
Abstract
Tilletia foetida (syn. T. laevis) leads to wheat common bunt, a worldwide disease that can lead to 80% yield loss and even total loss of production, together with degrading the quality of grains and flour by producing a rotten fish smell. To explore the potential microbial community that may contribute to the control of soil- and seed-borne pathogens, in this study, we analyzed the effects of the plant pathogenic fungus T. foetida on rhizosphere soil microorganisms in wheat seeds coated with different concentrations of a fungicide (Jianzhuang) used to control the disease. To analyze the bacterial and fungal abundance in T. foetida-infected and mock-infected plants, the microorganisms were sequenced using high-throughput HiSeq 2500 gene sequencing. The results showed that bacterial communities, including Verrucomicrobia, Patescibacteria, Armatimonadetes, Nitrospirae, Fibrobacteres, Chlamydiae, and Hydrogenedentes, and fungal communities, including Basidiomycota and Ciliophora, were more prevalent in the mock group than in the T. foetida-infected group, which may contribute to the control of wheat common bunt. Moreover, cluster and PCoA analysis revealed that replicates of the same samples were clustered together, and these results were also found in the distance index within-group analysis for bacterial and fungal communities in the T. foetida-infected and mock groups.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Tilletia foetida (syn. T. laevis) is a pathogen that can lead to wheat common bunt [1, 2]. Plants infected with T. foetida usually produce a lower yield, with low quality compared to healthy plants. Reductions in the quality and quantity of infected plants occur due to replacement of mature grains with bunt sori [1]. Additionally, wheat flour millers usually refuse grains infected by T. foetida, as very low infection rates can result in obvious unwanted odors in flour [3, 4]. The disease incidence can reach 70 to 80% using T. foetida-susceptible seeds, with 41% yield loss in Romania [5] and losses reaching 25–30% and 10–20% in Iran and Turkey, respectively [6].
Rhizosphere soil is a dynamic and complex environment; its biological activity is typically regulated by microorganisms, which play crucial roles in sustaining the health of agricultural and natural soil systems [7]. It is well known that fungal and bacterial communities are responsible for multifaceted biological functions in soils, and maintaining the biodiversity of microbes is crucial to soil fitness [8]. However, many factors affect rhizosphere soil microorganisms, such as plant pathogens, which are a critical component of rhizosphere microbial communities and play an important role in plant growth and health [9]. Some fungi are known for biocontrol activity against pathogenic microorganisms [10], which positively support plant productivity by enhancing plant growth. However, some fungi negatively influence plant health, such as some plant pathogens in the soil; for example, Fusarium graminearum can cause stalk rot disease of maize [11], Verticillium nonalfalfae can cause verticillium wilt on tree of heaven [12], and Macrophomina phaseolina can cause dry root rot disease [13]. T. foetida is a soil- and seed-borne pathogen that leads to wheat common bunt, which may cause 80% loss in China and degrade the quality of wheat seeds and flour by producing rotten fish smells. Moreover, the teliospores of the pathogen can live in soil for up to 10 years and can germinate or even undergo excretion by animals or people.
Seed treatments with fungicides are broadly used to control some fungal pathogens, such as Alternaria alternata and Fusarium sp. [14, 15]. T. foetida has the ability to penetrate locally into host plants, and seed treatment with fungicides is approved as an option for its control [16]. Common bunt symptoms appear during the boot stage under conducive environmental conditions and are difficult to control at this stage through fungicide spraying [17]. Therefore, fungicide application for seed treatment has been used for disease control. Previous studies showed that Jianzhuang fungicide has good results against different pathogens, such as Pseudomonas, compared to triazole, a common wheat fungicide, against different fungal pathogens [18,19,20]. However, pesticides often affect microorganisms in the soil based on the dose, properties of the soil, and environmental factors [21,22,23]. Some studies have shown that glyphosate application increased Proteobacteria concentrations in the rhizospheres of corn (Zea mays) and soybean (Glycine max) [24]. Ryan concluded that pesticide usage might affect some groups of microorganisms in the soil but has little effect on the soil community [25]. Gupta and Kalia [23] reported that the usage of pesticides affects all of the soil microbes, with reductions in the average populations of all groups studied in soil samples from fields with rice-wheat cropping. Regarding the use of Jianzhuang in controlling wheat common bunt, there is little information on the effects of fungicides on soil microorganisms.
To optimize the concentration of Jianzhuang applied on wheat seeds, based on Illumina HiSeq 2500 sequencing, this study characterized the rhizosphere fungal and bacterial communities in the context of infection with the plant pathogen T. foetida. Our work will lay a foundation for potential biocontrol activity with rhizosphere bacterial and fungal communities and provide insights into the interactions of plant pathogens, rhizosphere bacteria and fungi, and plant growth.
Materials and Methods
Plant Material and Pesticide Application
The wheat (Triticum aestivum L.) spring cultivar Morocco (highly susceptible) was obtained from the Institute of Plant Protection (IPP), Chinese Academy of Agricultural Sciences (CAAS), Beijing, China. Wheat seeds were sterilized with 30% sodium hypochlorite for 5 min, rinsed 5 times in ddH2O, and germinated for 30 days in an incubator (AUCMA, Qing Dao, China) to vernalize after fungicide application. The fungicide programs are illustrated in Table 1. After vernalization, seedlings were grown in a 1:2 mixture of organic matter (peat moss, Beijing, China) and soil (Beijing, China) in pots. Seedlings were grown in a 14-h light/10-h dark cycle at 15 °C to the tillering stage and at 25 °C at the boot stage.
Fungal Inoculation
T. foetida was grown on 2% agar media and incubated for 15 days at 15 °C under 24 h of light. The mycelium of T. foetida was harvested under laminar flow by adding 5 mL of ddH2O onto each T. foetida culture plate. Hyphae of T. foetida were injected 5 times into the root zone at 2-day intervals, while mock plants were treated with ddH2O. The suspension contained infectious hyphae at a concentration of 106 cfu/mL with an OD600 of 0.15. Rhizosphere soil was collected during the booting stage of wheat from T. foetida-infected and mock plants.
Molecular Detection of the Infection of T. foetida in Inoculated Wheat Leaves
DNA was extracted from wheat leaves 1 week after T. foetida inoculation using a Plant Genomic DNA Kit (TianGen, China). The primer sequences for T. foetida were (5′-TCACTTCAAGGTCGTTCCCG-3′)/L60R (5′-CGGGTCGAGGGGCGTAAACTTGA-3′). Polymerase chain reaction (PCR) and gel electrophoresis were performed to visualize the expected 660-bp band of T. foetida with high specificity, as described by Yao et al. [4].
Soil Sampling
Soil samples were collected from both T. foetida-infected and mock pots at 20 cm depth. For each sample, there were 6 T. foetida-infected plants and 6 mock plants, with 6 rhizosphere soil samples collected from each infected plant and 6 from mock plants. After shaking the roots to remove loose soil, a sterile brush was used to collect the residual soil from the roots. Equal amounts of rhizosphere soil from the three wheat plants were mixed and stored at − 80 °C. Ten grams of rhizosphere soil was collected and mixed into 90 mL of distilled water in a 150-mL tube (Houdior, China). These tubes were shaken for 10 min in an incubator shaker (ZHWY-200D, China) and incubated for 5 min. Soil solution (1 mL) for every sample was taken and sequenced after culturing.
DNA Extraction and PCR Amplification
Soil microbial DNA was extracted using a HiPure soil DNA kit (Magen, Guangzhou, China) following the manufacturer’s protocols. DNA was extracted from 0.5 g of soil suspension. The concentration and quality of every extracted DNA sample were evaluated based on the 260/280 nm and 260/230 nm absorbance ratios obtained using a NanoDrop 2000 (Thermo Scientific, USA). With the primers 341F (5′-CCTAGGGNGGCWGCAG-3′) and 806R (5′-GGACTACHVGGGTATCTAAT-3′) for V3–V4 of the 16S RNA gene and ITS3_KYO2 (5′-GATGAAGAACGYAGYRAA-3′) and ITS4 (5′-TCCTCCGCTTATTGATATGC-3′) for ITS2 region of ribosomal DNA, PCRs were performed in triplicate with a total volume of 50 μL, including 5 μL of 10× KOD buffer, 5 μL of 2.5 mM dNTPs, 1.5 μL of each primer (5 μM), 1 μL of KOD polymerase, and 100 ng of template DNA. The amplification program was 95 °C for 2 min, followed by 27 cycles at 98 °C for 10 s, 62 °C for 30 s, and a final extension of 68 °C for 10 min.
Illumina HiSeq 2500 Sequencing
PCR products were recovered using a 1.5% agarose gel and purified using an AxyPrep DNA Gel Extraction Kit (Axygen Biosciences, Union City, CA, USA) according to the manufacturer’s guidelines and quantified using an ABI Step OnePlus Real-time PCR system (Life Technologies, Foster City, USA). Purified amplicons were pooled in equimolar amounts and paired-end sequenced (2 × 250) on an Illumina platform according to standard protocols. Raw readings were stored in the NCBI Sequence Read Archive (SRA) database. Sequencing was performed in triplicate using samples from three independent experiments.
Data Analysis
After sequencing, all raw reads were analyzed using the QIIME standard pipeline [26] to trim the low-quality reads. Operational taxonomic units (OTUs) were clustered with a 97% similarity cutoff using UPARSE [27] (version 7.1 http://drive5.com/uparse/). The taxonomic classification was further checked using the Ribosomal Database Project (RDP) classifier (version 2.2) based on the Greengenes Database with 0.8 to 1 confidence [28, 29]. With the cluster file, alpha diversity statistics, including the Chao1 richness estimate, Shannon diversity index, and Simpson diversity index were calculated in MOTHUR for each sample [30, 31]. Beta diversity statistics based on UniFrac metrics were also carried out, including cluster analysis, weighted UniFrac distance metrics, and principal component analysis (PCoA) [31, 32]. Cluster analysis (CA) was used to group communities of twelve different samples. Weighted UniFrac distance metrics were used to estimate the community diversity of twelve samples. PCoA analysis to compare the discrepancies among individuals and communities was conducted based on (1) taxonomy results from the NCBI database; (2) OTUs described above; and (3) weighted UniFrac metrics.
Results
Phylotypes and Diversity of the Microbial Community
The soil samples were obtained from infected and mock plants with different concentrations of Jianzhuang, which were verified by specific primer markers of T. foetida (Figure S1). Using Illumina HiSeq 2500 sequencing, raw reads were produced for these samples. After filtering the raw reads using the QIME standard pipeline and removing all other chimeric tags, effective tags were used for further analysis. Average, total, minimum, and maximum ratios of all samples for raw data, clean data, raw tags, clean tags, and effective tags were calculated with the effective ratio. In total, 83,483–106,580 effective tags were obtained for these samples. Additionally, the percentage of valid data for each sample was more than 88%, indicating that at least 88% of tags of each sample met the requirements of sequencing length and quality (Table S1).
Furthermore, the number of OTUs was calculated for bacterial communities with three replications for each sample, and the changes in species abundance were also calculated from the domain to species level. OTU results showed that there were different species present during sequencing. The expression abundance levels of 10 species reached at least 2% in one sample, while other species were classified into unclassified categories (Table S2). Similarly, OTUs were calculated for the fungal species with three replications for every sample; the list of these species is provided in Table S3.
Bacterial Communities in Rhizosphere Soil After T. foetida Inoculation
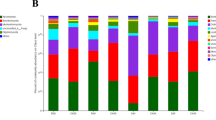
There were differences in the OTUs within T. foetida-infected and mock libraries of Jianzhuang-coated seeds. The relative abundance of different microbial communities varied in the T. foetida-infected and mock samples. The results showed that the IC (5% RD) library had maximum abundance, while the IF (no seed treatment) and NK (mock of 1.5 times RD) libraries had minimum relative abundance of Proteobacteria. However, the overall abundance of Proteobacteria was high in T. foetida-infected libraries. Similarly, the relative abundance of Planctomycetes was high in the mock group, including the NG (1.5% RD), NH (3% RD), NI (5% RD), and NJ (RD) groups, compared to the T. foetida-infected group, including the IA (1.5% RD), IB (3% RD), IC (5% RD), and ID (RD) samples. Additionally, the abundance levels of all other bacterial phyla were different under different treatments (Fig. 1).

Bacterial compositions of the different communities in rhizosphere soil. Relative abundance levels of different bacterial phyla within different libraries. IA (T. foetida + 1.5% RD of Jianzhuang), IB (T. foetida + 3% RD of Jianzhuang), IC (T. foetida + 5% RD of Jianzhuang), ID (T. foetida + RD of Jianzhuang), IE (T. foetida + 1.5 times RD of Jianzhuang), IF (T. foetida + no Jianzhuang), NG (mock + 1.5% RD of Jianzhuang), NH (mock + 3% RD of Jianzhuang), NI (mock + 5% RD of Jianzhuang), NJ (mock + % RD of Jianzhuang), NK (mock + 1.5 times RD of Jianzhuang), and NL (mock + no Jianzhuang). RD stands for recommended dose, and mock means ddH2O application
Fungal Communities in Rhizosphere Soil After T. foetida Infection
Fungal communities were analyzed after T. foetida infection in Jianzhuang-coated seeds using OTU analysis (Table S3). The T. foetida-infected samples from the IB (3% RD) library had the maximum number of Ascomycota. The relative abundance of Basidiomycota was higher in the mock-infected libraries than in the T. foetida-infected libraries. The maximum abundance of Basidiomycota was found in the NH (mock of 3% RD) library, and the minimum abundance of Basidiomycota was found in the infected IE (1.5% RD) library, suggesting that Jianzhuang (1.5% RD) was better for controlling T. foetida infection. Additionally, the abundance levels of all other fungal phyla were different under different treatments (Fig. 2).

Fungal compositions of the different communities in rhizosphere soil. Relative abundance levels of different fungal phyla within different samples. IA (T. foetida + 1.5% RD of Jianzhuang), IB (T. foetida + 3% RD of Jianzhuang), IC (T. foetida + 5% RD of Jianzhuang), ID (T. foetida + RD of Jianzhuang), IE (T. foetida + 1.5 times RD of Jianzhuang), IF (T. foetida + no Jianzhuang), NG (mock + 1.5% RD of Jianzhuang), NH (mock + 3% RD of Jianzhuang), NI (mock + 5% RD of Jianzhuang), NJ (mock + % RD of Jianzhuang), NK (mock + 1.5 times RD of Jianzhuang), and NL (mock + no Jianzhuang). RD stands for recommended dose, and mock means ddH2O application
Cluster and Principal Component Analysis for Bacterial Communities
As shown in Fig. 3a, comparing the distances between the six treatments and their control, it can be seen for the two groups, 5% “Jianzhuang” dressing (IC) and its control (NI) and 1 times “Jianzhuang” dressing (ID) and its control (NJ), the treatment group is the farthest from the control group. This was followed by 1.5% “Jianzhuang” seed dressing (IA) and its control (NG) and 1.5 times “Jianzhuang” seed dressing (IE) and its control (NK). Among the two groups of 3% “Jianzhuang” seed dressing (IB) treatment and its control (NH), and no seed treatment (IF) and its control (NL), the distance between the treatment and the control is relatively small.

Beta (β) diversity of twelve bacterial samples. a Phylogenetic tree. b Two-dimensional PCoA analysis. IA (T. foetida + 1.5% RD of Jianzhuang), IB (T. foetida + 3% RD of Jianzhuang), IC (T. foetida + 5% RD of Jianzhuang), ID (T. foetida + RD of Jianzhuang), IE (T. foetida + 1.5 times RD of Jianzhuang), IF (T. foetida + no Jianzhuang), NG (mock + 1.5% RD of Jianzhuang), NH (mock + 3% RD of Jianzhuang), NI (mock + 5% RD of Jianzhuang), NJ (mock + % RD of Jianzhuang), NK (mock + 1.5 times RD of Jianzhuang), and NL (mock + no Jianzhuang). RD stands for recommended dose, and mock means ddH2O application
Based on the unweighted UniFrac distance metrics, PCoA was performed to assess the similarities of different infected and mock libraries in Jianzhuang-coated seeds. The PCoA analysis results revealed maximum variations of 56.93% (PC1) and 11.96% (PC2), as shown in Fig. 3b. Similar results were also obtained between the 3% “Jianzhuang” seed dressing (IB) treatment and the control (NH), the no seed treatment group (IF) and the control (NL). The distance between these two groups was small, and the distance between the other groups was large. We also found that among all of the inoculated treatment groups, IA (1.5% RD), IC (5% RD), and ID (RD) were farther away from the other treatments, while the distances between IB (3% RD) and IE (1.5 times RD) were closer, indicating that the effects of these two concentrations of Jianzhuang seed dressings on the rhizosphere bacterial community were similar.
Cluster and Principal Component Analysis of Fungal Communities
In Fig. 4a, comparing the distances between the 6 treatments and their mock, the distance between the 3% “Jianzhuang” seed dressing (IB) treatment and the mock (NH) treatment was the largest. Among the three groups of 5% “Jianzhuang” seed dressing (IC) and its mock (NI), 1 time of “Jianzhuang” seed dressing (ID) and its mock (NJ), and no seed treatment (IF) and its mock (NL), the distance between the treatments and their mock was slightly shorter. Among 1.5% “Jianzhuang” seed dressing (IA) and its mock (NG) and 1.5 times “Jianzhuang” seed dressing (IE) and its mock (NK), the difference between the treatment group and the control group was relatively small.

Beta (β) diversity of twelve fungal samples. a Phylogenetic tree. b Two-dimensional PCoA analysis. IA (T. foetida + 1.5% RD of Jianzhuang), IB (T. foetida + 3% RD of Jianzhuang), IC (T. foetida + 5% RD of Jianzhuang), ID (T. foetida + RD of Jianzhuang), IE (T. foetida + 1.5 times RD of Jianzhuang), IF (T. foetida + no Jianzhuang), NG (mock + 1.5% RD of Jianzhuang), NH (mock + 3% RD of Jianzhuang), NI (mock + 5% RD of Jianzhuang), NJ (mock + % RD of Jianzhuang), NK (mock + 1.5 times RD of Jianzhuang), and NL (mock + no Jianzhuang). RD stands for recommended dose, and mock means ddH2O application
The results of PCoA revealed maximum variations of 59.34% (PC1) and 20.13% (PC2). It can be seen from Fig. 4b that both the treatment group and the mock group have good repeatability, and the sample is in a confidence circle. At the same time, the distance between each group and their mock was large, indicating that T. foetida has a relatively large impact on the fungal community of wheat rhizosphere soil.
Heatmap Analysis for Bacterial Communities
The abundance levels of eighteen bacterial phyla were characterized in T. foetida-inoculated and mock libraries. Community heatmap analysis was scaled to demonstrate the abundance levels of the various bacterial communities. The results showed that Proteobacteria (IC, T. foetida infected + 5% RD), Actinobacteria (IB, T. foetida infected + 3% RD), Firmicutes (IC, T. foetida infected + 5% RD), BRC1 (IB, T. foetida infected + 3% RD), Cyanobacteria (IC, T. foetida infected + 5% RD), and Elusimicrobia (IE, T. foetida infected + 1.5 times of RD) were abundant in the T. foetida infected compared to mock samples, while Planctomycetes (NG, mock + 1.5% RD), Bacteroidetes (NL, mock + no Jianzhuang treatment), Verrucomicrobia (NJ, mock + RD), Patescibacteria (NJ, mock + RD), Chloroflexi (NI, mock + 5% RD), Armatimonadetes (NG, mock + 1.5% RD), Nitrospirae (NI, mock + 1.5% RD), Fibrobacteres (NI, mock + 1.5% RD), Chlamydiae (NI, mock + 1.5% RD), and Hydrogenedentes (NG, mock + 1.5% RD) were abundant in the mock compared to T. foetida-infected libraries. However, Gemmatimonadetes (NK (1.5 times of RD), ID (RD), and IA (1.5% RD)) and Acidobacteria had similar abundances in T. foetida-infected and mock samples (Fig. 5).

Profiling results for the twelve samples. Richness heatmap of the 18 most abundant bacterial phyla. IA (T. foetida + 1.5% RD of Jianzhuang), IB (T. foetida + 3% RD of Jianzhuang), IC (T. foetida + 5% RD of Jianzhuang), ID (T. foetida + RD of Jianzhuang), IE (T. foetida + 1.5 times RD of Jianzhuang), IF (T. foetida + no Jianzhuang), NG (mock + 1.5% RD of Jianzhuang), NH (mock + 3% RD of Jianzhuang), NI (mock + 5% RD of Jianzhuang), NJ (mock + RD of Jianzhuang), NK (mock + 1.5 times RD of Jianzhuang), and NL (mock + no Jianzhuang). RD stands for recommended dose, and mock means ddH2O application
Heatmap Analysis for Fungal Communities
The abundance levels of seven fungal phyla were characterized into T. foetida-infected and mock libraries. The results revealed that only Ascomycota (IB = 3% RD) was abundant in the T. foetida-infected libraries, while Basidiomycota (NH = 3 RD), Ciliophora (NG, mock + 1.5% RD), Chytridiomycota (IF, T. foetida infected with no Jianzhuang treatment), Anthophyta (IF, T. foetida infected with no Jianzhuang treatment), and Chlorophyta (NI, mock + 5% RD) were more abundant in mock libraries than in T. foetida-infected libraries. However, Mortierellomycota had similar abundance in T. foetida-infected (ID, T. foetida + RD) and mock (NK, mock + 1.5 times RD) libraries (Fig. 6).

Profiling results of the twelve samples. Richness heatmap of the 7 most abundant fungal phyla. IA (T. foetida + 1.5% RD of Jianzhuang), IB (T. foetida + 3% RD of Jianzhuang), IC (T. foetida + 5% RD of Jianzhuang), ID (T. foetida + RD of Jianzhuang), IE (T. foetida + 1.5 times RD of Jianzhuang), IF (T. foetida + no Jianzhuang), NG (mock + 1.5% RD of Jianzhuang), NH (mock + 3% RD of Jianzhuang), NI (mock + 5% RD of Jianzhuang), NJ (mock + % RD of Jianzhuang), NK (mock + 1.5 times RD of Jianzhuang), and NL (mock + no Jianzhuang). RD stands for recommended dose, and mock means ddH2O application
Alpha Diversity Analysis of Bacterial and Fungal Communities in Wheat Rhizosphere Soil
PCoA analysis provided little variability information, and distance index within-group analysis was further used to assess the distance within groups. The results showed that the IB (3% RD), NI (mock of 5% RD), ID (RD), NJ (mock of RD), and NL (mock of no Jianzhuang treatment) libraries had lower distances within the groups (Fig. 7).

Alpha (α) diversity levels of twelve bacterial samples. IA (T. foetida + 1.5% RD of Jianzhuang), IB (T. foetida + 3% RD of Jianzhuang), IC (T. foetida + 5% RD of Jianzhuang), ID (T. foetida + RD of Jianzhuang), IE (T. foetida + 1.5 times RD of Jianzhuang), IF (T. foetida + no Jianzhuang), NG (mock + 1.5% RD of Jianzhuang), NH (mock + 3% RD of Jianzhuang), NI (mock + 5% RD of Jianzhuang), NJ (mock + % RD of Jianzhuang), NK (mock + 1.5 times RD of Jianzhuang), and NL (mock + no Jianzhuang). RD stands for recommended dose, and mock means no T. foetida application
Alpha (α) diversity analysis was also performed for fungal communities. The results showed that the IB (3% RD), NH (mock of 3% RD), IE (1.5 times RD), and NL (mock of no Jianzhuang treatment) libraries had minimum distances within the groups compared to the other samples (Fig. 8).

Alpha (α) diversity levels of twelve fungal samples. IA (T. foetida + 1.5% RD of Jianzhuang), IB (T. foetida + 3% RD of Jianzhuang), IC (T. foetida + 5% RD of Jianzhuang), ID (T. foetida + RD of Jianzhuang), IE (T. foetida + 1.5 times RD of Jianzhuang), IF (T. foetida + no Jianzhuang), NG (+ 1.5% RD of Jianzhuang), NH (mock + 3% RD of Jianzhuang), NI (mock + 5% RD of Jianzhuang), NJ (mock + % RD of Jianzhuang), NK (mock + 1.5 times RD of Jianzhuang), and NL (mock + no Jianzhuang). RD stands for recommended dose, and mock means application of ddH2O instead of T. foetida
Discussion
In this study, we investigated the effects of T. foetida on the microbial community with different concentrations of Jianzhuang pesticide-coated seeds and identified some communities that may contribute to the control of wheat common bunt, which will provide some important information on the interaction of the rhizosphere community with plant growth.
The raw reads, clean reads, raw tags, clean tags, effective tags, and effective ratio (%) were calculated for each treatment (Table S1) and were consistent with previous reports [33]. T. foetida-infected samples with Jianzhuang at different concentrations had slightly higher Proteobacteria and Gemmatimonadetes relative abundance levels than the mock libraries (Fig. 1). These findings suggest that Jianzhuang increased Proteobacteria diversity, which was affected by T. foetida. Previous studies revealed that Proteobacteria has roles in plant disease suppression and gene regulation, which have plant growth-promoting activities [34,35,36]. Interestingly, T. foetida belongs to the phylum Basidiomycota, and T. foetida-infected samples had a slightly lower abundance of Basidiomycota, possibly because of Jianzhuang application [37]. However, the relative abundance of the phylum Ascomycota was greater in the T. foetida-infected samples (Fig. 2) [38]. The observed results in this experiment revealed that T. foetida might increase the abundance levels of some fungal communities.
Based on the unweighted UniFrac distance metric, cluster and PCoA analyses were conducted to evaluate the similarities among the T. foetida-infected and mock libraries [39]. The results of cluster analysis (Fig. 3a) and PCoA analysis (Fig. 3b) showed that replicate samples of the same treatment under the same concentration of Jianzhuang were similar. Additionally, similar results were observed for the fungal phyla during cluster and PCoA analysis (Fig. 4a, b). The richness levels of eighteen bacterial phyla were checked in the T. foetida-infected and mock libraries. Actinobacteria and Cyanobacteria are able to suppress different plant pathogens and promote the morpho-physiological attributes of plants [40, 41]. Our results revealed that the richness levels of Actinobacteria and Cyanobacteria were high in T. foetida-infected plants, suggesting that Jianzhuang activates Actinobacteria and Cyanobacteria richness. Additionally, Nitrospirae abundance was high in mock samples (Fig. 5). Previous results reported that Oehmen played an important role in nitrogen fixation and removal [42]. In our results, Nitrospirae abundance was low, suggesting that T. foetida might have a negative impact on it. Similarly, the richness levels of seven fungal phyla were demonstrated in the T. foetida-infected and mock samples. The richness of Basidiomycota was abundant in the NH (3% RD) sample. T. foetida belongs to the phylum Basidiomycota, which suggests that Jianzhuang inhibits the proliferation of T. foetida (Fig. 6).
Advances in the knowledge of rhizosphere soil microbial diversity depend on improved approaches [43]. Soil microorganisms play important roles in soil fertility and soil biochemical processes and regulate soil temperature [44,45,46,47,48]. It is acknowledged that traditional molecular biological techniques such as simple PCR, terminal restriction fragment-length polymorphism (T-RFLP), and denaturing gradient gel electrophoresis (DGGE) have their own forms of reference and have intense labor requirements [49]. Next-generation sequencing has been described as easier and more accurate than traditional molecular techniques in characterizing rhizosphere soil microorganisms under controlled and natural conditions. Plant morpho-physiological attributes and performance are highly associated with plant-associated soil microorganisms under different conditions [50]. Up to a few thousand different species of bacteria, fungi, and various protists, including plant roots, act as biocontrol agents against various plant pathogens [50,51,52,53].
Previous studies have found that microbial abundance was greater in infected samples than in mock samples [7, 54]. The results of this study suggested that inoculation with T. foetida had negative effects on the rhizosphere soil microbial community by increasing or decreasing their relative abundance levels. The abundance levels of the bacterial communities Verrucomicrobia, Patescibacteria, Armatimonadetes, Nitrospirae, Fibrobacteres, Chlamydiae, and Hydrogenedentes and the fungal communities Basidiomycota and Ciliophora were higher in the mock group than in the T. foetida-infected group, which may contribute to the control of wheat common bunt.
References
Mourad A, Mahdy E, Bakheit BR et al (2018) Effect of common bunt infection on agronomic traits in wheat (Triticum aestivum L.). J Plant Genet Breed 2:1–7
El-Naimi M, Toubia-Rahme H, Mamluk OF (2000) Organic seed-treatment as a substitute for chemical seed-treatment to control common bunt of wheat. Eur J Plant Pathol 106:433–437
Lu ZX, Gaudet DA, Frick M, Puchalski B, Genswein B, Laroche A (2005) Identification and characterization of genes differentially expressed in the resistance reaction in wheat infected with Tilletia tritici, the common bunt pathogen. J Biochem Mol Biol 38:420–431. https://doi.org/10.5483/bmbrep.2005.38.4.420
Yao Z, Qin D, Chen D, Liu C, Chen W, Liu T, Liu B, Gao L (2019) Development of ISSR-derived SCAR marker and SYBR green I real-time PCR method for detection of teliospores of Tilletia laevis Kühn. Sci Rep 9:17651. https://doi.org/10.1038/s41598-019-54163-5
Calistru A-E, Jitareanu G (2016) Applications of x-ray CT for examining soil structure: a review. Bull UASVM Ser Agric 73:1843–5246. https://doi.org/10.15835/buasvmcn-agr
Parlak Y (1981) Seed borne pathogens on wheat (particularly smuts) in Turkey. Eppo Bull 11:83–86
Liu X, Cheng X, Wang H, Wang K, Qiao K (2015) Effect of fumigation with 1,3-dichloropropene on soil bacterial communities. Chemosphere 139:379–385. https://doi.org/10.1016/j.chemosphere.2015.07.034
Jackson CR, Randolph KC, Osborn SL, Tyler HL (2013) Culture dependent and independent analysis of bacterial communities associated with commercial salad leaf vegetables. BMC Microbiol 13:274. https://doi.org/10.1186/1471-2180-13-274
Pascale A, Proietti S, Pantelides IS, Stringlis IA (2020) Modulation of the root microbiome by plant molecules: the basis for targeted disease suppression and plant growth promotion. Front Plant Sci 10:1741. https://doi.org/10.3389/fpls.2019.01741
Shimazu M, Maehara N, Sato H (2002) Density dynamics of the entomopathogenic fungus, Beauveria bassiana Vuillemin (Deuteromycotina: Hyphomycetes) introduced into forest soil, and its influence on other soil microorganisms. Appl Entomol Zool 37:263–269. https://doi.org/10.1303/aez.2002.263
Zhang Y, He J, Jia LJ, Yuan TL, Zhang D, Guo Y, Wang Y, Tang WH (2016) Cellular tracking and gene profiling of Fusarium graminearum during maize stalk rot disease development elucidates its strategies in confronting phosphorus limitation in the host apoplast. PLoS Pathog 12:e1005485. https://doi.org/10.1371/journal.ppat.1005485
Rebbeck J, Malone MA, Short DPG, Kasson MT, O'Neal ES, Davis DD (2013) First report of Verticillium wilt caused by Verticillium nonalfalfae on tree-of-heaven (Ailanthus altissima) in Ohio. Plant Dis 97:999
Manjunatha SV, Naik MK, Khan MFR, Goswami RS (2013) Evaluation of bio-control agents for management of dry root rot of chickpea caused by Macrophomina phaseolina. Crop Prot 45:147–150. https://doi.org/10.1016/j.cropro.2012.09.003
Sharma KK, Singh US, Sharma P et al (2015) Seed treatments for sustainable agriculture—a review. J Appl Nat Sci 7:521–539. https://doi.org/10.31018/jans.v7i1.641
Monga D, Sain SK, Nakkeeran S et al (2018) Effectiveness of seed treatment with recommended fungicides on seed, soil borne diseases and productivity of cotton. J Mycol Pl Pathol 48(3):311–323
Shakoor MA, Ahmad M, Zia ullah Ghazali H et al (2014) Chemotherapy of Karnal bunt of wheat: a review. Int J Adv Res Biol Sci 1:163–188
Workneh F, Allen TW, Nash GH, Narasimhan B, Srinivasan R, Rush CM (2008) Rainfall and temperature distinguish between Karnal bunt positive and negative years in wheat fields in Texas. Phytopathol 98:95–100. https://doi.org/10.1094/PHYTO-98-1-0095
Slininger PJ, Shea-Andersh MA (2005) Proline-based modulation of 2,4-diacetylphloroglucinol and viable cell yields in cultures of Pseudomonas fluorescens wild-type and over-producing strains. Appl Microbiol Biotechnol 68:630–638. https://doi.org/10.1007/s00253-005-1907-4
Dorigan AF, de Carvalho G, Poloni NM et al (2019) Resistance to triazole fungicides in pyricularia species is associated with invasive plants from wheat fields in Brazil. Acta Sci Agron 41:1–10. https://doi.org/10.4025/actasciagron.v41i1.39332
Ribas e Ribas AD, Spolti P, Del Ponte EM et al (2016) Is the emergence of fungal resistance to medical triazoles related to their use in the agroecosystems? A mini review. Braz J Microbiol 47:793–799. https://doi.org/10.1016/j.bjm.2016.06.006
Ecobichon D (1991) Toxic effects of pesticides. In: Gupta PK (ed) (J) Fundamentals of toxicology, pp 185–202
Bending GD, Rodríguez-Cruz MS, Lincoln SD (2007) Fungicide impacts on microbial communities in soils with contrasting management histories. Chemosphere 69:82–88. https://doi.org/10.1016/j.chemosphere.2007.04.042
Kalia A, Gosal SK (2011) Effect of pesticide application on soil microorganisms. Arch Agron Soil Sci 57:569–596. https://doi.org/10.1080/03650341003787582
Newman MM, Hoilett N, Lorenz N, Dick RP, Liles MR, Ramsier C, Kloepper JW (2016) Glyphosate effects on soil rhizosphere-associated bacterial communities. Sci Total Environ 543:155–160. https://doi.org/10.1016/j.scitotenv.2015.11.008
Ryan M (1999) Is an enhanced soil biological community, relative to conventional neighbours, a consistent feature of alternative (organic and biodynamic) agricultural systems? Biol Agric Hortic 17:131–144. https://doi.org/10.1080/01448765.1999.9754832
Caporaso JG, Kuczynski J, Stombaugh J et al (2010) QIIME allows analysis of high-throughput community sequencing data intensity normalization improves color calling in SOLiD sequencing. Nat Publ Gr 7:335–336. https://doi.org/10.1038/nmeth0510-335
Edgar RC (2013) UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat Methods 10:996–998. https://doi.org/10.1038/nmeth.2604
Wang Q, Garrity GM, Tiedje JM, Cole JR (2007) Naïve Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol 73:5261–5267. https://doi.org/10.1128/AEM.00062-07
DeSantis TZ, Hugenholtz P, Larsen N et al (2006) Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol 72:5069–5072. https://doi.org/10.1128/AEM.03006-05
Bokulich NA, Subramanian S, Faith JJ, Gevers D, Gordon JI, Knight R, Mills DA, Caporaso JG (2013) Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nat Methods 10:57–59. https://doi.org/10.1038/nmeth.2276
Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, Sahl JW, Stres B, Thallinger GG, van Horn DJ, Weber CF (2009) Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 75:7537–7541. https://doi.org/10.1128/AEM.01541-09
Zhang T, Shao MF, Ye L (2012) 454 pyrosequencing reveals bacterial diversity of activated sludge from 14 sewage treatment plants. ISME J 6:1137–1147. https://doi.org/10.1038/ismej.2011.188
Hu M, Wang X, Wen X, Xia Y (2012) Microbial community structures in different wastewater treatment plants as revealed by 454-pyrosequencing analysis. Bioresour Technol 117:72–79. https://doi.org/10.1016/j.biortech.2012.04.061
Bruto M, Prigent-Combaret C, Muller D, Moënne-Loccoz Y (2014) Analysis of genes contributing to plant-beneficial functions in plant growth-promoting rhizobacteria and related Proteobacteria. Sci Rep 4:6261. https://doi.org/10.1038/srep06261
Spain AM, Krumholz LR, Elshahed MS (2009) Abundance, composition, diversity and novelty of soil Proteobacteria. ISME J 3:992–1000. https://doi.org/10.1038/ismej.2009.43
McLellan SL, Huse SM, Mueller-Spitz SR et al (2010) Diversity and population structure of sewage-derived microorganisms in wastewater treatment plant influent. Environ Microbiol 12:1376–1376. https://doi.org/10.1111/j.1462-2920.2010.02204.x
Liu J, Li C, Muhae-ud-din G et al (2020) Development of the droplet digital PCR to detect the teliospores of Tilletia controversa Kühn in the soil with greatly enhanced sensitivity. Front Microbiol 11:4. https://doi.org/10.3389/fmicb.2020.00004
Berbee ML (2001) The phylogeny of plant and animal pathogens in the Ascomycota. Physiol Mol Plant Pathol 59:165–187. https://doi.org/10.1006/pmpp.2001.0355
Hamady M, Lozupone C, Knight R (2010) Fast UniFrac: facilitating high-throughput phylogenetic analyses of microbial communities including analysis of pyrosequencing and PhyloChip data. ISME J 4:17–27. https://doi.org/10.1038/ismej.2009.97
Palaniyandi SA (2013) Effects of actinobacteria on plant disease suppression and growth promotion. Appl Microbiol Biotechnol 97:9621–9636. https://doi.org/10.1007/s00253-013-5206-1
Singh S (2014) A review on possible elicitor molecules of cyanobacteria: their role in improving plant growth and providing tolerance against biotic or abiotic stress 117:1221–1244. https://doi.org/10.1111/jam.12612
Oehmen A, Saunders AM, Vives MT, Yuan Z, Keller J (2006) Competition between polyphosphate and glycogen accumulating organisms in enhanced biological phosphorus removal systems with acetate and propionate as carbon sources. J Biotechnol 123:22–32. https://doi.org/10.1016/j.jbiotec.2005.10.009
Gans J, Wolinsky M, Dunbar J (2005) Computational improvements reveal great bacterial diversity and high toxicity in soil. Sci 309:1387–1390. https://doi.org/10.1126/science.1112665
Philippot L, Raaijmakers JM, Lemanceau P, Van Der Putten WH (2013) Going back to the roots: the microbial ecology of the rhizosphere. Nat Rev Microbiol 11:789–799. https://doi.org/10.1038/nrmicro3109
Keane BJ, Ineson P, Vallack HW, Blei E, Bentley M, Howarth S, McNamara NP, Rowe RL, Williams M, Toet S (2018) Greenhouse gas emissions from the energy crop oilseed rape (Brassica napus); the role of photosynthetically active radiation in diurnal N2O flux variation. GCB Bioenergy 10:306–319. https://doi.org/10.1111/gcbb.12491
Zhu X, Wang Y, Su Z, Lv L, Zhang Z (2018) Silencing of the wheat protein phosphatase 2A catalytic subunit TaPP2AC enhances host resistance to the necrotrophic pathogen rhizoctonia cerealis. Front Plant Sci 9:1437. https://doi.org/10.3389/fpls.2018.01437
Lin W, Liu W, Xue Q (2017) Spring maize yield, soil water use and water use efficiency under plastic film and straw mulches in the Loess Plateau. Sci Rep 6:38995. https://doi.org/10.1038/srep42455
Muñoz K, Buchmann C, Meyer M, Schmidt-Heydt M, Steinmetz Z, Diehl D, Thiele-Bruhn S, Schaumann GE (2017) Physicochemical and microbial soil quality indicators as affected by the agricultural management system in strawberry cultivation using straw or black polyethylene mulching. Appl Soil Ecol 113:36–44. https://doi.org/10.1016/j.apsoil.2017.01.014
Zhang M, Xu JH, Liu G, Yao XF, Li PF, Yang XP (2015) Characterization of the watermelon seedling infection process by Fusarium oxysporum f. sp. niveum. Plant Pathol 64:1076–1084. https://doi.org/10.1111/ppa.12355
Agler MT, Ruhe J, Kroll S, Morhenn C, Kim ST, Weigel D, Kemen EM (2016) Microbial hub taxa link host and abiotic factors to plant microbiome variation. PLoS Biol 14:e1002352. https://doi.org/10.1371/journal.pbio.1002352
Berendsen RL, Pieterse CMJ, Bakker PAHM (2012) The rhizosphere microbiome and plant health. Trends Plant Sci 17:478–486. https://doi.org/10.1016/j.tplants.2012.04.001
Bulgarelli D, Rott M, Schlaeppi K, ver Loren van Themaat E, Ahmadinejad N, Assenza F, Rauf P, Huettel B, Reinhardt R, Schmelzer E, Peplies J, Gloeckner FO, Amann R, Eickhorst T, Schulze-Lefert P (2012) Revealing structure and assembly cues for Arabidopsis root-inhabiting bacterial microbiota. Nature 488:91–95. https://doi.org/10.1038/nature11336
Muhae-ud-Din G, Moosa A, Ghummen UF et al (2018) Host status of commonly planted ornamentals to Meloidogyne incognita and management through endophytic bacteria. Pak J Zool 50:1393–1402. https://doi.org/10.17582/journal.pjz/2018.50.4.1393.1402
Xu J, Liu S, Song S, Guo H, Tang J, Yong JWH, Ma Y, Chen X (2018) Arbuscular mycorrhizal fungi influence decomposition and the associated soil microbial community under different soil phosphorus availability. Soil Biol Biochem 120:181–190. https://doi.org/10.1016/j.soilbio.2018.02.010
Funding
This work was supported by the National Natural Science Foundation of China (31761143011) and the National Key Research and Development Program of China (2018YFD0200406).
Author information
Authors and Affiliations
Contributions
Li Gao designed the experiment and wrote the manuscript; Han Zhang performed the experiment; Ghulam Muhae Ud Din and Zhenzhen Du revised and analyzed the manuscript; and Wanquan Chen, Taiguo Liu, and Sifeng Zhao provided the materials. All authors were involved in writing the paper and had final approval to publish the manuscript.
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare no competing interests.
Supplementary Information
Figure S1
Molecular detection of T. foetida from leaf samples with specific primers. Lines 1 and 25 negative controls; lines 2 and 26 positive controls; lines 3-24 and lines 27-40 T. foetida-infected leaf samples; M, DL2000 Marker (100, 250, 500, 750, 1000, 2000 bp); black arrows show the target band of 660 bp. (PNG 481 kb)
Table S1
Raw reads, clean reads, raw tags, clean tags, effective tags, and effective ratio (%) of twelve samples. (XLSX 13 kb)
Table S2
OTUs of bacterial communities of twelve samples with three replications (XLSX 4825 kb)
Table S3
OTUs of fungal communities of twelve samples with three replications (XLSX 691 kb)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Din, G.M.U., Du, Z., Zhang, H. et al. Effects of Tilletia foetida on Microbial Communities in the Rhizosphere Soil of Wheat Seeds Coated with Different Concentrations of Jianzhuang. Microb Ecol 82, 736–745 (2021). https://doi.org/10.1007/s00248-021-01696-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00248-021-01696-w