
Abstract
Local gene duplication is a prominent mechanism of gene copy number expansion. Elucidating the mechanisms by which local duplicates arise is necessary in understanding the evolution of genomes and their host organisms. Chromosome one of Arabidopsis thaliana contains an 81-gene array subdivided into 27 triplet units (t-units), with each t-unit containing three pre-transfer RNA genes. We utilized phylogenetic tree reconstructions and comparative genomics to order the events leading to the array’s formation, and propose a model using unequal crossing-over as the primary mechanism of array formation. The model is supported by additional phylogenetic information from intergenic spacer sequences separating each t-unit, comparative analysis to an orthologous array of 12 t-units in the sister taxa Arabidopsis lyrata, and additional modeling using a stochastic simulation of orthologous array divergence. Lastly, comparative phylogenetic analysis demonstrates that the two orthologous t-unit arrays undergo concerted evolution within each taxa and are likely fluctuating in copy number under neutral evolutionary drift. These findings hold larger implications for future research concerning gene and genome evolution.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Local gene duplication generates tandemly arrayed genes and is a ubiquitous occurrence during genome evolution. Local duplicates are presumed to contribute to genetic diversity through relaxation of selective pressure on one or both duplicates through redundancy of function. This relaxed selective pressure allows for gene evolution such as subfunctionalization (Force et al. 1999; Lynch and Conery 2000), where both the genes specialize in a subset of their original function, and neofunctionalization (Lewis 1953; Ohno 1970; Ohta 1988), where one of the copies evolves novel cellular functions while its counterpart retains ancestral functions. For duplication events that present a dominant negative dosage phenotype, subsequent selection may result in reversion back to the singlet condition, returning gene product dosage to a stoichiometric balance (Freeling 2009). Unequal crossing-over (UCO) between two duplex loci naturally forms a triplet and a singlet, so that there are no mechanical impediments to returning to the singlet state.
In contrast, some genomic loci can undergo copy number amplification under positive selection for increase in product dosage. Positive selection for increased dosage has been suggested to explain large tandem arrays of rRNA and histone genes (Hurles 2004) and is observed experimentally in bacteria to cope with nutrient availability (Horiuchi et al. 1962). A subset of genes that frequently occur in local tandem arrays is observed to have a high degree of sequence and functional diversity; some classes of plant disease resistance genes (e.g., NB LRR genes) are well-studied examples of this phenomenon. A single cluster of tandemly repeated NB-LRR genes may encode a diverse number of different pathogen specificities (Baumgarten et al. 2003). These arrays of high sequence diversity stand in contrast to the aforementioned rRNA arrays, for which sequence and functional homogenization (concerted evolution) is observed (Brown et al. 1972; Nei and Rooney 2005). Please see Sémon and Wolfe (2007) for an excellent review of the evolution and fates of gene duplicates.
Our study focuses on an unusual pre-tRNA gene array within the Arabidopsis lineage. In A. thaliana (At), this array consists of 81 pre-tRNA genes arranged into 27 repeating triplet gene units, which we call t-units. This pre-tRNA gene array was discovered in At by Beier et al. (1991) and shown to contain at least 20 copies of t-units. Further study provided data that a number of these pre-tRNA genes produce transcripts (Beier and Beier 1992; Stange et al. 1991). We show that Arabidopsis lyrata (Al), a sister taxon of At, contains an orthologous array of pre-tRNA genes at the syntenic locus to At. Using the complete genome sequences of both the At and Al genomes, our research has expanded upon the study of Beier et al. using a high-resolution comparative analysis of the t-unit array within two taxa of the Arabidopsis lineage to understand the evolution of these t-unit pre-tRNA gene arrays.
The evolutionary fate of duplicated genes has been the focus of a great deal of research (Zhang 2003; Sémon and Wolfe 2007; Freeling 2009); in contrast, detailed case studies on tandemly arrayed genes containing many repeat units are relatively few in number. While characterizations of the evolution of tandemly arrayed genes using comparative methods have been made (Jackson 2007), more studies are required in a wider range of taxa to gain an understanding of the general mechanisms that govern the evolution of tandem arrays in genomes. The Arabidopsis t-unit pre-tRNA gene array provides an excellent model system for gaining insight into the mechanisms of tandemly arrayed gene evolution due to its recent formation, its rapid expansion, and its conspicuous pattern of multi-gene duplication, the latter hinting at a higher order complexity to the local duplication process. Through a series of phylogenetic and comparative analyses, we demonstrate that complete t-units are almost without exception the basal units of the arrays’ expansion and evolve in concert within each taxon. In addition, we propose a generalized model of t-unit expansion within the array using unequal crossing-over (UCO) as the mechanism of copy number variation and sequence “homogenization,” and designed a custom computer program to stochastically simulate divergence between two orthologous arrays. These simulations demonstrate that relatively few crossover events within an array are necessary to cause lineage-specific divergence of orthologous arrays, as is observed in the At and Al t-unit arrays, leaving us to conclude that UCO is the primary mechanism of evolution within these tandem gene arrays and sufficient to explain their concerted evolution.
Materials and Methods
Many of the sequence comparisons used in this article were performed using the CoGe comparative genomics platform (Lyons and Freeling 2008; Lyons et al. 2008).
Nomenclature and Triplet Designation
A nomenclature was devised to denote both element type and location to maintain specificity in language for these highly repetitive genomic structures (Fig. 1). The pre-tRNA genes are denoted by the amino acid they transfer. A triplet unit, or t-unit, is defined as the genomic sequence from the start of a pre-tRNASer gene to the end of the second proximal 3′ pre-tRNATyr gene, with the t-unit oriented in the 5′ to 3′ direction. Each t-unit contains five elements in the following order: a pre-tRNASer gene, an intergenic region (IGR) B, a pre-tRNATyrA gene, an IGR C, and a pre-tRNATyrB gene. Exceptions to this rule are an incomplete t-unit on the 5′ end of the Al array, referred to as t-unit 0, and t-unit 1 in Al which contains an extra pre-tRNATyrB gene, referred to as pre-tRNATyrC, and an extra IGR C sequence, referred to as IGR D1; the At array contains no exceptions to the t-unit rule. Preceding each t-unit is an additional intergenic region, IGR A, defined as the sequence from the end of the prior (5′) t-unit to the start of the current t-unit. Suffix letters are used denote relative position within the t-unit—IGR B precedes IGR C, and pre-tRNATyrA precedes pre-tRNATyrB. Each element and t-unit is also given a number to indicate its absolute position from the 5′ end of the array. For example, t-unit 23 is the 23rd t-unit in the array, and IGR B12 is the intergenic region between pre-tRNASer and pre-tRNATyrA in t-unit 12. The number suffix of an IGR A sequence indicates its position is immediately 5′ of the t-unit of that number. The exception to this rule is IGR A28 in At, which is the IGR A sequence immediately 3′ of the last t-unit, t-unit 27. In addition, when t-units and array elements are referred to by the species from which they originate, At and Al refer to A. thaliana and A. lyrata, respectively.

A visualization of the triplet array in A. thaliana (At) and A. lyrata (Al). (a) The triplet array in At. The pre-tRNA genes are indicated by gray cylinders. The green background indicates G + C content; each G + C basepair is indicated by a single vertical green line. The entire array is 39.7 kb long, from the start of the first pre-tRNASer on the 5′ end to the final pre-tRNATyrB on the 3′ end (Chr. 1 21,272,452..21,312,137). (b) Two triplet units (t-units) in the array are shown, starting with the twelfth t-unit in the array on the left, and the thirteenth t-unit on the right. Each triplet indicates the two methods of nomenclature used throughout the article. Both letters and numbers are used to distinguish element position in different ways, with all nomenclature increasing from 5′ to 3′. Letters, such as IGR C, indicate relative position of an intergenic region within the t-unit, whereas numbers, such as IGR C13, indicate the element’s absolute position within the array. Thus, IGR C13 indicates it is the third IGR element, C, in the thirteenth triplet. T-unit 12 is indicated using the t-unit nomenclature, simply identifying the entire triplet’s location within the array. The t-unit sequence begins at the start of the pre-tRNASer gene, and ends at the termination of the pre-tRNATyrB gene, as indicated by the red, translucent rectangle. T-unit 13, on the right, designates its individual elements by their genetic type—the type of pre-tRNA gene they are (based on their designated amino acid), or the relative location of the intergenic sequence. IGR A lies between the t-units (inter-triplet). IGR A numbering indicates that it precedes the t-unit of the same number, such that IGR A13 is the sequence between t-unit 12 and t-unit 13; note that the exception to this rule is IGR A28 which immediately follows the terminal 3′ t-unit 27. (c) The triplet array in Al array. As the pre-tRNA genes in the array are not annotated, the genes are represented by colored boxes on top of the genomic G + C background (the orange background indicates an unsequenced region.) Pre-tRNASer genes are represented by red boxes, and pre-tRNATyr genes are represented by blue boxes. Note the violation of the three pre-tRNA gene rules in Al t-units 0 and 1
Gene and Intergenic Sequences
A complete list of the At pre-tRNA genes used in this study can be found at http://tinyurl.com/5bq3o9 as well as in a spreadsheet in the supplementary file; no annotations existed for the Al pre-tRNA genes at the time of this study. All At DNA sequences were obtained through The Arabidopsis Information Resource (TAIR), version eight of the Arabidopsis thaliana genome. A Perl script was written to parse the array’s DNA sequence, given the annotated gene start/stop positions and knowing the position of the IGRs relative to the pre-tRNA genes. The pre-tRNASer gene sequences were taken as annotated; however, the pre-tRNATyr gene models were incomplete. This was apparent through comparisons of the annotated pre-tRNATyr genes in the array to other, complete gene models both in the At genome, and an At gene stored in Genbank (M35958.1; http://tinyurl.com/n3f5vh). The pre-tRNATyr gene models were truncated before the gene’s intron sequence (Goodman et al. 1977). Ten incomplete gene sequences were aligned and compared using blastn (Altschul et al. 1990; E-value Cutoff: 0.001, Match: 1, Mismatch: −2, Gap Open Penalty: 5, Gap Extension Penalty: 2, Word Size: 7, No Filter on query sequence) to gain a consensus gene sequence and length, and we took the sequence of the most conservative high-scoring segment pair generated by the alignment algorithm. Complete gene models were also predicted using tRNAscan-SE 1.21 (Lowe and Eddy 1997) and these were concordant with the models obtained using blastn.
Version one of the Arabidopsis lyrata genome was obtained from the Joint Genome Institute (http://tinyurl.com/l9njsq). While no pre-tRNA genes were annotated in the region syntenic to the t-unit array in At, eleven full length and one partial t-units were found using blastn sequence similarity searches as well as tRNAscan-SE analysis as described above. As this is a draft genome using whole genome shotgun sequencing, we mapped the pair-end sequencing coverage of the region used in our analysis. We found overall coverage to be approximately 8× and had paired-end coverage spanning all parts of this region, but an increase in fold coverage on the 3′ end of the array. (Supplementary Fig. 1.) This may reflect sequencing sampling bias, or additional copies of t-units in unsequenced regions of the genome. All the raw DNA sequences used in this study (genes, intergenic regions, and triplet units) are available in FASTA format in the supplementary file.
Syntenic Alignments and Assessment
We evaluated synteny using the online comparative genomics package CoGe and the method outlined in Lyons et al. (2008). First, an approximately 165.4 kb sequence (http://tinyurl.com/6qghol) encompassing the At array (Chr. 1, 21190739-21356171) was retrieved, and annotated coding sequences within the region (5′ and 3′ of the pre-tRNA array) were catalogued (http://tinyurl.com/5z799u). Using the catalogued CDS sequences as queries, a tblastx search (Altschul et al. 1990) was then run against the Carica papaya (Cp) (Ming et al. 2008), Vitis vinifera (Vv) (Jaillon et al. 2007), Al, and At genomes using CoGeBlast (http://tinyurl.com/5udsyy); the genomic regions of the best blast hits were then analyzed for synteny using GEvo (http://tinyurl.com/d7mzhz) and the BlastZ alignment algorithm (Schwartz et al. 2003). Synteny was determined by identifying a collinear series of putatively homologous genes (Lyons and Freeling 2008). Four syntenic At and Al regions (including the region containing the array) were identified in addition to a single syntenic region from both grape and papaya (Supplementary Fig. 2). The ten syntenic genomic regions were as follows: Cp—supercontig_48: 1,128,741-1,287,100; Vv—Chr. 14: 3,198-528,677 (reverse complement); Al—scaffold_1: 3,597,899-3,652,421, scaffold_1: 32,768,291-32,878,607, scaffold_3: 2,260,368-2,319,437, scaffold_6: 12,044,518-12,265,938 (reverse complement); At—Chr. 1: 3,039,795-3,100,826, Chr. 1: 21,255,709-21,334,368, Chr. 3: 1,735,349-1,780,617, and Chr. 5: 9,269,098-9,473,551 (reverse complement).
Multiple Sequence Alignment
Sequences were initially aligned using ClustalW 1.83 (Thompson et al. 1994) using the default settings for a DNA multiple sequence alignment (Open Gap Penalty: 15, Gap Extension Penalty: 6.66, no Weight Transition, Matrix: IUB). The alignment generated by ClustalW was then manually refined in the program Mesquite (Maddison and Maddison 2009). The final alignment files used in subsequent phylogenetic reconstruction are available in the supplementary materials section in NEXUS format.
Phylogenetic Tree Reconstruction
Maximum likelihood (ML) trees were constructed using Paup* 4.0b10 (Swofford 2003). Evolutionary models for the likelihood analyses were selected by ModelTest 3.7 (Posada and Crandall 1998). Trees were visualized using FigTree (Rambaut 2006–2008). Unrooted ML trees were generated for distinct t-unit elements using a heuristic search with 1,000 repetitions, sequences added randomly, and bisection–reconnection (TBR) branch swapping.
Bayesian inference trees were also used to reconstruct t-unit phylogeny as well as the At IGR A phylogeny. Bayesian analysis was performed by MrBayes v3.1.2 (Ronquist and Huelsenbeck 2003) using a Markov Chain Monte Carlo (MCMC) run, with t-unit elements partitioned into five unlinked character-sets (genes and IGR sequence), and each partition using a distinct model selected as the best-fit by MrModelTest 2.2 (Nylander 2004). The simulation was run for ten million generations, distributed over six chains. Burn-in was set at 25% of the trees generated to calculate posterior probabilities. Bayesian inference trees were constructed using the Cyberinfrastructure for Phylogenetic Research (CIPRES) web portal (Miller et al. 2009).
Congruence of phylogenetic trees was tested by manually mapping trees for each t-unit element to their genomic locations, and further assessed using an incongruence length difference (ILD) test of homogeneity under a parsimony tree building criterion in Paup*, using 1,000 repetitions for the IGR A to t-unit tests, and 26 repetitions for the intra-t-unit test.
Array Pre-tRNA Gene Functionality Data
Data on functionality of pre-tRNA genes within the array was obtained from previously published data (Stange et al. 1991; Beier and Beier 1992). Cloned sequences generated from the previous literature (Beier et al. 1991), found in Genbank (X54368, X54369, X54370, X54371, X543672, X54373, X54374; http://tinyurl.com/kjo7ob), were compared to sequences in the array, and functionality data were assigned to identical sequences used in both the cloned gene in vitro analysis and in the array genomic sequence.
Tyrosine Usage Measurement
Data on prevalence of tyrosine codons in genomic coding sequence for both Arabidopsis species were obtained using the CoGe application OrganismView and analyzed via a two-sided binomial proportions test using the software package R (Team 2007).
Modeling and Simulating Unequal Crossing-Over
Unequal crossing-over (UCO) models were generated under the following assumptions:
-
(a)
UCO is the sole mechanism responsible for array copy number variation
-
(b)
The array structures were formed using a parsimonious number of expansion events
-
(c)
A given array becomes fixed in the population, such that the crossover events occur between identical arrays (i.e., no distinguishing crossover between homologous chromosomes versus sister chromatids).
Models were devised using these assumptions and the results from the t-unit Bayesian inference tree that t-units group exclusively by species. Only the t-unit phylogeny was taken into account when the models were devised, thus allowing non t-unit data (IGR A sequences) to provide independent validation of the models.
Using this model, a custom program was written in Perl to stochastically simulate the effects of UCO on orthologous array divergence, named “Array Divergence Simulation.pl” (Supplementary file). The program tracks the divergence of two hypothetical lineages with an initial identical tandem array of gene-unit elements over multiple generations. The stochastic simulation algorithm is as such
-
1.
Initialize two identical arrays of size n
-
2.
Choose two crossover sites at random in the set 0 to n, inclusive, for each array, giving a total of n + 1 possible crossover sites within an array of size n.
-
(a)
Crossover must have at least one positionally overlapping unit. If the sites chosen do not allow this, sites are reselected by returning to step 2.
-
(a)
-
3.
Generate crossover by swapping array units distal to their respective crossover sites.
-
4.
Randomly select one of the two resultant arrays for retention into the next cycle.
-
5.
Return to 2, unless
-
(a)
One of the two lineage-specific arrays has been truncated down to a single unit, and thus is unable to perform unequal crossover
-
(b)
Both lineage-specific arrays are homogenous for the same single unit (complete convergence).
-
(c)
Neither lineage-specific array contains any identical units (complete divergence).
-
(a)
Each start array consists of gene-units labeled 1 to n, where n is the initialized size of the array at the onset of the simulation. Each generation permits one crossing-over event (equal or unequal) within the array. The total number of crossover events (COT) possible in any given UCO step is
We can calculate the expected final array size after a recombination event E(A n )
where COE n is the possible number of crossover events given the start size of n, N n is the size of the array after the crossover event given start size of n, and P(I) is the probability that a particular product of UCO will be inherited into the next generation. This expands to
which can be further simplified
Thus, the expected post-recombination array size E(A n ) is equal to the initial array, providing our simulation with a parameter for neutral expansion and contraction (no bias toward either), neither mutagenesis of gene-unit elements in the arrays nor any process of gene conversion being permitted, so that simulated array divergence is solely due to gain / loss of gene-unit elements due to UCO events.
The simulation is repeated for 100,000 replicates for each starting array size of 2–40 elements, and the number of crossover events are computed and statistically analyzed for mean array divergence time. Note that only arrays that completely diverge are used to calculate divergence time, as the simulation is only concerned with the number of crossover events to array divergence and not the likelihood of two orthologous arrays reaching divergence. Another important point is that the two diverging arrays are kept isolated from one another; crossover only occurs between an array within a lineage, not across, as one expects in diverging lineages (i.e., post-speciation events).
Results
Date Boundaries of T-Unit Array Formation
A comparative genomics approach was utilized to determine the relative age of the t-unit array (Supplementary Fig. 2). Carica papaya (Cp) (same order as At, Brassicales) and Vitis vinifera (Vv) (basal eurosid) were chosen as outgroups to the Arabidopsis lineage. Subsequent to the divergence of Cp and the Arabidopsis lineage, the Arabidopsis lineage underwent two sequential whole genome duplication (WGD) events (Bowers et al. 2003; Ming et al. 2008). These WGD events resulted in ten syntenic genomic regions among these four taxa: four each from At and Al, and one each from Cp and Vv. Neither outgroup genome, Cp nor Vv, contains any pre-tRNA genes within their syntenic regions, nor the three other syntenic regions of At. The only other syntenic region containing the array is the orthologous Al genomic region. By parsimony, these results indicate that the pre-tRNA genes in the array are not ancestral to the array’s current locus, and likely transposed into the region after the most recent whole genome duplication event in the Arabidopsis lineage, and prior to the lineage divergence of At and Al.
In order to further refine these dates, a random t-unit was selected to query the NCBI whole-genome shotgun sequence database of Brassica rapa (http://tinyurl.com/nh8lgk) using the blastn alignment algorithm. No significant hits covering an entire t-unit, nor a series of hits covering the t-units’ tRNA constituents were found (data not shown), likely dating the array’s formation as subsequent to the divergence of Brassica and Arabidopsis lineages.
Origin of tRNATyr Doublet in T-Unit and Doublet Conservation
Owing to the tandem appearance of the pre-tRNATyr doublet, we hypothesized that the doublet in tail-to-head configuration formed from a simple, tandem duplication prior to t-unit expansion (perhaps by inexact reciprocal recombination). A Bayesian inference tree was constructed using all pre-tRNATyr genes from the At and Al arrays (Supplementary Fig. 3). The arrays’ pre-tRNATyr A and B genes cluster into distinct clades. The distinct clustering of the pre-tRNATyrA and the pre-tRNATyrB gene clades in both At and Al indicates that the doublet order is never violated during t-unit duplication; if it were, then intercalation between A and B genes would be observed in the phylogeny due to contradictions in gene locations versus gene homology. Furthermore, while pre-tRNATyrA genes intercalate between At and Al, the pre-tRNATyrB genes do not. The conservation of doublet order evidences that the pre-tRNATyr subsection of the t-unit is conserved during t-unit duplication in both lineages. The exceptions to this rule are the non-canonical t-units 0 and 1 in Al.
T-unit 0 in Al lacks a pre-tRNASer gene, and thus is in violation of the t-unit conservation rule. However, without further evidence, it is impossible to distinguish between an expansion event that failed to copy the pre-tRNASer gene and a gene loss event subsequent to duplication. T-unit 1 in Al has an additional pre-tRNATyr gene, and thus is also in violation of the t-unit conservation rule. The last two pre-tRNATyr genes in t-unit 1 both clade as B genes; however, they do not group together: pre-tRNATyrB1 clades basally with most of the other pre-tRNATyrB genes in the Al array, and pre-tRNATyrC1 clades with pre-tRNATyrB9.
Conservation of T-Unit During Expansion
Manual comparison of ML trees generated from each t-unit element in the At array (three pre-tRNA genes and two intergenic regions for a total of five distinct trees) showed that all the trees were congruent with one another, and without conflicts. However, the IGR A tree was not congruent with any of the other trees (Supplementary Fig. 4). This was further assessed using an ILD test of homogeneity. While At t-units partitioned into individual elements showed congruence (P > 0.25), IGR A sequences were found to be incongruent with t-unit phylogenies either 5′ (P = 0.001) or 3′ (P = 0.001) of the t-units. The congruence in t-unit element phylogenies, in conjunction with the data confirming conservation of the pre-tRNATyr doublet order (Supplementary Fig. 3), supports the hypothesis that the t-unit has been the array’s basal unit of duplication and expansion, and that recombination leading to copy number variation occurred in the IGR A sequences. While Al t-units exist in fewer copy number and have higher sequence identity to one another, Al’s pre-tRNATyr genes show a similar conservation of gene order to At’s. We inferred from this evidence, along with the similar t-unit structure and homology between the two arrays, that t-unit conservation occurs in the Al array as well, sans the two aforementioned exceptions in Al t-units 0 and 1.
Using blastn, we searched the At genome for additional t-unit loci; however, none were identified. Of the pre-tRNA genes in this array, seven additional loci in At were 100% identical to one or more of the pre-tRNASer genes. No other loci were 100% identical to any of the pre-tRNATyr genes. As in At, we failed to identify any non-array t-unit loci in the Al genome. Of the pre-tRNA genes in the Al array, five additional loci in Al were 100% identical to one or more of the pre-tRNASer genes, and none were 100% identical to any of the pre-tRNATyr genes.
Sequence Conservation Within T-Units and Tyrosine tRNA Gene Dosage
Phylogenetic analysis of all array t-units from both At and Al shows that t-units clade by paralogs within a taxa rather than orthologs between taxa (Fig. 2), concurrent with concerted evolution (Zimmer et al. 1980) occurring within each Arabidopsis lineage. Using the previous literature on functionality of t-unit pre-tRNA genes cloned from the At t-unit array (Stange et al. 1991; Beier and Beier 1992), we noted which t-unit genes were capable of producing a functional, mature tRNA product (Supplementary Fig. 5). A number of the genes in the t-unit array are reported not to produce a mature tRNA product in vitro, with non-functional and functional pre-tRNA genes concentrated toward the ends and central region of the At array, respectively.

Bayesian Inference tree of At and Al t-units. Bayesian posterior probabilities are indicated by numbers next to nodes. The root is arbitrarily drawn at the tree mid-point and should not be used to infer the actual root of the tree; the “Ancestral Array” and “Speciation” designations are to illustrate common ancestry. Branch lengths are substitutions per site
In order to test whether positive selection for pre-tRNATyr gene dosage was the driving force behind array expansion, we analyzed tyrosine amino acid usage across coding regions in At and Al. A total of 369,223 tyrosine codons were found in the At genome (out of 13,133,642 total codons), and 334,108 tyrosine codons were found in the Al genome (out of 11,804,715 codons total). A two-sided binomial proportions test showed a very slight lower usage of tyrosine residues in coding regions in At compared to Al (P < 0.005). Using CoGeBlast, we concluded that, aside from the t-unit array, both genomes contain an equal number of pre-tRNATyr genes (data not shown). As the At array contains over two times more pre-tRNATyr genes compared to the Al array (54 versus 24, respectively) without an increase in tyrosine residue usage in coding sequences, it is unlikely that the array’s expansion is due to positive selection for tRNA dosage. However, it should be noted that additional copies of t-units in Al could exist as evidenced by the increase in sequencing fold-coverage in the 3′ end of the array (Supplementary Fig. 1).
Varied Nature of Array Expansion
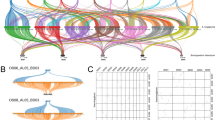
Figure 3 maps At’s t-unit phylogenetic tree onto the physical map of the array, revealing the varied nature of the array’s expansion. Multiple putative sites of expansion are observed, involving both single t-units and intercalated sets of two or more t-units. In the array center, t-units 8–14 cluster and, owing to the step-like pattern observed in the t-unit phylogeny, appear to have been duplicated through local tandem duplication events. Interestingly, the array ends show an intercalating pattern between potentially monophyletic clades based on the t-unit phylogeny. T-units 1–6 show two distinct doublets of t-units (t-units 1 and 3; t-units 5 and 7) intercalated by a triplet of t-units (t-units 2, 4, and 6), with the t-unit triplet clading with the t-unit 1 and 3 doublet. T-units 15–23 also show a similar pattern of intercalation, with the even t-units (16, 18, 20, and 22) clustering into a potentially monophyletic group distinct from the odd t-units (15, 17, 19, 21, and 23), which themselves form a potentially monophyletic group. These regions of the array showing intercalation of distinct phylogenetic clusters support expansion events involving multiple t-units as the basal unit of duplication.

Mapping of At t-unit Bayesian inference tree onto physical map of At array. Each triplet is encircled and its position noted numerically within the oval. Imposed on the physical map is the Bayesian inference tree of the triplet units, with each leaf of the tree linked to the appropriate t-unit in the physical array. The array is broken into four regions of duplication by the phylogenetic tree: A, B, C, and D, suggestive of four independent regions of expansion. The branch lengths are arbitrary in this tree
The Al array demonstrates a similar pattern of t-unit expansion (Fig. 4). T-unit 1 contains two pre-tRNATyrB genes, each with a different phylogenetic signal—the first clades basally with the other pre-tRNATyrB genes, and the second clades with pre-tRNATyrB9. In order to align t-unit 1 with the rest of the t-units in the array, only one of the two pre-tRNATyrB sequences was included. When the first pre-tRNATyrB is included in the alignment, t-unit 1 clades with t-unit 0; otherwise, if the second is included, then t-unit 1 clades with t-unit 9. In the Bayesian phylogenetic trees, only the second pre-tRNATyrB gene, pre-tRNATyrC1, was included in Al t-unit 1. As seen in At, Al’s array may also involve multiple expansion sites and t-units per expansion step.

Mapping of Al t-unit Bayesian inference tree onto physical map of Al array. Above is the array, with each t-unit encircled, and its position noted numerically below the oval. Imposed on the physical map is the Bayesian inference tree of the Al t-units, with each taxa in the tree linked to the appropriate t-unit in the physical array. An intercalated pattern of t-unit duplication, observed in the At array (Fig. 3), is observed here as well. The branch connecting t-units 0, 1, and 9 is drawn as a dotted line due to an inconsistent phylogenetic signal from the pre-tRNATyr genes in t-unit 1. In order to align the sequences, only two of the three pre-tRNATyr genes in the t-unit can be used to build the tree. If the last gene is excluded, then t-unit 1 clades with t-unit 0; however, if the middle gene is excluded, then t-unit 1 clades with t-unit 9. The branch lengths are arbitrary in this tree
Unequal Crossing-Over as a Model for Array Expansion and Validation by IGR A Phylogeny
Owing to the step-wise pattern of duplication observed, and prior evidence indicating that unequal crossing-over (UCO) can cause copy number variation in tandem gene arrays (Smith 1976; Tartof 1988; Achaz et al. 2000), we investigated whether a model using UCO as the sole mechanism of t-unit copy number expansion could yield the observed pattern of t-unit formation seen in Fig. 3. A model of UCO step-wise formation of t-units 1–7 in At (Supplementary Fig. 6) was devised under the following three assumptions: (i) UCO is the sole mechanism of t-unit copy number expansion (ii) a parsimonious model of array formation in which the fewest number of UCO events necessary to form the observed expansion patterns occurred, and (iii) crossing-over only occurred in the IGR A region, as the phylogenetic data suggest (namely, the incongruity between the t-unit phylogeny and the IGR A phylogeny). After the parsimonious UCO models were created, the predicted phylogenies of recombinant IGR A sequences created by UCO were compared to the actual IGR A phylogeny (Supplementary Fig. 4). The model-predicted phylogeny was congruent with the phylogenetic reconstruction.
In order to test whether the observed t-unit sequence divergence between At and Al could be a result of rapid t-unit turnover from UCO events, a computer program was written to stochastically simulate UCO events between two arrays, each starting with an identical array (common origin) and then evolving independently after a lineage divergence event. Crossover events could only occur between gene units, and equal crossover was permitted. Using array start sizes of 2–40 and with 100,000 repetitions of each simulation for a given start size, the mean number of crossover events to complete array divergence ranged from 7.62 to 16.9, with a local minimum at an array start size of four (Fig. 5). These simulations show that unequal crossover can cause tandem arrays in two species with a common ancestry to quickly diverge post-speciation, leading to the observed lineage-specific divergence of concerted evolution.

Correlation between start array size and number of crossover events until complete divergence of the array following a lineage divergence event, based on simulation data from Array Divergence Simulation.pl. Blue dots denote the mean number of crossover events to obtain complete divergence for 100,000 repetitions. The error bars represent one standard deviation from the mean
Discussion
Syntenic and phylogenetic examination of the At t-unit gene array (81 pre-tRNA genes organized into 27 t-units of three pre-tRNA genes each) resulted in an unexpected pattern of gene expansion that was also observed in a smaller, orthologous and syntenic Al array. These data suggest a complex series of t-unit duplications, and combined with empirical modeling and simulation data suggest further that unequal crossover events are the likely mechanism of expansion. Interestingly, the array does not appear to have expanded from a single locus within the array. Rather, the array expanded from multiple loci of sub-local duplication consisting of varying units of duplication, nearly all of which involved one or more of the intact t-units (Figs. 3 and 4).
Age of the Array and T-Unit Formation
Since the radiation of the eurosid super-order, Vv (basal rosid) and Cp (Brassicales) have experienced no WGD events while the Arabidopsis lineage has undergone two sequential WGD events (Ming et al. 2008). These two WGD events duplicated any ancestral region of the At genome twice, thereby generating three syntenic regions for any given genomic region of the At genome. By virtue of At and Al sharing a recent common ancestor, any intra-genomic syntenic regions found in At are likely to be shared in Al; this is the case for the genomic region in which the At t-unit array is found. Comparison of the genomic region in At containing the t-unit array to the three At intra-genomic syntenic regions, the four syntenic regions of Al, and the single syntenic regions of grape and papaya, respectively, revealed the existence of the t-unit array only on chromosome one of At and its orthologous syntenic region in Al (Supplementary Fig. 2). Neither outgroup syntenic regions, nor the other three syntenic regions in the At genome contain any pre-tRNA genes; this is also the case with the Al genome. Through parsimony, this indicates that all the pre-tRNA genes in the array did not exist in their current genomic location prior to the Arabidopsis lineage’s most recent WGD event. Furthermore, no orthologous t-unit array was found in the Brassica lineage, which diverged from the Arabidopsis lineage after the aforementioned most recent WGD event. However, until the B. rapa genome is completely sequenced and assembled, it remains possible that an orthologous t-unit array does exist in its genome. In addition, more genomic data on lineages diverging prior to and after the Arabidopsis–Brassica lineage split will aid in narrowing down the actual time of the array’s formation.
No other intact t-unit was found in either Arabidopsis genome indicating that the t-unit formed at this ancestral locus and was likely duplicated prior to the divergence of At and Al. As the t-unit arrays in At and Al are derived from the same ancestral array, their divergence time places a lower boundary on the age of the array. Thus, we may infer that the initial formation of the t-unit array likely occurred after the divergence of Arabidopsis and Brassica [14.5–20.4 million years ago (Yang et al. 1999; Bowers et al. 2003)], and necessarily prior to the divergence of At and Al [5–6 million years ago (Koch and Kiefer 2005)].
Unfortunately, a great unanswered question in this study is the origin of the t-unit prior to t-unit copy expansion. Presumably, the ancestral t-unit formed through a series of translocation events of pre-tRNA genes to the array locus, but there is no direct evidence to support this claim. Discovering an outgroup genome with a proto t-unit array may provide the data necessary to elucidate the apparent de novo formation of the ordered and conserved t-unit gene structure.
The Triplet Unit as the Basal Unit of Array Expansion
Apparent conservation of the t-unit during expansion is a fundamental property of the t-unit array. Although the combined At and Al pre-tRNATyr gene phylogeny demonstrated conservation of the pre-tRNATyr gene doublet within the t-units, these data are insufficient to state whether all elements within the t-units are conserved as a single unit during expansion events. By mapping the phylogeny of each sub-element of the t-unit onto their genomic positions within the array, each tree was shown to be congruent; homogeneity tests of congruence provided a statistical basis for using entire t-units in phylogenetic reconstruction. If elements within individual t-units were not conserved during expansion, different elements would show phylogenetic incongruence when mapped to their genomic locations. This conservation demonstrates that the t-unit is the basal unit of expansion in the At array, and also, with few exceptions, in the Al array. Even within the Al array, the gene order of the t-unit containing three pre-tRNATyr genes (Al t-unit 1) is apparently not violated, with the third gene clading with the pre-tRNATyrB genes. The incongruence of the IGR A phylogeny with that of the t-units is hypothesized to be a result of IGR A being used as the site of recombination during UCO resulting in recombinant inter-t-unit sequences that do not follow the other array element phylogenies.
Previous research on the At array has shown that a number of the pre-tRNA genes fail to produce a mature tRNA product in vitro (Stange et al. 1991; Beier and Beier 1992). While the nonfunctional pre-tRNAs appear to be a subset of the total pre-tRNA genes in the array, these putative pseudogenes argue that purifying selection is not acting on all the genes in the array, presumably due to redundancy of function. Compounded by the lack of correlation between pre-tRNATyr gene copy number in the t-unit array and tyrosine residue usage in coding sequences, the numerous deleterious mutations found in t-unit genes argues that the copy number expansions within the arrays is likely non-adaptive and fluctuations in array size are governed by neutral evolution such as drift. If accurate, we predict a range of copy number variation of t-units among individuals in Arabidopsis populations.
The conservation of the t-unit pattern in both At and Al argues for either selection against breaking up t-units, or an additional mechanism of duplication that preferably copies multi-gene units. Unlike protein coding genes, pre-tRNA genes contain internal promoters (Hofstetter et al. 1981), thus negating the necessity for gene duplication to carry additional sequence upstream of the gene to allow for proper transcription by RNA polymerase III. Pre-tRNA genes also require short repeating thymine nucleotides as a transcription termination signal (Stange et al. 1991). However, this termination sequence is only a few nucleotides 3′ of the gene, and is unlikely to provide sufficient selection against breaking up a t-unit during duplication.
An alternative hypothesis to direct selection for t-unit conservation is simply that the sequences flanking the t-units are hot-spots for recombination. In this model, recombination can occur within t-units, but the high affinity for molecular elements responsible for crossover competitively inhibits intra-t-unit recombination. Sequence analysis of the intergenic regions between t-units (IGR A) showed no significant homology to other loci in the At genome (data not shown). While this does not negate the possibility of a recombination hot-spot motif indirectly driving t-unit conservation, no current data supports this model. In addition, t-unit conservation may be driven by DNA topology in the form of nucleosome placement, higher order chromatin packaging, or epigenetic marks that would occlude recombination from occurring within the t-units. Future studies of the Arabidopsis arrays assessing the relationship between the chromatin state of the array and recombination rates within the array may aid in understanding the phenomenon of t-unit conservation during array expansion. In any case, we are currently unable to reconcile the neutral evolution of the array as a whole in terms of copy number variation with the apparent selection for conserved t-units.
In vitro assays of t-unit gene transcription showed that the presence of pre-tRNASer genes did not significantly affect the transcription or maturation of the pre-tRNATyr gene product adjacent to them (Stange et al. 1991). The presence of many putative pseudogenes within the array suggests that the high copy number allows mutated pre-tRNA gene sequences to fly “under the radar” of selection due to redundancy (Ganley and Kobayashi 2007). Interestingly, most of the noted pseudogenes are located at the ends of the array; given the nature of UCO, it is difficult to remove array ends, so it is no surprise that they contain the most divergent sequences. However, it remains unclear whether one would observe the same pattern of higher sequence divergence toward the array ends given gene conversion as the mechanism of sequence homogenization/concerted evolution. If the array centers are more prone to homogenization due to higher frequencies of UCO, perhaps selection is then acting upon the sites of unequal crossover instead of on the genes as a whole, where selection for expansion or contraction of t-units results in an array containing some minimum number of functional t-units. However, this could also be neutral and the result of UCO occurring with a higher likelihood near the center of the array than at the ends. A similar pattern of multi-gene duplicated units containing deleterious mutations is observed in tandem rRNA gene arrays (Ganley and Kobayashi 2007), suggesting a similar mechanism of multi-gene duplication.
Case Study for Modeling Array Formation Using Unequal Crossing-Over as Mechanism of Expansion
For a review of homologous recombination in plants and references to more in-depth reviews of recombination in general, please see Schuermann et al. (2005). Following an initial tandem gene duplication, UCO is a known mechanism of subsequent tandem gene copy number expansion (Smith 1976; Tartof 1988; Achaz et al. 2000) and, therefore, a plausible mechanism of t-unit array expansion. Experiments studying meiotic UCO in At have shown rates of gene copy number variation on the order of ~10−6 per array per plant per F1 meiosis (Jelesko et al. 2004), or at least one copy number variant in one out of approximately 700 seeds due solely to UCO (Gaut et al. 2007). Assuming a single meiotic event per year per organism that gives rise to a single progeny, a given array can expect a gene copy number variant of 1 array−1 Mya−1 per haploid genome. This is two orders of magnitude higher than the proposed rate of an initial tandem gene duplication arising out of 0.01 gene−1 Mya−1 (Lynch and Conery 2000). Note the former value does not correct for the increased frequency of unequal crossover as array size expands, which gives greater opportunity for misalignment (Eichler 2001), as has been observed in micro-satellite DNA (Richard and Dujon 2006). Given the ability of UCO to rapidly expand or contract pre-existing tandem gene arrays, UCO is a prime candidate for mechanistically explaining the formation of the t-unit array and its sequence homogenization in At and Al. In order to test the hypothesis of UCO as the mechanism of t-unit array expansion, a model was devised for the formation of t-units 1–7 (Supplementary Fig. 6). The model assumes that recombination occurs in the IGR A regions, with each duplication from UCO creating recombinant IGR A sequences. The model predicted specific phylogenetic relationships among the derived chimeric IGR A spacer regions, which were subsequently validated using the actual phylogeny of the IGR A sequences (Supplementary Fig. 4; Supplementary Fig. 6). While this parsimonious model does not presume to be an accurate recreation of the true evolutionary history of the array, it does demonstrate that the observed structure of the array can be reconstructed using UCO as the mechanism of copy number expansion.
If unequal crossing-over causes copy number variation via rapid expansion and contractions, and copy number is selectively neutral, then one can expect expansion events to occur at approximately the same rate as for contraction events. If so, then the model of UCO inherently predicts a continual loss of heterogeneity within the array, as t-units lost via array contraction are replaced by “homogenized” gene duplicates as the array re-expands. This would expand on the Birth-and-Death model of concerted evolution (Nei and Rooney 2005) by the explicit inclusion of a coupled birth/death mechanism by UCO. Our computer simulation of UCO in orthologous arrays demonstrated this phenomenon of concerted evolution through intra-array sequence homogenization. There appears to be little evidence to show that the copy number size of the array is adaptive given the variation in array size in the two Arabidopsis’ genomic regions, the apparent lack of positive selection for pre-tRNA dosage in relation to the codon usage across the genomes of At and Al, and the presence of many non-functional pre-tRNA genes within the At array. Thus, successive rounds of UCO inherently causes a loss of phylogenetic information during contraction events; when an expansion event occurs to increase copy number in a tandem array, the t-units replacing those lost will not replace the lost evolutionary information. Together this leads to the observed sequence homogenization between At and Al and the phylogenetic grouping of paralogous versus orthologous t-units, and is evidence of concerted evolution (Zimmer et al. 1980).
As the original discoverers of the tandem array astutely noted (Beier et al. 1991), due to the inferred rapid turnover of t-units, the true evolutionary history of the array will almost certainly prove impossible to determine due to erasure of evolutionary history (Gao and Innan 2004). However, our findings show that the recent evolutionary history of the array can be distilled, and its mechanisms of expansion can be understood. Furthermore, this comparative research between the At and Al arrays shows the highly dynamic nature of these arrays’ evolution, with new elements likely being added and removed independently in both lineages. Likewise, these arrays may be short-lived genomic phenomena. Without selective pressure to maintain multiple copies, these arrays can theoretically be reduced to a singlet state. More array sequences from closely related taxa will aid in understanding the mechanisms of tandem gene array evolution if a very recent expansion or contraction event is identified. Future genome projects involving both population level sequencing and strategic sequencing of taxa based on their phylogenetic relationships will further elucidate the mechanisms of evolution of the t-unit array, and tandem gene evolution in general. However, one outstanding feature of these arrays, which is not explained by our analyses is why UCO appears to occur only among IGR A regions, and perhaps additional taxa sampling will shed light into this mystery.
Conclusion
The triplet pre-tRNA (t-unit) gene arrays located on chromosome one of Arabidopsis thaliana and Arabidopsis lyrata have provided a high-resolution case study of the expansion of a unique triplet of tRNA genes. The t-unit formed after the divergence of Arabidopsis and Brassica lineages, and before the divergence of A. thaliana (At) and A. lyrata (Al) lineages. While the mechanism forming the ancestral t-unit almost certainly involved pre-tRNA gene transposition, it is otherwise an unknown saltation. Expansion from the initial t-unit into an array occurred from multiple origins, and unequal crossing-over (UCO) is likely responsible for the generation of at least part, if not the whole, of the array. UCO as the mechanism of t-unit duplication is supported by phylogentic analysis of intergenic sequences between t-units and comparison of t-unit arrays between At and Al. A consequence of UCO is that such arrays are subject to contraction as well as expansion, which will reduce overall sequence diversity and cause apparent sequence homogenization. This concerted evolution is evidenced by the apparent monophyly of t-units between At and Al, and is supported by computational simulation.
References
Achaz G, Coissac E, Viari A, Netter P (2000) Analysis of intrachromosomal duplications in yeast saccharomyces cerevisiae: a possible model for their origin. Mol Biol Evol 17:1268–1275
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215:403–410
Baumgarten A, Cannon S, Spangler R, May G (2003) Genome-level evolution of resistance genes in Arabidopsis thaliana. Genetics 165:309–319
Beier D, Beier H (1992) Expression of variant nuclear Arabidopsis tRNA(Ser) genes and pre-tRNA maturation differ in HeLa, yeast and wheat germ extracts. Mol General Genetics 233:201–208
Beier D, Stange N, Gross HJ, Beier H (1991) Nuclear tRNATyr genes are highly amplified at a single chromosomal site in the genome of Arabidopsis thaliana. Mol General Genetics 225:72–80
Bowers JE, Chapman BA, Rong J, Paterson AH (2003) Unravelling angiosperm genome evolution by phylogenetic analysis of chromosomal duplication events. Nature 422:433–438
Brown DD, Wensink PC, Jordan E (1972) A comparison of the ribosomal DNA’s of Xenopus laevis and Xenopus mulleri: the evolution of tandem genes. J Mol Biol 63:57–73
Eichler EE (2001) Recent duplication, domain accretion and the dynamic mutation of the human genome. Trends Genet 17:661–669
Force A, Lynch M, Pickett FB, Amores A, Yan YL, Postlethwait J (1999) Preservation of duplicate genes by complementary, degenerative mutations. Genetics 151:1531–1545
Freeling M (2009) Bias in plant gene content following different sorts of duplication: tandem, whole-genome, segmental, or by transposition. Annu Rev Plant Biol 60:433–453
Ganley ARD, Kobayashi T (2007) Highly efficient concerted evolution in the ribosomal DNA repeats: total rDNA repeat variation revealed by whole-genome shotgun sequence data. Genome Res 17:184–191
Gao L-Z, Innan H (2004) Very low gene duplication rate in the yeast genome. Science (New York, NY) 306:1367–1370
Gaut BS, Wright SI, Rizzon C, Dvorak J, Anderson LK (2007) Recombination: an underappreciated factor in the evolution of plant genomes. Nat Rev Genet 8:77–84
Goodman HM, Olson MV, Hall BD (1977) Nucleotide sequence of a mutant eukaryotic gene: the yeast tyrosine-inserting ochre suppressor SUP4-o. Proc Natl Acad Sci USA 74:5453–5457
Hofstetter H, Kressman A, Birnstiel ML (1981) A split promoter for a eucaryotic tRNA gene. Cell 24:573–585
Horiuchi T, Tomizawa JI, Novick A (1962) Isolation and properties of bacteria capable of high rates of beta-galactosidase synthesis. Biochim Biophys Acta 55:152–163
Hurles M (2004) Gene duplication: the genomic trade in spare parts. PLoS Biol 2: e206 EP–e206 EP
Jackson AP (2007) Tandem gene arrays in Trypanosoma brucei: comparative phylogenomic analysis of duplicate sequence variation. BMC Evol Biol 7:54
Jaillon O, Aury J-M, Noel B, Policriti A, Clepet C, Casagrande A, Choisne N, Aubourg S, Vitulo N, Jubin C, Vezzi A, Legeai F, Hugueney P, Dasilva C, Horner D, Mica E, Jublot D, Poulain J, Bruyère C, Billault A, Segurens B, Gouyvenoux M, Ugarte E, Cattonaro F, Anthouard V, Vico V, Fabbro CD, Alaux M, Gaspero GD, Dumas V, Felice N, Paillard S, Juman I, Moroldo M, Scalabrin S, Canaguier A, Clainche IL, Malacrida G, Durand E, Pesole G, Laucou V, Chatelet P, Merdinoglu D, Delledonne M, Pezzotti M, Lecharny A, Scarpelli C, Artiguenave F, Pè ME, Valle G, Morgante M, Caboche M, Adam-Blondon A-F, Weissenbach J, Quétier F, Wincker P (2007) The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature 449:463–467
Jelesko JG, Carter K, Thompson W, Kinoshita Y, Gruissem W (2004) Meiotic Recombination Between Paralogous RBCSB Genes on Sister Chromatids of Arabidopsis thaliana. Genetics 166:947–957
Koch MA, Kiefer M (2005) Genome evolution among cruciferous plants: a lecture from the comparison of the genetic maps of three diploid species–Capsella rubella, Arabidopsis lyrata subsp. petraea, and A. thaliana. Am J Bot 92:761–767
Lewis D (1953) A relationship between dominance, phenotypic stability and variability, and a theory of alternative genetic pathways. Nature 172:1136–1137
Lowe TM, Eddy SR (1997) tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucl Acids Res 25:955–964
Lynch M, Conery JS (2000) The evolutionary fate and consequences of duplicate genes. Science 290:1151–1155
Lyons E, Freeling M (2008) How to usefully compare homologous plant genes and chromosomes as DNA sequences. Plant J 53:661–673
Lyons E, Pedersen B, Kane J, Alam M, Ming R, Tang H, Wang X, Bowers J, Paterson A, Lisch D, Freeling M (2008) Finding and comparing syntenic regions among Arabidopsis and the outgroups papaya, poplar, and grape: CoGe with rosids. Plant Physiol 148:1772–1781
Maddison W, Maddison D (2009) Mesquite: a modular system for evolutionary analysis. Version 2.6 URL: http://mesquiteproject.org
Miller MA, Holder MT, Vos R, Midford PE, Liebowitz T, Chan L, Hoover P, Warnow T (2009) The CIPRES Portals URL: http://www.phylo.org/sub_sections/portal
Ming R, Hou S, Feng Y, Yu Q, Dionne-Laporte A, Saw JH, Senin P, Wang W, Ly BV, Lewis KLT, Salzberg SL, Feng L, Jones MR, Skelton RL, Murray JE, Chen C, Qian W, Shen J, Du P, Eustice M, Tong E, Tang H, Lyons E, Paull RE, Michael TP, Wall K, Rice DW, Albert H, Wang M-L, Zhu YJ, Schatz M, Nagarajan N, Acob RA, Guan P, Blas A, Wai CM, Ackerman CM, Ren Y, Liu C, Wang J, Wang J, Na J-K, Shakirov EV, Haas B, Thimmapuram J, Nelson D, Wang X, Bowers JE, Gschwend AR, Delcher AL, Singh R, Suzuki JY, Tripathi S, Neupane K, Wei H, Irikura B, Paidi M, Jiang N, Zhang W, Presting G, Windsor A, Navajas-Perez R, Torres MJ, Feltus FA, Porter B, Li Y, Burroughs AM, Luo M-C, Liu L, Christopher DA, Mount SM, Moore PH, Sugimura T, Jiang J, Schuler MA, Friedman V, Mitchell-Olds T, Shippen DE, dePamphilis CW, Palmer JD, Freeling M, Paterson AH, Gonsalves D, Wang L, Alam M (2008) The draft genome of the transgenic tropical fruit tree papaya (Carica papaya Linnaeus). Nature 452:991–996
Nei M, Rooney AP (2005) Concerted and birth-and-death evolution of multigene families. Annu Rev Genet 39:121–152
Nylander JAA (2004) MrModelTest v2. In: Program distributed by the author, Evolutionary Biology Centre, Uppsala University
Ohno S (1970) Evolution by gene duplication. Springer-Verlag, Heidelberg, Germany
Ohta T (1988) Time for acquiring a new gene by duplication. Proc Natl Acad Sci USA 85:3509–3512
Posada D, Crandall KA (1998) MODELTEST: testing the model of DNA substitution. Bioinformatics 14:817–818
Rambaut A (2006–2008) Tree Figure Drawing Tool Version 1.1.2. In: Institute of evolutionary biology, University of Edinburg
Richard G-F, Dujon B (2006) Molecular evolution of minisatellites in hemiascomycetous yeasts. Mol Biol Evol 23:189–202
Ronquist F, Huelsenbeck JP (2003) MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19:1572–1574
Schuermann D, Molinier J, Fritsch O, Hohn B (2005) The dual nature of homologous recombination in plants. Trends Genet 21:172–181
Schwartz S, Kent WJ, Smit A, Zhang Z, Baertsch R, Hardison RC, Haussler D, Miller W (2003) Human-mouse alignments with BLASTZ. Genome Res 13:103–107
Sémon M, Wolfe KH (2007) Consequences of genome duplication. Curr Opin Genet Dev 17:505–512
Smith GP (1976) Evolution of repeated DNA sequences by unequal crossover. Science (New York, NY) 191:528–535
Stange N, Beier D, Beier H (1991) Expression of nuclear tRNA(Tyr) genes from Arabidopsis thaliana in HeLa cell and wheat germ extracts. Plant Mol Biol 16:865–875
Swofford D (2003) PAUP*. Phylogenetic Analysis Using Parsimony (*and Other Methods). Version 4b10. In: Sinauer Associates, Sunderland, Massachusetts
Tartof KD (1988) Unequal crossing over then and now. Genetics 120:1–6
Team RDC (2007) R: a language and environment for statistical computing. R Foundation for Statistical Computing. Vienna, Austria
Thompson JD, Higgins DG, Gibson TJ (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucl Acids Res 22:4673–4680
Yang YW, Lai KN, Tai PY, Li WH (1999) Rates of nucleotide substitution in angiosperm mitochondrial DNA sequences and dates of divergence between Brassica and other angiosperm lineages. J Mol Evol 48:597–604
Zhang J (2003) Evolution by gene duplication: an update. Trends Ecol Evol 18:292–298
Zimmer EA, Martin SL, Beverley SM, Kan YW, Wilson AC (1980) Rapid duplication and loss of genes coding for the a chains of hemoglobin. PNAS 77:2158–2162
Acknowledgments
Arabidopsis lyrata sequence data were produced by the US Department of Energy Joint Genome Institute (http://www.jgi.doe.gov/) in collaboration with the user community. This study is supported by the UC Berkeley Undergraduate Research Apprentice Program, the UC Berkeley Summer Undergraduate Research Fellowships program, and by the National Science Foundation grant MCB-0820821. We are grateful to Kelly Nissen and Damon Lisch for their contributive discussions and insight, Brent Mishler for his expertise in phylogenetics, Detlef Weigel for his leading the A. lyrata genome sequencing project, Jeremy Schmutz for his help with validating the assembly of A. lyrata, and the CIPRES portal for their excellent suite of phylogenetic tools.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Kane, J., Freeling, M. & Lyons, E. The Evolution of a High Copy Gene Array in Arabidopsis . J Mol Evol 70, 531–544 (2010). https://doi.org/10.1007/s00239-010-9350-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00239-010-9350-2