2B, Fig. 3). Another picture emerges from the studies of Yi et al.: they showed that chol disruption from lipid “rafts” did not affect the distribution of CD4 and CCR5, but altered the interaction of these receptor molecules with gp120. This would then suggest that chol-rich microdomains are required for the formation of the ternary complex (Yi et al. 2006) (Hypothesis 2A, Fig. 3).

Hypotheses for the involvement of lipid rafts in HIV entry into its target cells. See text for details
A more specific relation between Retroviruses glycoproteins and the components of the lipid microdomains SPL or glycoSPL will be discussed below.
The entry into host cells of other Retroviruses such as the ecotropic Murine Leukemia virus (MLV) (Lu and Silver 2000; Lu et al. 2002) or the human T-cell leukemia virus (Niyogi and Hildreth 2001) was shown to depend upon these chol-rich microdomains, where their receptors are potentially located.
On the whole, the role of lipid microdomains enriched in chol and SPL in Retroviruses entry and fusion remains elusive and a matter of great dispute. One must also keep in mind that most of these studies are based upon the use of drugs that modify the composition of the cell membrane (methyl β-cyclodextrin, filipin, nystatin, inhibitors of enzymes of SPL metabolism...), and whose effect on other membrane components/interactants such as e.g., elements of the cytoskeleton and tetraspanins has not been assessed. The influence of such components might be of importance on viral membrane fusion.
Other viruses and cholesterol
The Ebola and Marburg viruses (Filoviruses), the Vaccinia virus (Orthopoxvirus), the murine Hepatitis virus (MHV, Coronavirus), the lymphocytic choriomeningitis virus (LCMV, Arenavirus) and the Herpes Simplex virus (HSV, Herpesvirus) were shown to enter into host cells via chol-rich microdomains (Aman et al. 2003; Bavari et al. 2002; Bender et al. 2003; Chung et al. 2005; Shah et al. 2006; Thorp and Gallagher 2004; Yonezawa et al. 2005). Indeed, when chol dispersion and disruption of lipid microdomains were achieved using drugs such as filipin and nystatin, virus entry was largely reduced (by more than 50% for Filoviruses), although virion attachment was unaffected, suggesting an effect at a post binding step.
A putative receptor for Filoviruses is the folate receptor-α, supposedly residing in chol-rich microdomains. As already shown for Retroviruses, cellular receptors could cluster in dense chol-rich microdomains (Harder and Simons 1997), suggesting that specific lipids could create a favorable environment around target membrane receptors and enhance their oligomerization or facilitate their recognition. These lipids could therefore participate in the conformational rearrangements of fusion proteins in the viral membrane required for fusion to occur. These microdomains are proposed as the “gateway” for filoviruses entry (Bavari et al. 2002).
In order to investigate the link between vaccinia virion entry and chol-rich microdomains, Chung et al. isolated microdomains after incubating virions with target cells (Chung et al. 2005). They noticed that some viral proteins were found exclusively in chol-rich microdomains whereas others were found in both microdomains and non-microdomains. The authors speculated that some of these proteins could have a specific microdomain localization and could be involved in the stabilization of these domains to enhance downstream signalling events.
The receptor for the murine hepatitis Coronavirus is not associated with these specific microdomains during the virus entry step (Thorp and Gallagher 2004). After binding of viral spike protein S to MHV cellular receptors, a redistribution of viral and/or cellular proteins and/or membranes occurs, involving the shift of viral/cellular components into chol-rich microdomains. Nevertheless, it is not yet clear whether chol only acts as a clustering agent of viral proteins/cellular receptors after binding or as a direct entry cofactor. A similar mechanism seems valid for other Coronaviruses such as the human coronavirus 229E (Nomura et al. 2004).
For Herpesviruses, microdomains may as well act as a platform allowing cell entry and potential coreceptors clustering. Indeed one of the five HSV glycoproteins, gB, associates with lipid microdomains (Bender et al. 2003). HSV would also require the presence of chol outside the microdomains. Thus, chol may modulate the HSV entry process regardless of its ability to promote lipids microdomains.
Specific interactions between viral envelope glycoproteins and lipids
Retroviruses and glycoSPL
As already discussed above, “rafts” are largely used by Retroviruses as platforms for their entry into target cells. Recently, Beer and Pedersen showed that early entry events of the amphotropic murine leukemia virus (A-MLV) into cells depended upon large lipid platforms or “rafts” enriched in the monosialoganglioside GM1 (Beer and Pedersen 2006). Concerning HIV, specific and direct interactions between gp120, a subunit of the fusion glycoprotein, and glycoSPL have been observed (Hammache et al. 1998). Fantini et al. measured an increase in the surface pressure of monolayers of glycoSPL such as GalCer, globotriosyl-ceramide (Gb3) and GM3, as induced by the insertion into the monolayer of either a full-length recombinant gp120 or part of gp120, the V3 loop. No effect was observed with lactosyl-ceramide (LacCer) or glucosyl-ceramide (GlcCer), nor when the fatty acyl chain length of GalCer was varied from C18 to C24, indicating that this insertion was mainly driven by the headgroup of the glycoSPL. Also, the density of GalCer into model bilayers had no effect on the binding affinity of GalCer for gp120, indicating that this binding was independent of the membrane distribution of GalCer (Viard et al. 2004). One must however keep in mind that these experiments were performed with monomeric gp120, and that HIV Env forms a trimer at the surface of the virus.
Recently, the Blumenthal’s group, working in the context of HIV-1 Env-expressing cells, glycoSPL-deficient cells and cells bearing HIV-1 receptors, demonstrated that optimal fusion conditions were achieved when gp120, the CD4 receptor and glycoSPL were associated in a trimolecular complex (Rawat et al. 2006). Interestingly, CD4 is found in GM3-enriched microdomains in the plasma membrane of lymphocytic cells, and plasma membrane chol is required to maintain the integrity of receptor pools. GlycoSPL would therefore promote HIV fusion by stabilizing association between HIV Env, CD4 and the coreceptors CXCR4 and CCR5.
We can infer that specific recognition between HIV gp120 and glycoSPL at the molecular level might induce conformational changes into gp41, the subunit of the HIV fusion protein that contains the fusion peptide. These conformational changes might lead to the proper exposure of the fusion peptide toward the target membrane. Gp41 was found to interact with GalCer, establishing a lectin-carbohydrate interaction (Alfsen and Bomsel 2002; Chazal and Gerlier 2003), whereas the interaction between gp120 and GalCer would rather be of the carbohydrate–carbohydrate type, involving the numerous glycosyl moieties of gp120. Since this interaction was not discussed in the context of membrane fusion but in the context of viral neutralization, we can only speculate that gp41/GalCer binding might be translated into fusion-relevant conformations.
A few studies using synthetic peptides derived from the fusion peptide sequence of HIV-1 further documented the link between lipid composition and fusion activity. Using the HIV-1 fusion peptide, Cladera et al. demonstrated that peptide membrane affinity and peptide-induced lipid mixing of model membranes was enhanced by the presence of chol in these membranes, due to the influence of chol on the membrane dipole potential (Buzon and Cladera 2006). Using the same fusion peptide sequence and studying its structure in a lipid environment by 13C solid-state NMR, Weliky et al. observed narrower line widths of the spectra for a lipid composition comparable to that found in the virus and its target T cells, than for a single PC composition (Yang and Weliky 2003). This is indicative of a greater peptide structural homogeneity for complex lipidic compositions, notably containing chol, SM and PI. This was translated at the functional level into a greater fusogenicity to these complex compositions. A trimer of this fusion peptide was obtained and studied by the same approach; this trimer was α-helical when associated with membranes devoid of chol, and β-stranded when associated with membranes which have a lipid headgroup and cholesterol composition similar to that of HIV target cells (Zheng et al. 2006). For Retroviruses the role of (specific) lipids or lipid compositions is therefore apparent on peptide/protein structure and fusion function.
Alphaviruses/flaviviruses and chol/SPL
It has long been described that SFV fusion is strictly cholesterol- and sphingolipid-dependent, both in in vitro and in vivo experiments (Kielian and Helenius 1984; Phalen and Kielian 1991). Similar strict lipid dependence was observed for the fusion of Sindbis virus with liposomes (Smit et al. 1999) and in vivo (Lu et al. 1999). The insertion of the fusion peptide of E1 into the target membrane requires the presence of chol and SPL, together with the application of low pH (Ahn et al. 2002). E1 then refolds to form a stable E1 homotrimer whose formation is also chol- and SPL-dependent (Klimjack et al. 1994). The dependence of Alphavirus fusion upon chol and SPL did not appear to correlate with the formation of lipid “rafts” in target liposome membranes, and SFV fusion required only very little amounts of SPL in the target membrane (Waarts et al. 2002), suggesting a direct protein–lipid interaction for the “ignition” of fusion. The influence of either lipid was investigated in SFV by selecting mutants grown on chol-depleted insect cells. Three mutants were isolated, srf-3, srf-4 and srf-5 (for sterol requirement in function) that exhibited a complete independence of the fusion process toward chol (for srf-3), or cholesterol and sphingolipids (for srf-4 and srf-5) (Chatterjee et al. 2002, 2000; Vashishtha et al. 1998) (Fig. 4). When positioning these mutations on the E1 glycoprotein structure, it appears that the mutation on a proline in position 226 (Pro226) in srf-3 affects a zone facing the fusion peptide loop, called the ij loop. The spatial environment in close vicinity around the fusion loop seems therefore to be essential for lipid recognition, specially since membrane binding facilitates trimerization (Klimjack et al. 1994). One can also conclude that this point mutation is in a “lipid-sensing” loop, and such a lipid sensor could act at an early step upstream to the trimerization, fusion peptide insertion and fusion steps (Kielian et al. 2000). The leucine (Leu44) and valine (Val178) residues mutated in srf-4 and srf-5, respectively, are in two distinct loops lying nearby the flexible hinge region between domains I and II, meaning that key regions for sphingolipid sensing are spatially close to this hinge domain whose flexibility is essential to fusion [reviewed in Kielian (2006; Kielian and Rey (2006)]. One can suggest that the presence of specific amino acids in the vicinity of this hinge domain is essential for preserving its flexibility. These amino acids could be involved in lipid recognition and undergo rearrangements after lipid binding that would open up this hinge region and lead to its upward motion. This fits particularly well with the observation on Dengue virus that in the presence of lipid-like molecules (detergent), the hinge displays an open conformation which accomodates one hydrophobic molecule (Modis et al. 2003).

Backbone 3D-structure of regions including the fusion peptide (FP) of SFV E1. FP is at the extreme right, Cys involved in disulphide bridges essential to the architecture of the FP region are shown as yellow balls (extreme right), Pro226 is shown as a red ball, and His230, Leu44 and Val178 are shown as blue balls. Central domain I is in magenta, the dimerization domain II in yellow (containing the FP) and the C-terminal domain III is in blue (see text for details; balls are symbols for C-alpha). The coordinates of these structures were retrieved from the PDB (protein data base) under accession number 1I9W (Lescar et al. 2001)
Interestingly, the Pro226 and Leu44 residues are not conserved among Alphaviruses (Roussel et al. 2006). In contrast, a histidine residue in position 230 (His230) is strictly conserved among Alphaviruses and Flaviviruses. His230 is in the ij loop (Roussel et al. 2006) (Fig. 4). A mutant SFV bearing a His230Ala mutation was totally blocked in fusion at a late stage, although the mutant E1 displayed normal conformational changes including E1/E2 dissociation, fusion loop exposure, cholesterol-dependent target membrane association, and homotrimer formation (Chanel-Vos and Kielian 2004). This points to an important role of this ij hairpin during fusion, although the His230 is not reported to be directly involved in lipid recognition. Recently, the picture emerged that the His230 could play a role on the hinge region and the orientation of the domain II tips to ensure correct interactions of the domain II tip with target membranes or with the tips of adjacent homotrimers (Chanel-Vos and Kielian 2006).
All these results point toward specific E1-lipid interactions rather than a requirement for “rafts” in Alphavirus fusion. Going into more details of the chol and SPL molecules revealed that fusion of SFV and Sindbis virus displayed a specific requirement for the 3-hydroxyl group of chol (Bron et al. 1993; Lu et al. 1999; Phalen and Kielian 1991; Smit et al. 1999; White and Helenius 1980) and SPL (Corver et al. 1995). Additional requirements for a C–C double bond on the SPL backbone (Corver et al. 1995), for the D-erythro over the L-erythro or threo series of SPL (Moesby et al. 1995) and supposedly for hydrogen bonding between hydroxyls of the SPL backbone and amino acids of E1 (Samsonov et al. 2002) revealed a fine tuning in lipid recognition by E1 at the molecular level, including stereospecificity.
The fusion reactions of the Flaviviruses TBE and West Nile are also enhanced by cholesterol (Corver et al. 2000; Gollins and Porterfield 1986; Stiasny et al. 2003), but no SPL requirement has been observed for their fusion step per se (Corver et al. 2000; Stiasny et al. 2003). As with Alphaviruses though, chol and possibly SPL had a strong promoting effect on the membrane binding and trimerization of the E fusion protein, involving the 3-hydroxyl group of lipids. However, these effects are much less pronounced than for Alphaviruses with respect to the overall fusion and can only be demonstrated when fusion is slowed down by lowering the temperature (Stiasny et al. 2003). Recent data suggested that Flaviviruses could enter their host cells by fusing with the plasma membrane at neutral pH, due to an interaction with a membrane patch of appropriate lipid composition (Koschinski et al. 2003). However, increasing in vivo evidence indicates that most Flaviviridae enter their host cells by endocytosis after receptor recognition at the plasma membrane (Stiasny and Heinz 2006). If lipids were therefore to play a role in Flaviviruses fusion, it would most probably be inside the endosome.
Hepacivirus and chol/SPL
The only representative of this viral genus is the hepatitis C virus (HCV). Increasing evidence indicates that HCV enters its host cells by endocytosis after receptor recognition at the plasma membrane (Bartosch and Cosset 2006). The role of chol in HCV entry has been addressed recently by the Chisari’s group (Kapadia et al. 2007), which showed that in vitro cell infection was chol-dependent. This sterol was also shown to facilitate HCV-mediated fusion in an HCV pseudoparticles/liposomes system, and this was dependent upon the presence of native and functional E1 and E2 proteins at the surface of the viral pseudoparticles (Lavillette et al. 2006). Additional results from our group indicated that the presence of the 3-hydroxyl group of chol was required to observe this facilitating effect, and that the highest rate and extent of fusion could be achieved when SM and chol were present in the target liposomal membranes (Nourrisson D., Bartosch B., Cosset FL. and Pécheur EI., unpublished observations). However, in the absence of 3D structural data on the E1 or E2 envelope proteins, it is not possible at present to discuss any lipid specificity or involvement in HCV membrane fusion.
Other viruses
No specific lipid requirement was reported for the fusion of Orthomyxoviruses such as the influenza virus, and the Sendai, NDV and SV5 Paramyxoviruses (Hawkes and Mak 2006; Rawat et al. 2003). As already developed above, several virus families use membranes as platforms for their entry, and lipids therefore act through their physico-chemical and mechanical properties.
Lipids as curvature modulators; consequences for fusion
The commonly accepted mechanism for membrane fusion involves the hemifusion intermediate state of negative curvature or stalk, where the distal leaflets and the aqueous inner contents remain distinct (Chernomordik and Kozlov 2005). The stalk structure, which has an hourglass shape, has been identified by Yang and Huang by X-ray diffraction (Yang et al. 2003). Nevertheless, the structure was observed at the equilibrium rather than as a transient structure.
Membrane curvature mainly depends on the molecular shape of lipids. Indeed, cone-shaped lipids with a spontaneous negative curvature such as PE, fatty acids and cholesterol promote hemifusion when present in the outer membrane leaflet, whereas inverted cone-shaped lipids with a spontaneous positive curvature such as lysolipids hinder the hemifusion process (Fig. 1). The energy required to induce hemifusion can be brought by fusion proteins in the case of viral fusion. They actually interfere with bilayer structures, and release energy from protein restructuring or protein–membrane interactions. This may produce the bilayer distortion required for hemifusion (Chernomordik and Kozlov 2005). Chernormordik et al. showed that fusion driven by either class I and class II fusion proteins gave rise to similar fusion intermediates (Zaitseva et al. 2005), that could be similarly modulated by lipids. This has also been shown by Gaudin for the fusion driven by the rabies glycoprotein (Gaudin 2000a), most probably belonging to the third class of fusion proteins described recently for VSV, another member of the Rhabdoviridae family (Roche et al. 2006). Moreover, subtle conformational changes could be locally induced by lipids modulating curvature in the structure of the fusion peptide at the onset of fusion, as shown on a model fusion peptide (Pécheur et al. 2000) and as suggested for TBEV (Stiasny and Heinz 2004). Also, the spacing between lipid head groups can modulate fusion, by facilitating or impairing the insertion of the fusion peptide into the target membrane (Pécheur et al. 1999).
Conclusions and perspectives
When considering lipids and fusion proteins as the minimal partners engaged in a fusion process, it appears that several features are common to different families of viruses. Although two, maybe three, main classes of fusion proteins could be distinguished upon structural and biochemical considerations, it seems as if these classes could induce fusion in a very similar way. For all classes of proteins, lipids appear to play a key role in the conformational changes that lead to the interaction of the fusion peptide with target membranes, and in the membrane deformation(s) that follow the initial peptide-induced curvature modification step. Lipids may directly induce conformational changes into the protein, or create a local environment around the proteins favorable to the expression of their fusion properties (receptor presentation, architecture around the fusion proteins into the viral membrane). Most recently, the novel concept of “lipid packing sensor” emerged. This lipid packing sensor is an helical motif present in cellular proteins, which has also been identified in viral proteins in a bioinformatics search (Drin et al. 2007). Such a sensor has been identified in the gL fusion glycoprotein of the Epstein-Barr virus and in the gB precursor protein of the human herpesvirus 6A, two Herpesviruses, and in the F fusion protein of a human parainfluenza virus, a Paramyxovirus (B. Antonny, personal communication). This exciting discovery opens new perspectives with respect to viral fusion proteins, that may contain such sensors. This would be of outstanding importance in our understanding of the subtle and complex interplay between protein-induced fusion and lipid modulation.
Moreover, lipids and in particular, lipids specifically recognized by viral fusion proteins on the fusion pathway can be targets for future antiviral therapies. Host sphingolipid biosynthesis has emerged as a target for HCV therapy (Sakamoto et al. 2005), as well as peroxisome proliferator-activated receptor-α, whose inhibition leads to misregulation of lipid metabolism events (Rakic et al. 2006). An inhibitor of HCV entry and membrane fusion interacting with membranes has also been recently described (Boriskin et al. 2006). HIV infection could be blocked by a soluble mimic of the glycoSPL Gb3 (Lund et al. 2006), and also by a combination of methyl-β-cyclodextrin and raft-inhibiting cholesterol analogues (Hawkes and Mak 2006).
Lipids are key actors on the pathway to membrane fusion. Delineating their intricacy with fusion proteins is an exciting challenge, and will help us better understand virus pathogenesis.
Abbreviations
- Chol:
-
Cholesterol
- HA:
-
Influenza hemagglutinin
- HCV:
-
Hepatitis C virus
- HIV:
-
Human immunodeficiency virus
- HSV:
-
Herpes simplex virus
- lysoPL:
-
Lysophospholipids
- MLV:
-
Murine leukemia virus
- PC:
-
Phosphatidylcholine
- PE:
-
Phosphatidylethanolamine
- PI:
-
Phosphatidyl inositol
- PIP:
-
Phosphatidyl inositol phosphates
- PS:
-
Phosphatidylserine
- SFV:
-
Semliki forest virus
- SM:
-
Sphingomyelin
- SPL:
-
Sphingolipids
- TBEV:
-
Tick-borne encephalitis virus
- TMD:
-
Transmembrane domains
- VSV:
-
Vesicular stomatitis virus
References
Ahn A, Gibbons DL, Kielian M (2002) The fusion peptide of Semliki Forest virus associates with sterol-rich membrane domains. J Virol 76:3267–3275
Alazard-Dany N, Ottmann Terrangle M, Volchkov V (2006) Ebola and Marburg viruses: the humans strike back. Med Sci (Paris) 22:405–410
Alfsen A, Bomsel M (2002) HIV-1 gp41 envelope residues 650-685 exposed on native virus act as a lectin to bind epithelial cell galactosyl ceramide. J Biol Chem 277:25649–25659
Aman MJ, Bosio CM, Panchal RG, Burnett JC, Schmaljohn A, Bavari S (2003) Molecular mechanisms of filovirus cellular trafficking. Microbes Infect 5:639–649
Bartosch B, Cosset FL (2006) Cell entry of hepatitis C virus. Virology 348:1–12
Bavari S, Bosio CM, Wiegand E, Ruthel G, Will AB, Geisbert TW, Hevey M, Schmaljohn C, Schmaljohn A, Aman MJ (2002) Lipid raft microdomains: a gateway for compartmentalized trafficking of Ebola and Marburg viruses. J Exp Med 195:593–602
Becher A, McIlhinney RA (2005) Consequences of lipid raft association on G-protein-coupled receptor function. Biochem Soc Symp 72:151–164
Beer C, Pedersen L (2006) Amphotropic murine leukemia virus is preferentially attached to cholesterol-rich microdomains after binding to mouse fibroblasts. Virol J 3:21
Bender FC, Whitbeck JC, Ponce de Leon M, Lou H, Eisenberg RJ, Cohen GH (2003) Specific association of glycoprotein B with lipid rafts during herpes simplex virus entry. J Virol 77:9542-9552
Blumenthal R, Clague MJ, Durell SR, Epand RM (2003) Membrane fusion. Chem Rev 103:53–69
Boriskin YS, Pécheur EI, Polyak SJ (2006) Arbidol: a broad-spectrum antiviral that inhibits acute and chronic HCV infection. Virol J 3:56
Bressanelli S, Stiasny K, Allison SL, Stura EA, Duquerroy S, Lescar J, Heinz FX, Rey FA (2004) Structure of a flavivirus envelope glycoprotein in its low-pH-induced membrane fusion conformation. Embo J 23:728–738
Bron R, Wahlberg JM, Garoff H, Wilschut J (1993) Membrane fusion of Semliki Forest virus in a model system: correlation between fusion kinetics and structural changes in the envelope glycoprotein. Embo J 12:693–701
Bullough PA, Hughson FM, Skehel JJ, Wiley DC (1994) Structure of influenza haemagglutinin at the pH of membrane fusion. Nature 371:37–43
Buzon V, Cladera J (2006) Effect of cholesterol on the interaction of the HIV GP41 fusion peptide with model membranes. Importance of the membrane dipole potential. Biochemistry 45:15768–15775
Chanel-Vos C, Kielian M (2004) A conserved histidine in the ij loop of the Semliki Forest virus E1 protein plays an important role in membrane fusion. J Virol 78:13543–13552
Chanel-Vos C, Kielian M (2006) Second-site revertants of a Semliki Forest virus fusion-block mutation reveal the dynamics of a class II membrane fusion protein. J Virol 80:6115–6122
Chatterjee PK, Vashishtha M, Kielian M (2000) Biochemical consequences of a mutation that controls the cholesterol dependence of Semliki Forest virus fusion. J Virol 74:1623–1631
Chatterjee PK, Eng CH, Kielian M (2002) Novel mutations that control the sphingolipid and cholesterol dependence of the Semliki Forest virus fusion protein. J Virol 76:12712-12722
Chazal N, Gerlier D (2003) Virus entry, assembly, budding, and membrane rafts. Microbiol Mol Biol Rev 67:226–237, table of contents
Chernomordik LV, Kozlov MM (2005) Membrane hemifusion: crossing a chasm in two leaps. Cell 123:375–382
Chernomordik LV, Zimmerberg J, Kozlov MM (2006) Membranes of the world unite! J Cell Biol 175:201–207
Chu VC, McElroy LJ, Chu V, Bauman BE, Whittaker GR (2006) The avian coronavirus infectious bronchitis virus undergoes direct low-pH-dependent fusion activation during entry into host cells. J Virol 80:3180–3188
Chung CS, Huang CY, Chang W (2005) Vaccinia virus penetration requires cholesterol and results in specific viral envelope proteins associated with lipid rafts. J Virol 79:1623–1634
Colman PM, Lawrence MC (2003) The structural biology of type I viral membrane fusion. Nat Rev Mol Cell Biol 4:309–319
Corver J, Moesby L, Erukulla RK, Reddy KC, Bittman R, Wilschut J (1995) Sphingolipid-dependent fusion of Semliki Forest virus with cholesterol-containing liposomes requires both the 3-hydroxyl group and the double bond of the sphingolipid backbone. J Virol 69:3220–3223
Corver J, Ortiz A, Allison SL, Schalich J, Heinz FX, Wilschut J (2000) Membrane fusion activity of tick-borne encephalitis virus and recombinant subviral particles in a liposomal model system. Virology 269:37–46
Da Poian AT, Carneiro FA, Stauffer F (2005) Viral membrane fusion: is glycoprotein G of rhabdoviruses a representative of a new class of viral fusion proteins? Braz J Med Biol Res 38:813–823
Drin G, Casella JF, Gautier R, Boehmer T, Schwartz TU, Antonny B (2007) A general amphipathic alpha-helical motif for sensing membrane curvature. Nat Struct Mol Biol 14:138–146
Durrer P, Gaudin Y, Ruigrok RW, Graf R, Brunner J (1995) Photolabeling identifies a putative fusion domain in the envelope glycoprotein of rabies and vesicular stomatitis viruses. J Biol Chem 270:17575–17581
Fackler OT, Krausslich HG (2006) Interactions of human retroviruses with the host cell cytoskeleton. Curr Opin Microbiol 9:409–415
Ferlenghi I, Clarke M, Ruttan T, Allison SL, Schalich J, Heinz FX, Harrison SC, Rey FA, Fuller SD (2001) Molecular organization of a recombinant subviral particle from tick-borne encephalitis virus. Mol Cell 7:593–602
Fuller SD, Berriman JA, Butcher SJ, Gowen BE (1995) Low pH induces swiveling of the glycoprotein heterodimers in the Semliki Forest virus spike complex. Cell 81:715–725
Gaibelet G, Planchenault T, Mazeres S, Dumas F, Arenzana-Seisdedos F, Lopez A, Lagane B, Bachelerie F (2006) CD4 and CCR5 constitutively interact at the plasma membrane of living cells: a confocal fluorescence resonance energy transfer-based approach. J Biol Chem 281:37921–37929
Garry RF, Dash S (2003) Proteomics computational analyses suggest that hepatitis C virus E1 and pestivirus E2 envelope glycoproteins are truncated class II fusion proteins. Virology 307:255–65
Gaudin Y (2000a) Rabies virus-induced membrane fusion pathway. J Cell Biol 150:601–612
Gaudin Y (2000b) Reversibility in fusion protein conformational changes. The intriguing case of rhabdovirus-induced membrane fusion. Subcell Biochem 34:379–408
Gibbons DL, Vaney MC, Roussel A, Vigouroux A, Reilly B, Lepault J, Kielian M, Rey FA (2004) Conformational change and protein-protein interactions of the fusion protein of Semliki Forest virus. Nature 427:320–325
Gollins SW, Porterfield JS (1986) pH-dependent fusion between the flavivirus West Nile and liposomal model membranes. J Gen Virol 67(Pt 1):157–166
Hammache D, Pieroni G, Yahi N, Delezay O, Koch N, Lafont H, Tamalet C, Fantini J (1998) Specific interaction of HIV-1 and HIV-2 surface envelope glycoproteins with monolayers of galactosylceramide and ganglioside GM3. J Biol Chem 273:7967–7971
Harder T, Simons K (1997) Caveolae, DIGs, and the dynamics of sphingolipid-cholesterol microdomains. Curr Opin Cell Biol 9:534–542
Hawkes DJ, Mak J (2006) Lipid membrane; a novel target for viral and bacterial pathogens. Curr Drug Targets 7:1615–1621
Heldwein EE, Lou H, Bender FC, Cohen GH, Eisenberg RJ, Harrison SC (2006) Crystal structure of glycoprotein B from herpes simplex virus 1. Science 313:217–220
Ito H, Watanabe S, Sanchez A, Whitt MA, Kawaoka Y (1999) Mutational analysis of the putative fusion domain of Ebola virus glycoprotein. J Virol 73:8907–8912
Kahya N (2006) Targeting membrane proteins to liquid-ordered phases: molecular self-organization explored by fluorescence correlation spectroscopy. Chem Phys Lipids 141:158–168
Kapadia SB, Barth H, Baumert T, McKeating JA, Chisari FV (2007) Initiation of hepatitis C virus infection is dependent on cholesterol and cooperativity between CD81 and scavenger receptor B type I. J Virol 81:374–383
Kemble GW, Danieli T, White JM (1994) Lipid-anchored influenza hemagglutinin promotes hemifusion, not complete fusion. Cell 76:383–391
Kielian M (2006) Class II virus membrane fusion proteins. Virology 344:38–47
Kielian MC, Helenius A (1984) Role of cholesterol in fusion of Semliki Forest virus with membranes. J Virol 52:281–283
Kielian M, Rey FA (2006) Virus membrane-fusion proteins: more than one way to make a hairpin. Nat Rev Microbiol 4:67–76
Kielian M, Chatterjee PK, Gibbons DL, Lu YE (2000) Specific roles for lipids in virus fusion and exit. Examples from the alphaviruses. Subcell Biochem 34:409–455
Klimjack MR, Jeffrey S, Kielian M (1994) Membrane and protein interactions of a soluble form of the Semliki Forest virus fusion protein. J Virol 68:6940–6946
Kobayashi M, Bennett MC, Bercot T, Singh IR (2006) Functional analysis of hepatitis C virus envelope proteins, using a cell-cell fusion assay. J Virol 80:1817–1825
Koschinski A, Wengler G, Repp H (2003) The membrane proteins of flaviviruses form ion-permeable pores in the target membrane after fusion: identification of the pores and analysis of their possible role in virus infection. J Gen Virol 84:1711–1721
Kuhn RJ, Zhang W, Rossmann MG, Pletnev SV, Corver J, Lenches E, Jones CT, Mukhopadhyay S, Chipman PR, Strauss EG, Baker TS, Strauss JH (2002) Structure of dengue virus: implications for flavivirus organization, maturation, and fusion. Cell 108:717–725
Kunz S, Borrow P, Oldstone MB (2002) Receptor structure, binding, and cell entry of arenaviruses. Curr Top Microbiol Immunol 262:111–137
Lavillette D, Bartosch B, Nourrisson D, Verney G, Cosset FL, Penin F, Pécheur EI (2006) Hepatitis C Virus Glycoproteins Mediate Low pH-dependent membrane fusion with liposomes. J Biol Chem 281:3909–3917
Lemmon MA (2004) Pleckstrin homology domains: not just for phosphoinositides. Biochem Soc Trans 32:707–711
Lescar J, Roussel A, Wien MW, Navaza J, Fuller SD, Wengler G, Rey FA (2001) The Fusion glycoprotein shell of Semliki Forest virus: an icosahedral assembly primed for fusogenic activation at endosomal pH. Cell 105:137–148
Lu X, Silver J (2000) Ecotropic murine leukemia virus receptor is physically associated with caveolin and membrane rafts. Virology 276:251–258
Lu YE, Cassese T, Kielian M (1999) The cholesterol requirement for sindbis virus entry and exit and characterization of a spike protein region involved in cholesterol dependence. J Virol 73:4272–4278
Lu X, Xiong Y, Silver J (2002) Asymmetric requirement for cholesterol in receptor-bearing but not envelope-bearing membranes for fusion mediated by ecotropic murine leukemia virus. J Virol 76:6701–6709
Lund N, Branch DR, Mylvaganam M, Chark D, Ma XZ, Sakac D, Binnington B, Fantini J, Puri A, Blumenthal R, Lingwood CA (2006) A novel soluble mimic of the glycolipid, globotriaosyl ceramide inhibits HIV infection. Aids 20:333–343
Manes S, Lacalle RA, Gomez-Mouton C, del Real G, Mira E, Martinez AC (2001) Membrane raft microdomains in chemokine receptor function. Semin Immunol 13:147–157
McIntosh TJ, Simon SA (2006) Roles of bilayer material properties in function and distribution of membrane proteins. Annu Rev Biophys Biomol Struct 35:177–198
Modis Y, Ogata S, Clements D, Harrison SC (2003) A ligand-binding pocket in the dengue virus envelope glycoprotein. Proc Natl Acad Sci USA 100:6986–6991
Moesby L, Corver J, Erukulla RK, Bittman R, Wilschut J (1995) Sphingolipids activate membrane fusion of Semliki Forest virus in a stereospecific manner. Biochemistry 34:10319–10324
Monath T, Heinz F (1996) Flaviviruses. In: Fields B, Knipe D, Howley P (eds) Fields virology, vol 1. Lippincott-Raven, Philadelphia, pp 961–1034
Mukhopadhyay S, Kim BS, Chipman PR, Rossmann MG, Kuhn RJ (2003) Structure of West Nile virus. Science 302:248
Niyogi K, Hildreth JE (2001) Characterization of new syncytium-inhibiting monoclonal antibodies implicates lipid rafts in human T-cell leukemia virus type 1 syncytium formation. J Virol 75:7351–7361
Nomura R, Kiyota A, Suzaki E, Kataoka K, Ohe Y, Miyamoto K, Senda T, Fujimoto T (2004) Human coronavirus 229E binds to CD13 in rafts and enters the cell through caveolae. J Virol 78:8701–8708
Pécheur EI, Sainte-Marie J, Bienvenue A, Hoekstra D (1999) Lipid headgroup spacing and peptide penetration, but not peptide oligomerization, modulate peptide-induced fusion. Biochemistry 38:364–373
Pécheur EI, Martin I, Bienvenue A, Ruysschaert JM, Hoekstra D (2000) Protein-induced fusion can be modulated by target membrane lipids through a structural switch at the level of the fusion peptide. J Biol Chem 275:3936–3942
Percherancier Y, Lagane B, Planchenault T, Staropoli I, Altmeyer R, Virelizier JL, Arenzana-Seisdedos F, Hoessli DC, Bachelerie F (2003) HIV-1 entry into T-cells is not dependent on CD4 and CCR5 localization to sphingolipid-enriched, detergent-resistant, raft membrane domains. J Biol Chem 278:3153–3161
Phalen T, Kielian M (1991) Cholesterol is required for infection by Semliki Forest virus. J Cell Biol 112:615–623
Rakic B, Sagan SM, Noestheden M, Belanger S, Nan X, Evans CL, Xie XS, Pezacki JP (2006) Peroxisome proliferator-activated receptor alpha antagonism inhibits hepatitis C virus replication. Chem Biol 13:23–30
Rawat SS, Viard M, Gallo SA, Rein A, Blumenthal R, Puri A (2003) Modulation of entry of enveloped viruses by cholesterol and sphingolipids (review). Mol Membr Biol 20:243–254
Rawat SS, Johnson BT, Puri A (2005) Sphingolipids: modulators of HIV-1 infection and pathogenesis. Biosci Rep 25:329–343
Rawat SS, Viard M, Gallo SA, Blumenthal R, Puri A (2006) Sphingolipids, cholesterol, and HIV-1: a paradigm in viral fusion. Glycoconj J 23:189–197
Rey FA (2006) Molecular gymnastics at the herpesvirus surface. EMBO Rep 7:1000–1005
Rey FA, Heinz FX, Mandl C, Kunz C, Harrison SC (1995) The envelope glycoprotein from tick-borne encephalitis virus at 2 A resolution. Nature 375:291–298
Roche S, Gaudin Y (2002) Characterization of the equilibrium between the native and fusion-inactive conformation of rabies virus glycoprotein indicates that the fusion complex is made of several trimers. Virology 297:128–135
Roche S, Bressanelli S, Rey FA, Gaudin Y (2006) Crystal structure of the low-pH form of the vesicular stomatitis virus glycoprotein G. Science 313:187–191
Roche S, Rey FA, Gaudin Y, Bressanelli S (2007) Structure of the prefusion form of the vesicular stomatitis virus glycoprotein G. Science 315:843–848
Roussel A, Lescar J, Vaney MC, Wengler G, Rey FA (2006) Structure and interactions at the viral surface of the envelope protein E1 of Semliki Forest virus. Structure 14:75–86
Roux KH, Taylor KA (2007) AIDS virus envelope spike structure. Curr Opin Struct Biol 17:244–252
Russell CJ, Luque LE (2006) The structural basis of paramyxovirus invasion. Trends Microbiol 14:243–246
Sakamoto H, Okamoto K, Aoki M, Kato H, Katsume A, Ohta A, Tsukuda T, Shimma N, Aoki Y, Arisawa M, Kohara M, Sudoh M (2005) Host sphingolipid biosynthesis as a target for hepatitis C virus therapy. Nat Chem Biol 1:333–337
Samsonov AV, Chatterjee PK, Razinkov VI, Eng CH, Kielian M, Cohen FS (2002) Effects of membrane potential and sphingolipid structures on fusion of Semliki Forest virus. J Virol 76:12691–12702
Schlesinger S, Schlesinger M (1996) Togaviridae: the viruses and their replication. In: Fields B, Knipe D, Howley P (eds) Fields virology, vol 1. Lippincott-Raven, Philadelphia, pp 825–841
Shah WA, Peng H, Carbonetto S (2006) Role of non-raft cholesterol in lymphocytic choriomeningitis virus infection via alpha-dystroglycan. J Gen Virol 87:673–678
Siegel DP, Epand RM (1997) The mechanism of lamellar-to-inverted hexagonal phase transitions in phosphatidylethanolamine: implications for membrane fusion mechanisms. Biophys J 73:3089–3111
Smit JM, Bittman R, Wilschut J (1999) Low-pH-dependent fusion of Sindbis virus with receptor-free cholesterol- and sphingolipid-containing liposomes. J Virol 73:8476–8484
Steven AC, Spear PG (2006) Biochemistry. Viral glycoproteins and an evolutionary conundrum. Science 313:177–178
Stiasny K, Heinz FX (2004) Effect of membrane curvature-modifying lipids on membrane fusion by tick-borne encephalitis virus. J Virol 78:8536–8542
Stiasny K, Heinz FX (2006) Flavivirus membrane fusion. J Gen Virol 87:2755–2766
Stiasny K, Koessl C, Heinz FX (2003) Involvement of lipids in different steps of the flavivirus fusion mechanism. J Virol 77:7856–7862
Thorp EB, Gallagher TM (2004) Requirements for CEACAMs and cholesterol during murine coronavirus cell entry. J Virol 78:2682–2692
Tong S, Compans RW (2000) Oligomerization, secretion, and biological function of an anchor-free parainfluenza virus type 2 (PI2) fusion protein. Virology 270:368–376
van Meer G (1998) Lipids of the Golgi membrane. Trends Cell Biol 8:29–33
van Meer G (2005) Cellular lipidomics. Embo J 24:3159–3165
Vashishtha M, Phalen T, Marquardt MT, Ryu JS, Ng AC, Kielian M (1998) A single point mutation controls the cholesterol dependence of Semliki Forest virus entry and exit. J Cell Biol 140:91–99
Viard M, Parolini I, Rawat SS, Fecchi K, Sargiacomo M, Puri A, Blumenthal R (2004) The role of glycosphingolipids in HIV signaling, entry and pathogenesis. Glycoconj J 20:213–222
Waarts BL, Bittman R, Wilschut J (2002) Sphingolipid and cholesterol dependence of alphavirus membrane fusion. Lack of correlation with lipid raft formation in target liposomes. J Biol Chem 277:38141–38147
Waarts BL, Smit JM, Aneke OJ, McInerney GM, Liljestrom P, Bittman R, Wilschut J (2005) Reversible acid-induced inactivation of the membrane fusion protein of Semliki Forest virus. J Virol 79:7942–7948
Weiss CD, Barnett SW, Cacalano N, Killeen N, Littman DR, White JM (1996) Studies of HIV-1 envelope glycoprotein-mediated fusion using a simple fluorescence assay. Aids 10:241–246
White J, Helenius A (1980) pH-dependent fusion between the Semliki Forest virus membrane and liposomes. Proc Natl Acad Sci USA 77:3273–3277
Yagnik AT, Lahm A, Meola A, Roccasecca RM, Ercole BB, Nicosia A, Tramontano A (2000) A model for the hepatitis C virus envelope glycoprotein E2. Proteins 40:355–366
Yang J, Weliky DP (2003) Solid-state nuclear magnetic resonance evidence for parallel and antiparallel strand arrangements in the membrane-associated HIV-1 fusion peptide. Biochemistry 42:11879–11890
Yang L, Ding L, Huang HW (2003) New phases of phospholipids and implications to the membrane fusion problem. Biochemistry 42:6631–6635
Yi L, Fang J, Isik N, Chim J, Jin T (2006) HIV gp120-induced interaction between CD4 and CCR5 requires cholesterol-rich microenvironments revealed by live cell fluorescence resonance energy transfer imaging. J Biol Chem 281:35446–35453
Yonezawa A, Cavrois M, Greene WC (2005) Studies of ebola virus glycoprotein-mediated entry and fusion by using pseudotyped human immunodeficiency virus type 1 virions: involvement of cytoskeletal proteins and enhancement by tumor necrosis factor alpha. J Virol 79:918–926
Zaitseva E, Mittal A, Griffin DE, Chernomordik LV (2005) Class II fusion protein of alphaviruses drives membrane fusion through the same pathway as class I proteins. J Cell Biol 169:167–177
Zhang W, Mukhopadhyay S, Pletnev SV, Baker TS, Kuhn RJ, Rossmann MG (2002) Placement of the structural proteins in Sindbis virus. J Virol 76:11645–11658
Zhang W, Chipman PR, Corver J, Johnson PR, Zhang Y, Mukhopadhyay S, Baker TS, Strauss JH, Rossmann MG, Kuhn RJ (2003) Visualization of membrane protein domains by cryo-electron microscopy of dengue virus. Nat Struct Biol 10:907–912
Zheng Z, Yang R, Bodner ML, Weliky DP (2006) Conformational flexibility and strand arrangements of the membrane-associated HIV fusion peptide trimer probed by solid-state NMR spectroscopy. Biochemistry 45:12960–12975
Zhu P, Liu J, Bess J Jr, Chertova E, Lifson JD, Grise H, Ofek GA, Taylor KA, Roux KH (2006) Distribution and three-dimensional structure of AIDS virus envelope spikes. Nature 441:847–852
Acknowledgments
We wish to thank the Société Française de Biophysique (SFB) for the opportunity to write this review, Patrice Gouet and Antoine Corbin for critical reading of the manuscript, and Marie Perrault for editing.
Author information
Authors and Affiliations
Corresponding author
Additional information
Presented at the joint biannual meeting of the SFB-GEIMM-GRIP, Anglet France, 14–19 October, 2006.
Rights and permissions
About this article
Cite this article
Teissier, É., Pécheur, EI. Lipids as modulators of membrane fusion mediated by viral fusion proteins. Eur Biophys J 36, 887–899 (2007). https://doi.org/10.1007/s00249-007-0201-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00249-007-0201-z