
Abstract
The oligopeptide transporter (OPT) family of peptide and iron-siderophore transporters includes members from both prokaryotes and eukaryotes but with restricted distribution in the latter domain. Eukaryotic members were found only in fungi and plants with a single slime mold homologue clustering with the fungal proteins. All functionally characterized eukaryotic peptide transporters segregate from the known iron-siderophore transporters on a phylogenetic tree. Prokaryotic members are widespread, deriving from many different phyla. Although they belong only to the iron-siderophore subdivision, genome context analyses suggest that many of them are peptide transporters. OPT family proteins have 16 or occasionally 17 transmembrane-spanning α-helical segments (TMSs). We provide statistical evidence that the 16-TMS topology arose via three sequential duplication events followed by a gene-fusion event for proteins with a seventeenth TMS. The proposed pathway is as follows: 2 TMSs → 4 TMSs → 8 TMSs → 16 TMSs → 17 TMSs. The seventeenth C-terminal TMS, which probably arose just once, is found in just one phylogenetic group of these homologues. Analyses for orthology revealed that a few phylogenetic clusters consist exclusively of orthologues but most have undergone intermixing, suggestive of horizontal transfer. It appears that in this family horizontal gene transfer was frequent among prokaryotes, rare among eukaryotes and largely absent between prokaryotes and eukaryotes as well as between plants and fungi. These observations provide guides for future structural and functional analyses of OPT family members.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Transport proteins have been classified in the Transporter Classification Database (TCDB, www.tcdb.org; Saier 2000a, b; Saier et al. 2006, 2009). The first class is composed of channels/pores which catalyze facilitated diffusion by an energy-independent process. Electrochemical potential–driven transporters, comprising the second class, are stereospecific carriers catalyzing uniport, antiport and/or symport (Saier 2000c; Busch and Saier 2004). Primary active transporters, class 3, utilize a primary source of energy (chemical, electrical and/or solar) to drive transport of a solute against a concentration gradient (Saier 2000a). Group translocators, class 4, utilize a primary energy source to chemically alter a substrate in a process coupled to transport across a membrane (Mitchell and Moyle 1958).
The oligopeptide transporter (OPT, TC 2.A.67) family consists of electrochemical potential–driven transporters (class 2). All functionally characterized members of this family catalyze uptake of their solutes by a cation:solute symport mechanism (Hauser et al. 2001; Lubkowitz 2006; Yen et al. 2001). Functionally characterized members consist of transporters specific for oligopeptides (three to eight amino acids) and iron-siderophores (Yen et al. 2001). Characterized peptide transporters transport oligopeptides, glutathione and glutathione conjugates (Kaur et al. 2009; Lubkowitz et al. 1998). Characterized “yellow stripe” (YS) homologues, on the other hand, mediate the uptake of metal-chelating phytosiderophores, including iron-nicotinamine and complexes of iron with secondary amino acid derivatives such as mugineic acid and deoxymugineic acid (Kaur et al. 2009). The biochemical and physiological characteristics of several OPT homologues have been studied (Lubkowitz 2006; Osawa et al. 2006; Stacey et al. 2008; Thakur et al. 2008). Two highly conserved motifs (NPG and KIPPR) have been found in many OPT family proteins (Koh et al. 2002). The OPT family is not to be confused with the proton-dependent oligopeptide transporter (POT or PTR, TC 2.A.17) family (Paulsen and Skurray 1994), the peptide transporters (PepTs) of the ATP-binding cassette (ABC, TC 3.A.1.5) superfamily (Saier 2000a; Busch and Saier 2004) or the peptide/acetyl-CoA transporters (PATs) of the major facilitator superfamily (MFS, TC 2.A.1.25) (Pao et al. 1998).
Oligopeptide transport plays important roles in nitrogen storage and mobilization, quorum sensing, differentiation, sexual induction, mating and pheromone sensing. One of the yeast homologues is the sexual differentiation process (ISP4) protein of Schizosaccharomyces pombe. In yeast, OPT family homologues transport oligopeptides, which are commonly tri-, tetra- and/or pentapeptides (Wiles et al. 2006). Recently, it has been found that high-affinity Saccharomyces cerevisiae and Sc. pombe glutathione transporters, Hgt1p and OPT1, respectively, belong to the OPT family (Dworeck et al. 2009; Kaur et al. 2009).
In Candida albicans, eight OPT genes have been identified, encoding putative OPTs. Almost all are represented by polymorphic alleles (Reuss and Morschhauser 2006). OPT1,2,3∆ triple mutants were found to have a severe growth defect, which could be rescued by reintroduction of a single copy of OPT1, OPT2 or OPT3. The various transporters differ in their substrate preferences as shown by the ability of strains expressing specific OPT genes to grow on peptides of defined length and sequence (Reuss and Morschhauser 2006).
In plants, many OPTs appear to be plasma membrane–embedded proteins that import substrates from the apoplasm (the aqueous phase of the cell wall) and the external environment. They may play roles in plant growth and development (Lubkowitz 2006). Unlike many other OPTs, which function in long-distance transport of peptides or metals, YS1, an Fe3+-phytosiderophore uptake system of Zea mays, is known to translocate substrates from the rhizosphere (the region of the soil that is directly influenced by root secretions and associated with soil microbes) (Yen et al. 2001; Curie et al. 2001). Expression of the YS1 gene is increased in roots and shoots under iron-deficient conditions (Curie et al. 2001). When YS1 is expressed in mutant yeast lacking its native iron uptake system, it is able to correct the defect, specifically in Fe3+ phytosiderophore–containing media.
In Arabidopsis, nine OPT paralogues have been identified (Koh et al. 2002), seven of which mediate transport of tetra- and pentapeptides. Cagnac et al. (2004) showed that AtOPT6 can mediate uptake of glutathione derivatives and metal complexes, which led them to suggest that it may also be involved in stress resistance.
Bacterial and archaeal homologues of the OPT family have yet to be characterized biochemically, but as shown here, they are prevalent throughout the prokaryotic world (Kaur et al. 2009). A high-resolution three-dimensional X-ray structure of an OPT family homologue has yet to be solved. We therefore carried out detailed bioinformatic analyses of these transporters, showing that the family is far more widespread than previously recognized and demonstrating the evolutionary relationships of the members of this family to each other. Most surprisingly, we found that these 16-TMS proteins arose from a two-TMS precursor–encoding genetic element which duplicated three times sequentially: 2 TMSs → 4 TMSs → 8 TMSs → 16 TMSs. Although this finding is in principle similar to the origin of animal Na+ and Ca2+ channel proteins of the voltage-gated ion channel (VIC, TC 1.A.1) family, where a six-TMS precursor twice duplicated to give 24-TMS proteins (Nelson et al. 1999), this is the first demonstration of such an event occurring from a two-TMS element and involving three successive intragenic duplication events.
Methods
PSI-BLAST (Altschul et al. 1997) searches were performed to screen the National Center for Biotechnology Information (NCBI) nonredundant protein database using C. albicans Opt1 (gi 74582040), Sc. pombe Isp4 (gi 19859374), Sa. cerevisiae Opt1 (gi 731969), Z. mays YS1 (gi 75168533) and Myxococcus xanthus EspB (gi 75421577). The corresponding TinySeq XML format (NCBI) of these proteins was obtained and modified using the script MakeTable5 (Yen et al. 2009) to generate a FASTA file for all of the sequences and a table containing each protein’s abbreviation, description, organismal source, size, gi number, organismal kingdom or phylum and organismal domain. MakeTable5 was also used to remove fragments and protein sequences with >90% sequence identity to an included protein.
Multiple alignments of homologous proteins and phylogenetic trees were generated using the CLUSTAL X program (Thompson et al. 1997) followed by the TreeView program (Zhai et al. 2002) with default settings. The WHAT (Zhai and Saier 2001a) and TMHMM (Kall et al. 2007) programs were used to perform topological analyses on single protein sequences. The AveHAS program (Zhai and Saier 2001b) with default settings was used to generate average hydropathy, amphipathicity and similarity plots for multiply aligned sequences. Internal homologous repeat segments in all OPT proteins examined were statistically compared using the IC(Faa2) program (Yen et al. 2009). Segments giving the best comparison scores were further examined using the GAP program with default settings and 500 random shuffles with comparison scores expressed in standard deviations (SDs) (Devereux et al. 1984). A value of 10 SD corresponds to a probability of 10−24 that the observed degree of similarity occurred by chance (Dayhoff et al. 1983). To optimize, nonaligned segments were removed, numbers of identities were maximized and numbers of gaps were minimized, maintaining a length of at least 60 residues. The comparison score was then determined again as before. For a stretch of at least 60 amino acyl residues, corresponding to a typical, average-sized protein domain, 10 SD is deemed sufficient to establish homology (Saier 1994; Saier et al. 2009; Yen et al. 2009).
The GGSEARCH (http://fasta.bioch.virginia.edu/gasta_www2/fasta_list2.shtml), HMMER (http://hmmer.janelia.org; Eddy, 2008) and SAM (Yen et al. 2009; Wang et al. 2009) programs were subsequently used to provide confirmatory evidence for homology. The halves, quarters and eighths of these homologues, which showed significant sequence similarity using IC/GAP (Table 2), were subsequently used to generate a profile and a database for each program.
The hmmbuild program was first used to build an HMM profile for each eight- or four-TMS segment. This profile was then calibrated using the hmmcalibrate program to obtain more accurate e-values. The resulting calibrated profile was then used to search a corresponding eight- or four-TMS segment database (FASTA-formatted sequence file) with the hmmsearch program. The resulting output file showed the domain and alignment annotation for each sequence. The HMMER commands used were
-
hmmbuild <hmm file> <alignment file>
-
hmmcalibrate <hmm file>
-
hmmsearch <hmm file> <sequence file>
The same essential procedures were used for SAM and GGSEARCH. Using the SAM program, the sequence files from the halves and quarters were first trained to build models. The models were subsequently used to search against a database consisting of the corresponding untrained halves and quarters. The SAM commands used were
-
buildmodel <model name> -train <training set> -randseed0
-
hmmscore <output> -I <model file> -db <target sequence file? –sw 2 –calibrate 1
GGSEARCH of the FASTA package from the University of Virginia (http://fasta.bioch.virginia.edu/fasta_www2?fasta_www.cgi?rm=selectandpgm=gnw) was similarly used to compare the eight-TMS halves and the four-TMS quarters.
Results
Phylogenetic Analysis of OPT Family Members
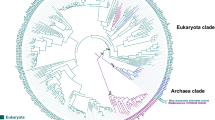
The 325 proteins included in this study are listed alphabetically in supplementary Table S1 (http://biology.ucsd.edu/~msaier/supmat/OPT/index.html) and according to cluster and position in the phylogenetic tree (Fig. 1) in Table 1. The dendogram corresponding to the tree shown in Fig. 1 can be viewed in supplementary Fig. S2. The tree shown in Fig. 1 reveals five clusters subdivided as follows. Cluster 1 includes three subclusters, 1A–1C; clusters 2 and 3 have two subclusters each, A and B; cluster 4 includes seven subclusters, labeled 4A–4G; and cluster 5 contains four subclusters, 5A–5D (Fig. 1).

Phylogenetic tree of 325 OPT superfamily proteins based on the ClustalX multiple alignment shown in Fig. S1 and drawn using the FigTree program. Clusters 1–5 are labeled with their respective subclusters. Subclusters 1A–3B are putative peptide transporters, while some members of subclusters 4A–5D are known to be iron-siderophore transporters. Protein abbreviations are presented in Table 1 in the same order as shown in the tree, together with the characteristics of these proteins. The positions of the individual proteins are revealed in the dendrogram shown in Fig. S2
The data presented in Table 1 reveal the organismal types and size distributions of these proteins according to subcluster. Thus, for example, subclusters 1A (56 proteins) and 1B (48 proteins) are derived exclusively from fungi, but subcluster 1C (27 proteins) is derived exclusively from plants. Subcluster 1C is more distantly related to 1A and 1B than these latter two subclusters are to each other (Fig. 1). The average sizes of the proteins in subclusters 1A–1C are 825 ± 103 amino acids (aas), 893 ± 41 aas and 761 ± 105 aas, respectively. These size differences are statistically significant and suggest fundamental differences between these three groups of proteins. Plant proteins on average are 11% smaller than fungal proteins. This corresponds to the same average size differences observed between plant and fungal homologues of several other ubiquitous families of transporters, as reported by Chung et al. (2001).
The variations in size within each of these subclusters are also of considerable interest. For example, in subcluster 1A, the four proteins Ncr6, Cgl3, Ssc1 and Gze5 cluster tightly together and are roughly 250 aas larger than most of the other homologues. BLAST searches revealed that the extra amino acids in these proteins are at the N termini, do not comprise a domain recognized by the Conserved Domain Database (CDD) and, although probably homologous, are very diverse in sequence. Another protein of even greater size is Cci3, with 1,292 aas. This protein also exhibits a long N-terminal extension that proved to similarly represent a CDD nonrecognizable domain. It showed similarity to only a few other fungal proteins. Finally, two moderately large fungal proteins, Cne3 and Uma1, have 961–985 aas. The extensions again proved to be at the N termini, and these sequences showed little similarity to other protein sequences in the NCBI database. When these large homologues were removed from the list of subcluster 1A proteins, the average size proved to be 790 ± 30 aas. Thus, we conclude that the basic size of these proteins is about 790 aas, and all of the larger homologues have extra N-terminal hydrophilic extensions.
The variation in size within subcluster 1B is minimal. Several proteins have sizes within the range 900–967 aas, but one protein, Yli7, contains 1,032 aas. This protein was also examined and proved to have an N-terminal extension that was not homologous to anything in the NCBI database. When this protein was removed from subcluster 1B proteins, the average size was 890 ± 36 aas.
Subcluster 1C includes proteins with sizes that vary between 689 and 771 aas with one exception, Osa16. This plant protein shows a long C-terminal hydrophilic extension of about 530 aas. CDD recognized this domain as a member of the pepsin (protease) superfamily. It makes physiological sense that a protease would be fused to a peptide transporter, and thus, it appears likely that this fusion is not artifactual. Two programs, TMHMM (Krogh et al. 2001) and HMMTOP (Tusnady and Simon 2001), were used to determine the orientation of this protein in the membrane. Both programs indicated that the protease domain is located on the cytoplasmic side of the membrane. In fact, these programs showed agreement that most 16-TMS members of the OPT family have both their N and C termini on the inside. Excluding Osa16, the average size for all remaining proteins in this subcluster is 742 ± 20 aas.
Clusters 2 (11 proteins) and 3 (16 proteins) are close together on the phylogenetic tree, and both derive exclusively from fungi. Both clusters can be subdivided into two subclusters; the subclusters in cluster 2 are deep-branching, while those in cluster 3 are not. Cluster 3 proteins have an average size of 788 ± 30 aas, and all proteins occur within the range 746–860 aas. Cluster 2 is of even greater size uniformity except for one protein (Ncr4), which is about twice as large (1,619 aas) as the others. The OPT family homology region begins at about residue 920 with the expected ~16 TMSs, while the first 900 residues exhibit characteristics of a water-soluble protein. A BLAST search against the NCBI database of this region retrieved fungal peptidases from the S41 family. It was therefore clear that Ncr4 is the second OPT family protein identified which has a fused protease domain. However, in contrast to Osa16, which had a C-terminal pepsin fusion, Ncr4 has an N-terminal peptidase S41 homologue fusion. Again, the two programs, TMHMM and HMMTOP, were used to estimate the orientation of this protein in the membrane. Surprisingly, and contrary to results of most other members of the OPT family, these two programs predicted that the N terminus of Ncr4 is on the outside. We therefore examined the distribution of lysine and arginine residues within the transmembrane domain of this protein as well as all members present in the multiple alignment shown in Supplementary Figure S1, which can be viewed on our Web site. In both cases, the results clearly suggested that the N termini are on the cytoplasmic side of the membrane. The mistake made by the two programs may have resulted from incorrect assignments of four cytoplasmic regions that the programs considered transmembrane. Once again, fusion of a peptidase with a peptide transporter makes excellent physiological sense. As expected, based on topological and charge distribution analyses, the cytoplasmic peptidase would hydrolyze the peptides brought in by the transporter in a sequential or coupled process (Saier et al. 2005; Merdanovic et al. 2005; Black and DiRusso 2007).
Cluster 4 (84 proteins) and cluster 5 (83 proteins) are the two largest clusters of OPT family members (about half of the total proteins included), as shown in the top half of the tree in Fig. 1. While cluster 4 can be conveniently divided into seven subclusters, we have divided cluster 5 into 4 subclusters. All cluster 4 proteins are derived from prokaryotes, very few of which are derived from archaea (two in subcluster 4A, one in subcluster 4B, two in subcluster 4F and one in subcluster 4G). Only subcluster 4F lacks bacterial homologues. Within each of these subclusters there is little size variation; thus, the average sizes of subclusters 4A–4D vary between 642 and 665 aas. By contrast, the proteins in subclusters 4E–4G are much smaller (average subcluster size of 529–553 aas). Not even a single protein within these seven subclusters is substantially outside of its subcluster size range. The difference in size between these two groups of subclusters, about 110 residues, proved to be due to a C-terminal extension present in every one of the former proteins but lacking in the latter as well as the loss of several short sequences within the loop regions between transmembrane domains of the latter. This 110-aa extension proved to be unrelated to anything else in the NCBI nr-protein databank.
Cluster 5 is much more divergent with respect to organismal type and size, but each of the four subclusters exhibits a surprising degree of uniformity. Thus, subcluster 5A (15 proteins) derives exclusively from δ- and γ-proteobacteria, and these proteins exhibit an average size of 589 ± 29 aas; no protein is appreciably outside of this range. Subcluster 5B (27 proteins) derives from fungi with one exception, a protein from the slime mold Dictyostelium discoideum. The average size is 742 ± 45 aas, and two Aspergillus proteins are substantially larger than the others (Afu3, 843 aas; Aor6, 851 aas). Examination of the multiple alignment revealed that these latter two proteins have neither N- nor C-terminal extensions. Instead, both have internal insertions near their N termini immediately preceding TMS 1. These inserts are found only in these two proteins. The other insert is near the C termini of these proteins, immediately preceding the last TMS. Homologous sequences are found in a few other proteins, mostly from species of Aspergillus. Neither of these 40-residue inserts shows appreciable sequence similarity with other proteins in the NCBI Protein Database.
Subcluster 5C (four proteins) derives from three β-proteobacteria and one δ-proteobacterium. The average size is 606 ± 20 aas, similar to that of subcluster 5A, also derived from proteobacteria. These proteins are much shorter than the eukaryotic proteins of subclusters 5B and 5D. Subcluster 5D (37 proteins) is derived exclusively from plants and has an average size of 697 ± 40 aas. Only one protein is substantially larger than the others, Osa13 (882 aas). It has an approximately 150-residue C-terminal hydrophilic extension found in no other member of this subcluster. This region of the protein showed a low degree of sequence similarity with chloride transporters of the ClC family (TC 2.A.49). However, the functional significance of this observation is questionable.
One member of each subcluster was used as the query sequence to search TCDB using TC-BLAST. All subclusters in clusters 1–3 (lower half of the tree) proved to bring up peptide transporters, while all of the subclusters from clusters 4 and 5 brought up the iron-complex transporters. The phylogenetic segregation between these two functional types is considerable, suggesting that, in general, function correlates with phylogeny. However, genome context analyses reported below suggest otherwise.
Orthologous Relationships Within Subclusters of the OPT Family Tree
The phylogenetic tree for the 16S/18S rRNAs is shown in Fig. 2. The bacteria appear at the top of this tree, the archaea in the small cluster on the right-hand side and the eukaryotes at the bottom. Every genus included in our study of OPT family members is represented in this tree with the exceptions of Acidobacteria, Ashbya, Cryptococcus and Thlaspi. The tree shows that all of the γ- and β-proteobacteria cluster most closely together followed by the α-, δ- and ε-proteobacteria on the upper left-hand side. Surprisingly, in this tree, the ε-proteobacteria cluster loosely with the bacteroidetes, distantly from the other proteobacteria. The cluster on the upper right-hand side of the tree includes a single member of the acidobacteria, a single cluster of actinobacterial rRNAs and two distinct clusters of firmicutes. The eukaryotic branch of the tree shows the slime mold Dictyostelium closer to the center of the tree, with the fungal and plant RNAs clustering more closely to each other but much more distantly from the slime mold at the bottom of the tree.

Phylogenetic tree of 16S/18S rRNAs from all genera represented in this study with the exceptions of Acidobacteria, Ashbya, Cryptococcus and Thlaspi. All bacterial rRNAs appear at the top of the tree, the eukaryotic rRNAs are at the bottom of the tree and the three archaeal genera are positioned on the central branch on the right-hand side of the tree. The phylum/kingdom is indicated for each of the clusters, while the genus is shown at the end of each branch
Orthologues are defined as homologues (derived from a single common ancestor) that arose purely as a result of speciation. That is, they arose via vertical, rather than horizontal, transmission, from parent cell to daughter cell throughout their evolutionary histories. This is reflected by approximately the same phylogenetic relationships observed for the proteins under consideration and the 16S rRNAs. The 16S rRNAs are assumed to have arisen solely by vertical transmission. Any set of proteins that exhibit the same relationships to each other as to the 16S rRNAs that were derived from the same species are considered to exhibit orthologous relationships.
Comparing the protein tree (Figs. 1 and S2) with the RNA tree (Fig. 2), we see that in some, but not other, cases orthologous relationships are difficult to establish. This is true for the large cluster 1. For example, subcluster 1C can be subdivided into five sub-subclusters, all but one of which contain paralogues from a single organism. In the largest sub-subcluster, for example, we find five paralogues from Vitis vinifera, two from Oryza sativa of the Indica group and two from Arabidopsis thaliana. The only sub-subcluster that lacks paralogues is the uppermost sub-subcluster with four proteins from four different organisms. Based on the comparison between Figs. 1 and 2, only in this sub-subcluster are the results consistent with orthology.
In the adjacent sub-subcluster, where we find three proteins, one from rice (Oryza) and two from thale cress (Arabidopsis), it appears that the two thale cress proteins arose by gene duplication after these two organisms diverged from each other. The same situation is observed for the next sub-subcluster, where three Arabidopsis proteins cluster tightly together, with a single V. vinifera protein being the outlier. We interpret these results to mean that after Arabidopsis diverged from Vitis, two gene-duplication events in the former organism gave rise to the three paralogues Ath9, Ath16 and Ath17. Similar observations were made for subclusters 1A and 1B.
Cluster 2 shows relationships which suggest orthology. Thus, in both trees, we find the proteins and rRNAs from Neosartorya, Aspergillus and Sclerotinia clustering together; Candida, Lodderomyces and Pichia clustering together; and Neurospora and Botryotinia clustering together. Even within each of these three groups, the phylogenetic order in both trees is the same. We conclude that cluster 2 probably represents a collection of pure orthologues, with no evidence for paralogues or horizontal gene transfer. This observation suggests that these proteins all serve a single unified function in all of these organisms.
In contrast to cluster 2, cluster 3 contains a number of nonadjacent paralogues and shows clear nonorthologous relationships. The obvious paralogues include two proteins each from Gibberella zeae and Ustilago maydis in two different subclusters that are shared by this pair of paralogues from these two organisms. Additionally, based on the comparison between Figs. 1 and 2 (see also the dendogram in Fig. S2), Uma4 from U. maydis does not show orthologous relationships with the other members of this subcluster. Furthermore, the two Neurospora crassa proteins, Ncr5 and Ncr7, are two paralogues within the same sub-subcluster. On the other hand, the three Aspergillus proteins and the one from Neosartorya fisceri form a sub-subcluster on the protein tree as well as the RNA tree, and the same is true for the two Schizophyllum and Laccaria proteins and RNAs which form a distinct sub-subcluster in both trees. The relationships of all of these proteins are similar to the corresponding relationships in the rRNA tree and are therefore consistent with orthology.
The prokaryotic proteins were similarly analyzed. Starting with subcluster 4A, we find seven distinct sub-subclusters. Progressing in the clockwise direction, sub-subcluster 1 includes proteins from α- and β-proteobacteria as well as actinobacteria. As a single β-proteobacterial protein is flanked by α-proteobacterial proteins, it is possible that this one β-proteobacterial protein (Neu1) was obtained by horizontal transfer. However, the α-proteobacterial proteins do not show orthologous relationships. The actinobacterial proteins show relationships consistent with orthology.
Sub-subcluster 2 is derived exclusively from Campylobacter species. Sub-subcluster 3 contains β-proteobacterial proteins with a single outlier (Pae1) from a γ-proteobacterium. The members of this small sub-subcluster could be orthologous. However, in sub-subclusters 4, 6 and 7, orthology is not possible. For example, in sub-subcluster 4 Haemophilus and Actinobacillus proteins are interspersed, while in sub-subcluster 7 γ-proteobacterial and archaeal proteins are interspersed. It would appear that the precursor of the two archaeal proteins were obtained from γ-proteobacteria via horizontal transfer, but this remains speculative.
Analyses of subclusters 4B–4G allowed us to come to similar conclusions. Thus, for example, subcluster 4B contains proteins from highly divergent organisms including δ-proteobacteria, acidobacteria, firmicutes and archaea; subcluster 4C includes proteins from two different bacterial phyla, the bacteroidetes and the acidobacteria; subcluster 4E includes just two proteins from two different bacterial phyla; subcluster 4G contains proteins from firmicutes, β- and γ-proteobacteria, and an archaeon. It seems likely that in all of these subclusters horizontal gene transfer was rampant during the evolution of these proteins.
The four cluster 5 subclusters (A–D) were similarly analyzed. Subcluster 5A, derived from δ- and γ-proteobacteria, includes paralogues with little indication of orthology. Subcluster 5B derives from fungi with the exception of one slime mold protein. It also exhibits relationships suggestive of horizontal gene transfer (especially the slime mold protein Ddi1, which probably derived from a fungus) as well as distant paralogues from three different genera. Even the small subcluster 5C shows signs of the existence of horizontal gene transfer since the δ-proteobacterial protein (Sau3) is unexpectedly closely related to the β-proteobacterial proteins. Finally, subcluster 5D shows many paralogous proteins (e.g., at least 12 probable O. sativa [Japonica group] paralogues and at least seven A. thaliana paralogues). In this case, it is difficult to know if horizontal gene transfer has occurred as all of these proteins could have arisen by vertical transmission from multiple precursor paralogues in the primordial plant.
Topological Analyses of OPT Family Proteins
Figure 3 shows the average hydropathy (top) and average similarity (bottom) plots for all 325 members of the OPT family included in this study. This plot reveals 16 peaks of hydropathy that in general correspond to peaks of similarity. The first four TMSs (labeled 1–4) cluster loosely together. TMSs 4 and 5 are separated by a substantial hydrophilic loop, but again, the next four TMSs (5–8) cluster together. Between TMSs 8 and 9 is an even larger hydrophilic loop, but the remaining eight TMSs cluster tightly together. It is interesting to note that peak 3 and peak 11 appear to divide into two small peaks, possibly due to a misalignment. In fact, there appears to be a gap within the region designated as peak 3 and a smaller gap within the region designated as peak 11. Based on the appearance of this plot, it seemed possible that TMSs 1–8 are repeated in TMSs 9–16. Further, the clustering pattern suggested that these proteins might have arisen from a four-TMS precursor peptide that duplicated twice to give the present-day 16-TMS proteins. In this regard, it should be noted that in all four apparent quadrants the first two TMSs (1–2, 5–6, 9–10 and 13–14) are always close together, while the subsequent two TMSs in each quadrant are separated by greater distances. Following TMS 16 is a poorly conserved region that exhibits moderate hydrophobicity.

Average hydropathy, amphipathicity and similarity plots for the 325 OPT family proteins included in this study. The majority of OPT proteins contain 16 TMSs, which correspond to the 16 conserved peaks labeled 1–16. The central portion of this plot includes all 16 peaks of hydrophobicity which comprise the transporter domain. Functional assignments for the N- and C-terminal hydrophilic domains are discussed in the text. Upper graphs show average hydropathy (dark line) and average amphipathicity (light line), while the bottom graph shows average similarity (continuous line) as well as average hydropathy using a different program (vertical lines)
When the individual subclusters shown in Fig. 1 were analyzed for average hydropathy and average similarity as shown in Fig. 3 for all members of the family, we found that almost all subclusters exhibit the typical 16-TMS topology. However, the proteins within subclusters 4A–4D appeared to have a seventeenth transmembrane segment that was not part of the C-terminal four-TMS repeat. Also, in these four subclusters TMS 13 showed only moderate hydrophobicity as revealed by the AveHAS program. The origin of putative TMS 17 in these proteins is unknown, but it could have arisen as a result of a gene-fusion event. The long N- and C-terminal hydrophilic extensions have been discussed above, and two of them proved to be homologues of functionally recognizable proteases.
Establishment of Internal Repeats in OPT Family Proteins
As noted above, most members of the OPT family contain 16 putative TMSs, although a few appear to have 17 TMSs, the extra one being at the C terminus of each of the cluster 4A–4D proteins. In order to confirm TMS assignment and establish the evolutionary origins of these proteins, we conducted analyses of potential internal repeats. Although initially analyzed assuming different numbers of TMSs per repeat unit, we were able to show with relative ease that these proteins include an eight-TMS duplication. Thus, when using the IC/GAP programs to compare the first halves of these proteins with the second halves, comparison scores of up to 12.6 SD were obtained (see Table 2, Fig. 4). This value is substantially greater than that required to establish homology (Saier 1994; Yen et al. 2009; Wang et al. 2009; Matias et al. 2010).

Alignment of OPT TMSs 1–8 of Spr1 (Serratia proteamaculans, gi 157369266) with OPT TMSs 9–16 of Lsa1 (Lactobacillus sakei, gi 81427933). The IC program was used to identify the two internal segments exhibiting the greatest statistical similarity. The GAP program was used to generate the alignment with default settings and 500 random shuffles. Numbers at the beginning and end of each line indicate the residue numbers in the proteins. The vertical line represents an identity, the colon represents a close similiarity and the period represents a more distant similarity. This convention of presentation is also used in Figs. 5 and 6. In all three figures, positions of the TMSs were predicted using the TMHMM program. This alignment gave a comparison score of 12.6 SD
We next examined the possibility that the eight-TMS halves themselves arose by an earlier intragenic duplication event from a four-TMS precursor. The results from these analyses are also presented in Table 2, and the alignment upon which the best comparison score was based is shown in Fig. 5. In Table 2, we summarize the results obtained using the IC and GAP programs with 500 random shuffles and default settings. All four quarters of these proteins were compared with each other. Only the top two scores are reported, and these were averaged. For all comparisons, values in excess of 10 SD were obtained, clearly indicating homology. However, the best scores were obtained when A vs. C and B vs. D were compared (12.2 and 13.2 SD, respectively). The fact that higher values were obtained for these two comparisons than for any of the others provides evidence that these two duplication events, giving rise to the 16-TMS proteins, were separated by a substantial period of evolutionary time. Thus, we suggest that the primordial four-TMS-encoding genetic element duplicated once to give the eight-TMS precursor and then, later, the second duplication occurred, giving rise to the 16-TMS proteins. Alternatively, segments A and C may share a structure/function that is substantially different from the structure/function shared by segments B and D (see “Discussion” section).

Alignment of OPT TMSs 1–4 of Mth1 (Moorella thermoacetica, gi 83589078) with OPT TMSs 9–12 of Mgr3 (Magnaporthe grisea, gi 39955178). This alignment gave a comparison score of 11.9 SD
As the final step, we examined the possibility that within each of the four-TMS quadrants of these proteins we could detect two two-TMS repeat sequences. Much to our surprise and delight, this possibility could be demonstrated. As shown in Table 2 and Fig. 6, comparing the first two TMSs with the second two TMSs of the first of these four four-TMS repeats gave a maximal value of 8.9 SD, which was insufficient to establish homology. However, when comparing the two two-TMS segments of the second of these four repeats, we were able to get comparison scores in excess of 10 SD, thus establishing homology. In this case, the alignment giving this value included all of TMS 5 compared to TMS 7. When the same was done with the third of these four repeats, a maximal value of 8.6 SD was obtained. The same procedure with the fourth of these four repeats did not give values above 7 SD. Applying the superfamily principle, the values obtained clearly indicate that these proteins arose from an initial two-TMS precursor. We therefore conclude that members of the OPT superfamily arose in three steps: duplication of two TMSs to give four, duplication of four-TMSs to give eight and duplication of eight-TMSs to give 16. The addition of a seventeenth TMS to a small fraction of these proteins presumably occurred as a result of a late gene-fusion event in just one phylogenetic cluster of these proteins.

Alignment of OPT TMSs 1 and 2 of Cim2 (Coccidioides immitis, gi 119190959) with OPT TMSs 3 and 4 of Pgu9 (Pichia guilliermondii, gi 146422868). This alignment gave a comparison score of 8.7 SD
Use and Evaluation of Programs to Detect Similarity and Establish Homology
To confirm the results obtained using the IC/GAP programs, three other programs capable of identifying sequence similarity between repeat segments were used. These programs were GGSEARCH, HMMER and SAM (Table 3). All three programs substantiated the conclusions obtained with IC/GAP. For example, when the two halves were compared with GGSEARCH, a value of 1.7e−8 was obtained. The best value resulting from the use of the HMMER program was 4e−4. When SAM was used, the best value was 4e−3. All of these values confirm our conclusion of homology.
When the four quarters of the OPT family proteins were compared, again the best values were usually obtained when segments A were compared with segments C and when segments B were compared with segments D. Thus, when using GGSEARCH, the values for these two comparisons were 8.6e−6 and 3.9e−8. When using HMMER, the best values were 0.03 and 0.006. With SAM, the best values were 0.002 and 0.001, respectively (Table 3). As revealed by the data in Table 3, only in two instances were values obtained in the other comparisons comparable to these. These results confirm that (1) the four four-TMS quarters of OPT family proteins are all homologous and therefore derive from a common origin, (2) the first and third four-TMS segments are more similar to each other than they are to the second and fourth TMS segments and (3) the second and fourth TMS segments are more similar to each other than they are to the first and third segments.
Functional Predictions Based on Genome Context
Each subcluster was examined using the SEED database in order to allow prediction of potential substrates. These analyses were conducted only for prokaryotic clusters found in clusters 4 and 5. These subclusters will be analyzed sequentially.
Ccr1 in subcluster 4A (see Table 1) is present within a gene cluster that includes an acetyl transferase of the GNAT family (position 2), many members of which are aminoacyl and aminoglycoside transferases. Also present is a PhoH-like ATPase with a Rossman fold similar to ArsA of Escherichia coli. In the same operon with the oligopeptide transporter gene, we also find a glutathione S-transferase-like protein, which undoubtedly uses glutathione as a substrate for nucleophilic addition reactions involving electrophiles. Another nearby gene encodes a protein with a peptidoglycan-binding domain, presumably to anchor the protein or a protein complex to the cell wall. These observations suggest that this particular OPT family permease may be a peptide transporter specific for glutathione. Also within subcluster 4A, Nmo1 was examined and proved in several genomes to be directly upstream of and transcribed in the same direction as genes encoding dipeptidyl aminopeptidases (position 2). Again, it appears likely that Nmo1 is a peptide uptake porter. A third protein examined was Reu3, which brought up operons in various organisms, several of which encoded peptidases of different designations. Again, the most likely function for this protein appears to be a peptide transporter. We therefore conclude that many or perhaps all of the OPT family members in subcluster 4A are peptide transporters.
Cno1 within subcluster 4B proved to be related to operons which encode ornithine carbamoyl transferases, alanine symporters, potential N-acetyl muramoyl-l-alanine amidases and enzymes involved in glutamate metabolism. Because of the association of amino acid metablic enzymes, we again predict that these proteins take up peptides. When Aba2 was examined, several operons appeared to encode dipeptidyl aminopeptidases downstream of the OPT family transporter. Thus, we conclude that subcluster 4B proteins also transport peptides.
When Bfr1 of subcluster 4C was examined using SEED, a frequently cotranscribed gene encodes an endo-1,4-β-xylanase, which may be anchored to the outer membrane. The transcriptional regulator for this operon appears to be a member of the AraC family. Similar results were obtained when Pdi1 was examined. We interpret these results to suggest that subcluster 4C OPT family proteins may be concerned with uptake of xylan-oligosaccharides.
Lca1 of subcluster 4D proved to be present in a gene cluster which also includes genes encoding catabolic threonine dehydrotase, a dipeptidase and an alanine dehydrogenase. In fact, these proteins appear to be in a single operon in the closely related species Pediococcus pentosaceus. Similar results were obtained when Cbe1 was used as the query sequence. We therefore conclude that these proteins are peptide transporters.
Only two proteins comprise subcluster 4E. In the gene cluster with Cce1, we identified genes encoding a pantothenate kinase as well as phospholipases. The other member of this subcluster is from an organism that is not included in the SEED database. These results may suggest that the substrate of this and related transporters could be a phospholipid, but the data are insufficient to make such a prediction with confidence.
Subcluster 4F could not be examined as representation was not present in SEED. However, subcluster 4G included Vei1 in a gene cluster that appeared to be involved in aromatic amino acid metabolism. While we might therefore predict that these transporters are also peptide uptake systems, we again do not believe the evidence is sufficient to make this prediction with confidence.
Cluster 5 proteins include four subclusters. Subclusters 5A and 5C include proteins derived from prokaryotes, while subclusters 5B and 5D include proteins only from eukaryotes. We therefore examined the former two clusters. Examining Ade2 of subcluster 5A, we observed a probable regulatory serine/threonine kinase (position 2) as well as components of a pyruvate/α-ketoglutarate dehydrogenase complex. We also identified an octanoate-[acyl-carrier-protein]-protein-n-octanoyl transferase, a deoxyribonuclease, a protein that recognizes phosphothreonine residues in proteins as well as an aspartokinase involved in threonine and homoserine biosynthesis. Another protein in this subcluster, Ppu1, brought up in position 2 a glutathione S-transferase as well as a putative transcriptional regulator of the LysR type. Finally, Asp4 brought up a glycosyl transferase as well as an NADPH-dependent reductase. We are therefore hesitant to make predictions for the members of this subcluster.
Subcluster 5C includes Reu2, which proved to be encoded by a gene that colocalizes with a zinc-binding protein encoding gene (position 2) and a mutT mutator protein (7,8-dihydro-8-oxoguanine-triphosphatase), with all three probably in a single operon. This operon may be regulated by an AsnC-type transcriptional regulator. Nearby genes also encode a putative ATP/GTP-binding protein, a dephospho-CoA kinase and components of either pyruvate or α-ketoglutarate dehydrogenase complexes. We tentatively suggest that these transporters might be nucleoside or oligonucleotide transporters.
Discussion
In this article, we have described the OPT family of peptide and iron-siderophore uptake transporters and have defined the evolutionary pathway by which these proteins arose. This pathway is illustrated in Fig. 7. A genetic element encoding a two-TMS precursor duplicated to give four TMSs, this duplicated again to give eight TMSs and this also duplicated to give the final 16-TMS topology. In few instances has it been possible to trace back the evolutionary history as far as we have done for the OPT family (Saier 2003). Furthermore, in no other instance has this particular pathway been demonstrated for any other family of transport proteins (Saier 2003 and unpublished observations).

Proposed pathway for the evolutionary appearance of present-day OPT family proteins. Evidence is presented that the ultimate precursor of the 16- (and sometimes 17-) TMS proteins was a two-TMS hairpin structure (top). This then duplicated three times: first to give the four-TMS intermediate, second to give the eight-TMS intermediate and last to give the present-day 16-TMS proteins. Evidence was presented that either the duplication of four TMSs to give eight TMSs occurred substantially before the duplication of eight-TMSs that gave rise to the 16-TMS permeases or segments 1 and 3 share functional/structural features not shared by segments 2 and 4 (see “Discussion” section). In the 17-TMS proteins, the extra TMS is at the C termini of these homologues
We could demonstrate greater similarities between TMSs 1–4 and TMSs 9–12, as well as between TMSs 5–8 and TMSs 13–16, than for other quadrants compared, suggesting that there was a reasonable period of evolutionary time between these two last duplication events. However, the fact that similar maximal values were obtained for the eight-TMS halves, the four-TMS quarters and the two-TMS eighths suggests that all three of these duplication events happened in a relatively short period of evolutionary time. These two apparent inconsistencies could be resolved if the first and third quadrants serve a common structure/function that differs from that of quadrants 2 and 4. In an analogous situation where a six-TMS voltage-gated ion channel has four six-TMS repeats, this last possibility seemed unlikely (Nelson et al. 1999).
A similar situation has been suggested for members of the mitochondrial carrier family which underwent triplication of a primordial two-TMS-encoding genetic element (Kuan and Saier 1993a, b). This family of proteins appears to have undergone rapid intragenic and extragenic duplication events, giving rise not only to the six-TMS porters but also to the main functional types or subfamilies within a relatively short period of time (Kuan and Saier 1993a). Interestingly, in the mitochondrial carriers, the third thirds of these proteins diverged in sequence more than the first two thirds (Kuan and Saier 1993a). The explanation for this observation is not yet clear, but possibly, the last two TMSs are of less functional importance than the first four.
Many transporters have been shown to arise from a two-TMS precursor, but in no case has it been possible to demonstrate three sequential duplication events. Other families in which a two-TMS element duplicated to give four TMSs include the voltage-gated ion channel (VIC, TC 1.A.1) family, the c-subunits of F-type ATPases (F-ATPase, TC 3.A.2) which both duplicated and triplicated and the YiaAB family (TC 9.B.44) (Saier 2003d). Several examples of 4 TMS transmembrane proteins that arose from duplication of a simple 2 TMS hairpin structure have been documented (Sawhney M, Tamang DG and Saier MH Jr., unpublished observations).
A surprising observation was that all members of the OPT family have either 16 or 17 TMSs. The vast majority have 16 TMSs, while a smaller fraction (subclusters 4A–4D in the phylogenetic tree shown in Fig. 1) have 17 putative TMSs. In fact, no 17-TMS protein was found outside of subclusters 4A–4D, and only 17-TMS proteins were found in these four subclusters. The extra TMS at the C termini of these proteins most probably arose only once during the evolution of this family. The only additional variations apparently resulted from the fusion of these integral membrane proteins with soluble domains, two of which could be recognized on the basis of homology searches. In these two cases the fused domains proved to correspond to two different families of peptidases. Since the transporters were predicted to function in peptide uptake and the peptidase domains were predicted to be localized to the cytoplasmic side of the membrane, the fusion of these two catalytic proteins made excellent physiological sense. The peptidase domain probably hydrolyzes peptides upon entry into the cell, possibly in a tightly or loosely coupled process. If tightly coupled, this could be a novel example of group translocation where chemical modification of the substrate is coupled to its transport (Herbert et al. 2003; Hirsch et al. 1998; Merdanovic et al. 2005; Saier et al. 2005).
Uniformity of topology is found in some families, while others show tremendous variation. For example, all recognized proteins in the mitochondrial carrier family (TC 2.A.29) have six TMSs, and no exception has yet been reported (Kuan and Saier 1993a and unpublished results). Another example is the largest superfamily of secondary carriers, the major facilitator superfamily (TC 2.A.1). All recognized members of this superfamily have either 12 or 14 TMSs, where the extra two TMSs in the 14-TMS proteins are present in the center between the two six-TMS repeat units, and they occur only in three of the 70 currently recognized MFS families. This situation is to be contrasted with families that show tremendous topological variations. These include the integral membrane cytochrome c biogenesis proteins of the heme handling protein family (TC 9.B.14) (Lee et al. 2007) and the SdpI family of receptor/signal-transduction proteins (TC 9.A.32) (Povolotsky et al. 2010). In both of these cases, the families include proteins having a wide variety of topological types with numbers of TMSs ranging anywhere from three to 12. Further, they can have segments present in inverted order in some of the proteins relative to other members of the same family. In the SdpI family, this is understood because the different three-TMS repeat segments within these proteins probably serve distinct subfunctions (Povolotsky et al. 2010).
OPT family members were found in both eukaryotes and prokaryotes. The vast majority of the eukaryotic proteins were derived from fungi (subclusters 1A, 1B and 5B as well as clusters 2 and 3) and plants (subclusters 1C and 5D). The only exception is a single slime mold homologue found in subcluster 5B, a cluster otherwise derived entirely from fungi. We hypothesize that this one homologue from D. discoideum was acquired by horizontal transfer from a fungus, a suggestion that is not surprising since slime molds eat other microorganisms (Eichinger et al. 2005). However, we obtained no evidence for horizontal transfer within and between fungi and plants. In view of the fact that homologues of these proteins are found in many bacterial and archaeal phyla, it is surprising that these proteins are not found within the animal kingdom or any of the unicellular eukaryotes except for slime molds.
Prokaryotic homologues of the OPT family are found in subclusters 4A–4G as well as 5A and 5C. In contrast to the situation with eukaryotes, apparent horizontal transfer within and between prokaryotic phyla has been rampant. For example, in subcluster 4A, proteins are derived from four of the five common classes of proteobacteria, the only exception being the δ-proteobacteria. However, this subcluster also contains proteins from actinobacteria and even euryarchaeota. Similarly, subcluster 4B includes proteins from δ-proteobacteria, firmicutes, acidobacteria and euryarchaeota. Subcluster 4C has protein representation only from bacteroidetes and acidobacteria. Subcluster 4D is one of the few “pure” prokaryotic subclusters where all of the proteins derive from firmicutes. Subcluster 4G, a small subcluster of seven proteins, is surprisingly diverse, having members from firmicutes, β- and γ-proteobacteria and euryarchaeota. Finally, subcluster 5A has representation only from γ- and δ-proteobacteria, while subcluster 5C has representation only from β- and δ-proteobacteria. These observations can be interpreted to suggest that horizontal transfer between phyla has occurred in all but two of the prokaryotic subclusters identified in this study.
The large OPT family consists of peptide and iron-siderophore uptake porters, and based on functionally characterized eukaryotic members of this family, iron-siderophore transporters (clusters 4 and 5) segregate from peptide transporters (clusters 1–3). Our operon and genomic context analyses, however, suggest that prokaryotic members of the OPT family are often peptide transporters. This was true for subclusters 4A, 4B and 4D and possibly for 4G and 5A. However, the small subcluster 4C appears more likely to be specific for oligosaccharides, specifically for β-xylan-oligosaccharides. Furthermore, weak evidence suggests that subcluster 5C proteins might be nucleoside or oligonucleotide transporters. At least one eukaryotic OPT can transport both peptides and iron-siderophores. Further, some of the phytosiderophores and mugineic acids resemble peptides in structure. Thus, although the OPT family includes members capable of taking up both types of substrates, there is a need to provide functional analyses of prokaryotic OPTs of the various subclusters in order to establish the range of substrates transported by members of this family.
We have no clear explanation as to why OPT family members appear to be lacking in the animal kingdom as well as many eukaryotic protists. It is possible that these proteins entered the eukaryotic domain from prokaryotes late by horizontal transfer rather than early by vertical descent and that they were either obtained only by fungi and plants (our preferred explanation) or lost from the animal kingdom as well as many eukaryotic protists. If further genome sequencing reveals the presence of these homologues in other types of eukaryotes, this will raise the question of whether these arose by horizontal gene transfer from fungi, plants or slime molds. This may be an important question since in this study we found very little evidence for horizontal transfer between eukaryotic phyla. Future functional analyses and further sequencing efforts are likely to provide eventual answers to these questions. We hope that the analyses reported here will serve as useful guides for molecular biological and bioinformatic analyses of this important family of transporters.
References
Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402
Black PN, DiRusso CC (2007) Vectorial acylation: linking fatty acid transport and activation to metabolic trafficking. Novartis Found Symp 286:127–138 (discussion 138–141, 162–163, 196–203)
Busch W, Saier MH Jr (2004) The IUBMB-endorsed transporter classification system. Mol Biotechnol 27:253–262
Cagnac O, Bourbouloux A, Chakrabarty D, Zhang MY, Delrot S (2004) AtOPT6 transports glutathione derivatives and is induced by primisulfuron. Plant Physiol 135:1378–1387
Chung YJ, Krueger C, Metzgar D, Saier MH Jr (2001) Size comparisons among integral membrane transport protein homologues in bacteria, Archaea, and Eucarya. J Bacteriol 183:1012–1021
Curie C, Panaviene Z, Loulergue C, Dellaporta SL, Briat JF, Walker EL (2001) Maize yellow stripe1 encodes a membrane protein directly involved in Fe(III) uptake. Nature 409:346–349
Dayhoff MO, Barker WC, Hunt LT (1983) Establishing homologies in protein sequences. Methods Enzymol 91:524–545
Devereux J, Haeberli P, Smithies O (1984) A comprehensive set of sequence analysis programs for the VAX. Nucleic Acids Res 12:387–395
Dworeck T, Wolf K, Zimmermann M (2009) SpOPT1, a member of the oligopeptide family (OPT) of the fission yeast Schizosaccharomyces pombe, is involved in the transport of glutathione through the outer membrane of the cell. Yeast 26:67–73
Eddy SR (2008) A probabilistic model of local sequence alignment that simplifies statistical significance estimation. PLoS Comput Biol 4:e1000069
Eichinger L, Pachebat JA, Glockner G, Rajandream MA, Sucgang R, Berriman M, Song J, Olsen R, Szafranski K, Xu Q, Tunggal B, Kummerfeld S, Madera M, Konfortov BA, Rivero F, Bankier AT, Lehmann R, Hamlin N, Davies R, Gaudet P, Fey P, Pilcher K, Chen G, Saunders D, Sodergren E, Davis P, Kerhornou A, Nie X, Hall N, Anjard C, Hemphill L, Bason N, Farbrother P, Desany B, Just E, Morio T, Rost R, Churcher C, Cooper J, Haydock S, van Driessche N, Cronin A, Goodhead I, Muzny D, Mourier T, Pain A, Lu M, Harper D, Lindsay R, Hauser H, James K, Quiles M, Madan Babu M, Saito T, Buchrieser C, Wardroper A, Felder M, Thangavelu M, Johnson D, Knights A, Loulseged H, Mungall K, Oliver K, Price C, Quail MA, Urushihara H, Hernandez J, Rabbinowitsch E, Steffen D, Sanders M, Ma J, Kohara Y, Sharp S, Simmonds M, Spiegler S, Tivey A, Sugano S, White B, Walker D, Woodward J, Winckler T, Tanaka Y, Shaulsky G, Schleicher M, Weinstock G, Rosenthal A, Cox EC, Chisholm RL, Gibbs R, Loomis WF, Platzer M, Kay RR, Williams J, Dear PH, Noegel AA, Barrell B, Kuspa A (2005) The genome of the social amoeba Dictyostelium discoideum. Nature 435:43–57
Hauser M, Narita V, Donhardt AM, Naider F, Becker JM (2001) Multiplicity and regulation of genes encoding peptide transporters in Saccharomyces cerevisiae. Mol Membr Biol 18:105–112
Herbert M, Sauer E, Smethurst G, Kraiss A, Hilpert AK, Reidl J (2003) Nicotinamide ribosyl uptake mutants in Haemophilus influenzae. Infect Immun 71:5398–5401
Hirsch D, Stahl A, Lodish HF (1998) A family of fatty acid transporters conserved from mycobacterium to man. Proc Natl Acad Sci USA 95:8625–8629
Kall L, Krogh A, Sonnhammer EL (2007) Advantages of combined transmembrane topology and signal peptide prediction—the Phobius web server. Nucleic Acids Res 35:W429–W432
Kaur J, Srikanth CV, Bachhawat AK (2009) Differential roles played by the native cysteine residues of the yeast glutathione transporter, Hgt1p. FEMS Yeast Res 9:849–866
Koh S, Wiles AM, Sharp JS, Naider FR, Becker JM, Stacey G (2002) An oligopeptide transporter gene family in Arabidopsis. Plant Physiol 128:21–29
Krogh A, Larsson B, von Heijne G, Sonnhammer EL (2001) Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol 305:567–580
Kuan J, Saier MH Jr (1993a) The mitochondrial carrier family of transport proteins: structural, functional, and evolutionary relationships. Crit Rev Biochem Mol Biol 28:209–233
Kuan J, Saier MH Jr (1993b) Expansion of the mitochondrial carrier family. Res Microbiol 144:671–672
Lee JH, Harvat EM, Stevens JM, Ferguson SJ, Saier MH Jr (2007) Evolutionary origins of members of a superfamily of integral membrane cytochrome c biogenesis proteins. Biochim Biophys Acta 1768:2164–2181
Lubkowitz M (2006) The OPT family functions in long-distance peptide and metal transport in plants. Genet Eng (NY) 27:35–55
Lubkowitz MA, Barnes D, Breslav M, Burchfield A, Naider F, Becker JM (1998) Schizosaccharomyces pombe isp4 encodes a transporter representing a novel family of oligopeptide transporters. Mol Microbiol 28:729–741
Matias MG, Gomolplitinant KM, Tamang DG, Saier MH Jr (2010) Animal Ca2+ release-activated Ca2+ (CRAC) channels are homologous to and derived from the ubiquitous cation diffusion facilitators. BCM Res Notes 3(1):158
Merdanovic M, Sauer E, Reidl J (2005) Coupling of NAD+ biosynthesis and nicotinamide ribosyl transport: characterization of NadR ribonucleotide kinase mutants of Haemophilus influenzae. J Bacteriol 187:4410–4420
Mitchell P, Moyle J (1958) Group-translocation: a consequence of enzyme-catalysed group-transfer. Nature 182:372–373
Nelson RD, Kuan G, Saier MH Jr, Montal M (1999) Modular assembly of voltage-gated channel proteins: a sequence analysis and phylogenetic study. J Mol Microbiol Biotechnol 1:281–287
Osawa H, Stacey G, Gassmann W (2006) ScOPT1 and AtOPT4 function as proton-coupled oligopeptide transporters with broad but distinct substrate specificities. Biochem J 393:267–275
Pao SS, Paulsen IT, Saier MH Jr (1998) Major facilitator superfamily. Microbiol Mol Biol Rev 62:1–34
Paulsen IT, Skurray RA (1994) The POT family of transport proteins. Trends Biochem Sci 19:404
Povolotsky TL, Orlova E, Tamang DG, Saier MH Jr (2010) Defense against cannibalsim: the SpdI family of bacterial immunity/signal transduction proteins. J Membr Biol 235:145–162
Reuss O, Morschhauser J (2006) A family of oligopeptide transporters is required for growth of Candida albicans on proteins. Mol Microbiol 60:795–812
Saier MH Jr (1994) Computer-aided analyses of transport protein sequences: gleaning evidence concerning function, structure, biogenesis, and evolution. Microbiol Rev 58:71–93
Saier MH Jr (2000a) A functional-phylogenetic classification system for transmembrane solute transporters. Microbiol Mol Biol Rev 64:354–411
Saier MH Jr (2000b) Families of proteins forming transmembrane channels. J Membr Biol 175:165–180
Saier MH Jr (2000c) Vectorial metabolism and the evolution of transport systems. J Bacteriol 182:5029–5035
Saier MH Jr (2003) Tracing pathways of transport protein evolution. Mol Microbiol 48:1145–1156
Saier MH Jr, Hvorup RN, Barabote RD (2005) Evolution of the bacterial phosphotransferase system: from carriers and enzymes to group translocators. Biochem Soc Trans 33:220–224
Saier MH Jr, Tran CV, Barabote RD (2006) TCDB: the Transporter Classification Database for membrane transport protein analyses and informaiton. Nuceic Acids Res 34:181–186
Saier MH Jr, Yen MR, Noto K, Tamang DG, Elkan C (2009) The Transporter Classification Database: recent advances. Nucleic Acids Res 37:D274–D278
Stacey MG, Patel A, McClain WE, Mathieu M, Remley M, Rogers EE, Gassmann W, Blevins DG, Stacey G (2008) The Arabidopsis AtOPT3 protein functions in metal homeostasis and movement of iron to developing seeds. Plant Physiol 146:589–601
Thakur A, Kaur J, Bachhawat AK (2008) Pgt1, a glutathione transporter from the fission yeast Schizosaccharomyces pombe. FEMS Yeast Res 8:916–929
Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG (1997) The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res 25:4876–4882
Tusnady GE, Simon I (2001) The HMMTOP transmembrane topology prediction server. Bioinformatics 17:849–850
Wang B, Dukarevich M, Sun EI, Yen MR, Saier MH Jr (2009) Membrane porters of ATP-binding cassette transport systems are polyphyletic. J Membr Biol 231:1–10
Wiles AM, Cai H, Naider F, Becker JM (2006) Nutrient regulation of oligopeptide transport in Saccharomyces cerevisiae. Microbiology 152:3133–3145
Yen MR, Tseng YH, Saier MH Jr (2001) Maize Yellow Stripe1, an iron-phytosiderophore uptake transporter, is a member of the oligopeptide transporter (OPT) family. Microbiology 147:2881–2883
Yen MR, Choi J, Saier MH Jr (2009) Bioinformatic analyses of transmembrane transport: novel software for deducing protein phylogeny, topology, and evolution. J Mol Microbiol Biotechnol 17:163–176
Zhai Y, Saier MH Jr (2001a) A Web-based program (WHAT) for the simultaneous prediction of hydropathy, amphipathicity, secondary structure and transmembrane topology for a single protein sequence. J Mol Microbiol Biotechnol 3:501–502
Zhai Y, Saier MH Jr (2001b) A Web-based program for the prediction of average hydropathy, average amphipathicity and average similarity of multiply aligned homologous proteins. J Mol Microbiol Biotechnol 3:285–286
Zhai Y, Tchieu J, Saier MH Jr (2002) A Web-based Tree View (TV) program for the visualization of phylogenetic trees. J Mol Microbiol Biotechnol 4:69–70
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Gomolplitinant, K.M., Saier, M.H. Evolution of the Oligopeptide Transporter Family. J Membrane Biol 240, 89–110 (2011). https://doi.org/10.1007/s00232-011-9347-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00232-011-9347-9