
Abstract
We have previously shown that protein kinase C (PKC) activation has distinct effects on the structure and barrier properties of cultured epithelial cells (HT29 and MDCK I). Since the claudin family of tight junction (TJ)-associated proteins is considered to be crucial for the function of mature TJ, we assessed their expression patterns and cellular destination, detergent solubility and phosphorylation upon PKC stimulation for 2 or 18 h with phorbol myristate acetate (PMA). In HT29 cells, claudins 1, 3, 4 and 5 and possibly claudin 2 were redistributed to apical cell–cell contacts after PKC activation and the amounts of claudins 1, 3 and 5, but not of claudin 2, were increased in cell lysates. By contrast, in MDCK I cells, PMA treatment resulted in redistribution of claudins 1, 3, 4 and 5 from the TJ and in reorganization of the proteins into more insoluble complexes. Claudins 1 and 4 were phosphorylated in both MDCK I and HT29 cells, but PKC-induced changes in claudin phosphorylation state were detected only in MDCK I cells. A major difference between HT29 and MDCK I cells, which have low and high basal transepithelial electrical resistance, respectively, was the absence of claudin 2 in the latter. Our findings show that PKC activation targets in characteristic ways the expression patterns, destination, detergent solubility and phosphorylation state of claudins in epithelial cells with different capacities to form an epithelial barrier.
Similar content being viewed by others

Introduction
Tight junctions (TJs) form a continuous cellular seal between the apical and basolateral compartments of epithelial cells. They create a selective barrier to the diffusion of solutes across the cell layer and prevent the intermixing of proteins and lipids between the two domains, thereby helping to maintain cell polarization (Gumbiner 1987; Schneeberger and Lynch 1992; Anderson and Van Itallie 1995; Balda and Matter 1998; Tsukita et al. 2001). They comprise a complex of many proteins, including transmembrane proteins like occludin (Furuse et al. 1993; Ando-Akatsuka et al. 1996), JAM (Martin-Padura et al. 1998), claudins (Furuse et al. 1998) and tricellulin (Ikenouchi et al. 2005). These transmembrane proteins interact with a cytoplasmic plaque of adaptors, e.g., the ZO proteins (Stevenson et al. 1986), providing a link to the cytoskeleton. In total, around 40 proteins of the TJ fulfill roles in scaffolding, cytoskeletal attachment, cell polarity, signaling and vesicle trafficking (Gonzalez-Mariscal et al. 2003; Chiba et al. 2008; Guillemot et al. 2008). The paracellular fence is regulated by a wide range of physiological stimuli and cell signals, including protein kinase C (PKC) activation (Tsukamoto and Nigam 1999; Andreeva et al. 2001; Matter and Balda 2003; Sjo et al. 2003, 2005).
The epithelial barriers vary widely among different tissues and even among epithelial cells in the same tissue. Functionally, they behave as though equipped with pores with distinct charges and size selectivities (Powell 1981; Reuss et al. 1981; Sundqvist and Magnusson 1985; Van Itallie et al. 2003, 2008; Van Itallie and Anderson 2004), which are critically dependent on the presence of different claudins (Furuse et al. 1998; Morita et al. 1999; Tsukita et al. 2001; Van Itallie and Anderson 2006). The claudin family has at least 24 members with variable expression patterns in different cell types and tissues. Some, like claudin-1, are ubiquitously expressed, whereas others are restricted to specific cell types or periods of development (Simon et al. 1999; Turksen and Troy 2001, 2004; Tamagawa et al. 2003). In rat intestinal epithelium, claudins 1–5 display a specific distribution; claudin 2 is detected only in the crypts of the small and large intestine, and claudin 4 is restricted to the villi (Rahner et al. 2001). A complex segment-specific pattern of claudin 2 expression in human intestinal epithelial cells indicates a potential correlation with intestinal differentiation (Escaffit et al. 2005), where claudin 2 was found only in undifferentiated cells. The variation in TJ paracellular permeability of different epithelia might thus be explained by distinct combinations of claudin isoforms. There may furthermore be “housekeeping” and “specialized” claudins expressed widely and at high levels or exclusively and at low levels, respectively (Van Itallie and Anderson 2006). The paired claudin strands display a dynamic behavior with retained overall structural integrity, but they could, perhaps during reorganization of the strands, allow some solutes to pass across the TJ (Sasaki et al. 2003; Krause et al. 2008).
Detergent insolubility of proteins is considered to indicate their integration into macromolecular complexes such as intercellular junctions (Sakakibara et al. 1997; Fujibe et al. 2004) or lipid rafts (Nusrat et al. 2000). The latter have been suggested to have a close relationship to the TJ. Consequently, TJ proteins could be expected to be found in the detergent-insoluble fraction.
Based on this proposal, we investigated effects of phorbol 12-myristate 13-acetate (PMA)-induced PKC activation on expression, destination, detergent solubility and phosphorylation of individual claudins in cultured epithelial cells with different barrier properties of their TJ, i.e., MDCK I and HT29 cells. These have distinct basal permeability characteristics and different origins, i.e., kidney and gut epithelium, respectively. MDCK I cells form a very tight monolayer with high transepithelial electric resistance (TER) and low permeability to uncharged molecules, while HT29 cells present a leaky barrier with low TER and high permeability. First, in HT29 cells, PKC activation resulted in a redistribution of claudins 1, 3, 4 and 5 and possibly claudin 2 to apical cell–cell contacts and an increase in the amount of claudins 1, 3 and 5, but not of claudin 2, in cell lysates. By contrast, in MDCK I cells, PKC activation yielded a decrease of claudins 1, 3, 4 and 5 from the TJ and a shift for these claudins from a detergent-soluble to a detergent-resistant fraction. Second, comparing leaky HT29 and tight MDCK I cells, there was a complete absence of claudin 2 in the latter, corroborating findings by other investigators (Furuse et al. 2001; Amasheh et al. 2002). Third, phosphorylated claudins were demonstrated in MDCK I and HT29 cells, but only in MDCK I cells did PKC activation induce detectable changes in the claudin phosphorylation state. This suggests that PKC targets in distinct ways both the expression patterns and destination of claudins in epithelial cells with different capacities to form a tight epithelial barrier.
Materials and Methods
Cell Culture
As epithelial cell models, the Madin-Darby canine kidney I cell line (MDCK I) and the human colon carcinoma cell line HT29 (Fogh and Trempe 1975) were used. Cells were grown in Dulbecco′s modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum, 100 U/ml penicillin, 100 μg/ml streptomycin and 4 mM l-glutamine (all from Invitrogen, Stockholm, Sweden). To promote a small intestinal, epithelial phenotype of HT29 cells, a medium with 25 mM galactose (Sigma, St. Louis, MO) instead of glucose was used (Pinto et al. 1982; Zweibaum et al. 1985; Neutra and Louvard 1989). Cells were maintained in tissue culture flasks (Falcon VWR, Stockholm, Sweden) at 37°C in a humidified 5% CO2 atmosphere. For experiments, cells were plated on collagen-coated coverslips (Biocoat; Becton–Dickinson, Franklin Lakes, NJ) or on six-well tissue culture plates (Falcon). The culture medium was exchanged every second day, and cells were used after 5–7 days for MDCK I and after 7–10 days for HT29. PKC activation was achieved by incubation with 100 nM PMA (Sigma), diluted in culture medium.
Immunofluorescence
Cells were grown on collagen-coated glass coverslips and stimulated with 100 nM PMA for 0, 2 or 18 h; rinsed in Krebs-Ringers glucose buffer (KRG; in mM: 120 NaCl, 4.9 KCl, 1.2 MgSO4, 1.7 KH2PO4, 8.3 Na2HPO4, 10 glucose and 1.0 CaCl2); and fixed for 15 min in 2.5% paraformaldehyde (Sigma) at room temperature. After washing in phosphate-buffered saline (PBS; in mM: 147 NaCl, 2.7 KCl, 1.5 KH2PO4 and 6.7 Na2HPO4), cells were permeabilized in 0.2% Triton X-100 (Sigma) for 5 min at room temperature and washed in PBS. After blocking nonspecific antibody binding with 1% bovine serum albumin (BSA; Roche, Mannheim, Germany), 1 mM glycine (GE Healthcare BioSciences, Uppsala, Sweden) and 10% normal swine serum (Dako, Glostrup, Denmark) in PBS, cells were incubated for 1 h at 37°C with monoclonal mouse antibodies against claudin 2, polyclonal rabbit antibodies against claudin 3 and monoclonal mouse antibodies against claudins 1, 4 and 5, conjugated with AlexaFluor 488 (all from Invitrogen/Zymed Laboratories, San Fransisco, CA). Thereafter, they were washed in PBS and unconjugated antibodies reacted with AlexaFluor 488-labeled anti-mouse or anti-rabbit antibodies for 1 h at 37°C. Cells were washed in PBS and mounted upside down on a three-well microscope slide in ProLong mounting medium to prevent photobleaching (Invitrogen/Molecular Probes, Eugene, OR). The three-dimensional localization of claudins was assessed with confocal laser scanning microscopy (Radiance 2100 MP; Bio-Rad Zeiss, Jena, Germany). The 488 nm wavelength of the argon laser was used for excitation of AlexaFluor 488. The primary dichroic mirror with a cut-off wavelength of 535 nm was used to separate excitation light from emitted light. For specific detection of the green signal, a 545DF30 nm bandpass filter was used. Vertical sections and series of horizontal sections were collected, using a 60× oil immersion objective (NA 1.4). With horizontal scanning, we displayed one section, representing the most apical part of the cell layer. Representative images from multiple experiments (three or more) are shown in the figures.
Detergent Extraction of Cell Monolayers
Cell monolayers were grown on tissue culture plates and first exposed to 100 nM PMA for 0, 2 or 18 h and then subjected to detergent extraction with non-ionic NP40 according to Sakakibara and coworkers (1997). Immediately after the PMA treatment, cells were placed on ice, washed with KRG supplemented with 1 mM Na3VO4 and lysed in 500 μl NP40 buffer (1% NP40, 0.15 M NaCl, 25 mM HEPES [pH 7.4], 25 mM NaF and 4 mM EDTA). The lysis buffer contained 0.01% benzonase (Merck, Spånga, Sweden), 1 mM Na3VO4 and a mixture of protease inhibitors (Complete; Roche Diagnostics, Stockholm, Sweden). Cells were scraped into microcentrifuge tubes and turned for 30 min. After centrifugation at 15,000×g for 10 min, the supernatant was collected as the NP40-soluble fraction (NPsol). The pellet was resuspended in 100 μl of sodium dodecyl sulfate (SDS) lysis buffer (1% SDS, 25 mM HEPES [pH 7.5], 4 mM EDTA, 25 mM NaF) and homogenized with a Kontes (Vineland, NY) homogenizer. The homogenate was combined with 900 μl of NP40 buffer to further wash the homogenizer and the lysate was passed 10 times through a 27G needle, rotated again for 30 min and centrifuged. The supernatant was collected as the NP40-insoluble fraction (NPins). To determine the total amount of claudins, cells were lysed in NP40 buffer with 0.2% SDS, homogenized, passed through a needle, rotated and centrifuged and the supernatants were collected. All procedures were carried out at 4°C.
Immunoprecipitation
For immunoprecipitation, the NPsol and the NPins were precleared with protein-G/A agarose beads (Calbiochem EMD Chemicals Merck, Darmstadt, Germany) and incubated with polyclonal antibodies against claudin 1 or monoclonal antibodies against claudin 1 and 4, respectively, for 1 h and again with agarose beads. The beads were collected by centrifugation at 1,000×g for 5 min and washed four times in lysis buffer supplemented with 0.01% benzonase, 1 mM Na3VO4 and protease inhibitors. Immunoprecipitation was carried out at 4°C. Precipitates were then subjected to electrophoresis and immunoblotting.
Immunoblotting Analyses
For electrophoresis, equal amounts of total protein within each fraction were loaded onto the gel. The total protein concentration was approximately fourfold higher in NPsol than in NPins. Proteins were dissolved in NuPAGE®LDS sample buffer (Invitrogen) and heated for 10 min at 70°C. SDS polyacrylamide gel electrophoresis (SDS-PAGE) (Laemmli 1970) was performed using a 4–12% gel (Invitrogen), and the fractionated proteins were electroblotted (Towbin et al. 1979) onto PVDF membranes (Millipore, Stockholm, Sweden). After blocking with 3% BSA in PBS with 0.1% Tween 20 (PBS-T), membranes were incubated with polyclonal antibodies against claudins 1 and 3 (Invitrogen/Zymed) and phosphothreonine (Cell Signaling Technology, Danvers, MA) or with monoclonal antibodies against claudins 1, 2, 4 and 5 (Invitrogen/Zymed) and phosphotyrosine (Cell Signaling), respectively, washed in PBS-T and further incubated with HRP-conjugated secondary antibodies (Dako). The ECL Western blot analysis system (GE Healthcare) was used to detect the antibodies. The density ratio of the specific bands was quantified using ImageJ (National Institutes of Health, Bethesda, MD). Representative blots from multiple experiments (three or more) are shown in the figures. Blots were digitally contrasted with preserved relative intensity of specific claudin bands.
Results
Initial Localization of Claudins
MDCK I Cells
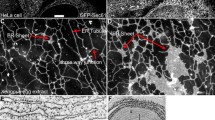
Cells formed a confluent layer with a regular hexagonal cell shape. Conventional immunofluorescence and confocal microscopic horizontal sections showed distinct staining patterns for claudins 1, 3, 4 and 5 at the apical part of the cell–cell contact (Fig. 1a, x-y sections) and vertical sections displayed additional expression at basolateral cell–cell contacts (Fig. 1a, z sections). Claudin 4 was more apparent at the apical parts (Fig. 1a, z section). Claudin 3 expression was less distinct than that of claudins 1, 4 and 5. Claudin 2 could not be detected at all in the cells.

In MDCK I and HT29 control cells, claudins were apically and laterally localized. a In MDCK I cells, PKC activation by PMA treatment resulted in redistribution of claudins 1, 3, 4, 5 and possibly 2 from the TJ. b In HT29 cells, PKC activation increased the localization of claudins 1, 3, 4 and 5 at the TJ. Cells were treated with PMA for 2 and 18 h and subjected to immunostaining with anticlaudin antibodies. Apical localization of claudins was assessed with confocal laser scanning microscopy, represented by the most apical part of the cell layer. For lateral localization, z-section confocal images were collected; the apical part of cells is facing upward. Scale bar = 10 μm
HT29 Cells
A confluent layer of irregular cells was formed. Diffuse staining of claudins 1, 2, 3, 4 and 5 was seen at apical and lateral cell–cell contacts but also at the apical and basolateral membranes (Fig. 1b). Claudins 4 and 5 also showed more distinct staining in certain cells.
Effect of PMA-Induced PKC Activation on the Distribution of Claudins
MDCK I Cells
A 2-h PMA stimulation resulted in weaker staining of claudin 1 at apical cell–cell contacts (Fig. 1a). After 18 h with PMA the claudin 1 staining at the apical cell borders was diffuse and partially fragmented. Claudin 3 was weaker and scattered after 2 h with PMA, and after 18 h the staining became totally fragmented. Claudin 4 staining was enhanced at the apical cell borders after 2 h with PMA, but after 18 h it became fragmented. Claudin 5 expression showed uneven staining after 2 h with PMA; some areas with enhanced staining became fragmented after 18 h. Claudin 2 remained undetectable in the cells after both 2-h and 18-h PMA incubation.
HT29 Cells
By PMA treatment for 2 or 18 h, light microscopy showed that the morphology changed from an irregular to a hexagonal shape, which confirms previous findings (Sjo et al. 2003). Confocal imaging showed increased and more distinct apical staining of claudins 1, 3, 4 and 5 after 2 and 18 h with PMA. However, for claudin 2, only some cells displayed clearly increased fragmented staining at apical cell–cell contacts after 2 and 18 h (Fig. 1b).
Expression and Detergent Solubility of Claudins
Western blot was used to examine the expression of claudins in MDCK I and HT29 cells during PKC-induced TJ reorganization and the distribution of claudins between the NPsol and NPins fractions. The relative intensity of the protein bands was measured.
Claudins 1, 3, 4 and 5 were expressed in both cell lines, whereas claudin 2 was seen only in HT29 cells (Fig. 2), which is consistent with the confocal images. The apparent molecular mass of all claudins was approximately 18 kDa. Claudin 4 expressed in HT29 cells showed a lower molecular mass (16 kDa) compared to claudin 4 in MDCK I cells (18 kDa). The claudins were recovered from both fractions of MDCK I and HT29 cells.

PMA-induced PKC activation induced a shift of 18-kDa claudins from the NPsol to the NPins fraction in MDCK I cells, and it increased the amount of claudin 5 in both fractions of HT29 cells and of claudins 1 and 3 in NPins. In MDCK I cells, the total expression (Tot) of claudins 1, 4 and 5 was unchanged, whereas a slight decrease in claudin 3 expression was noted. In HT29 cells, PMA did not change the total expression of claudins 1, 2, 3 and 4 but claudin 5 showed a possible increase after 18 h. Cells were stimulated with PMA for 2 and 18 h. NPsol, NPins and nonfractionated lysates (Tot) were separated by SDS-PAGE and immunoblotted with the appropriate antibodies. The approximate estimated molecular mass of the claudins was 16–18 kDa. Mean values and standard error of the mean (SEM) are shown, n ≥ 3. c = control cells
Both 2- and 18-h PMA treatments of confluent MDCK I cells reduced the amount of claudins 1, 3, 4 and 5 in NPsol, with a parallel increase of claudins 4 and 5 in NPins. Claudin 1 and 3 proteins in NPins increased after 2 h PMA, followed by a decrease after 18 h PMA (Fig. 2a). PMA stimulation of HT29 cells resulted in increased levels of claudin 5 in both NPsol and NPins and of claudins 1 and 3 in the insoluble fractions after 18-h, but not 2-h, incubation (Fig. 2b). The expression of claudin 2 was not changed in either fraction. In nonfractionated cell lysates of MDCK I, the expression of claudins 1, 4 and 5 was unchanged after either 2- or 18-h PMA incubation, whereas for claudin 3 a possible decrease was noted after 18 h. In HT29 cells, no detectable changes were found in the total expression of claudins 1, 2, 3 and 4 after 18 h with PMA (Fig. 2a, b) but claudin 5 showed a possible increase after 18 h.
In lysates from MDCK I and HT29 cells, several immunoreactive bands of higher molecular weight were detected, especially for claudin 1 with both monoclonal and polyclonal antibodies and for claudin 4 with monoclonal antibodies (not shown). Thus, in MDCK I, claudin 1 antibodies recognized proteins at 56 kDa in NPsol and at 45 and 150 kDa in NPins; with claudin 4 antibodies, protein bands at 56 kDa were detected in NPsol. In HT29 cells, claudin 1 antibodies reacted with a 47-kDa protein in NPsol and a 150-kDa protein in NPins and claudin 4 antibodies reacted with a 56-kDa protein in NPsol and a 150-kDa protein in NPins. PMA incubation increased the relative expression of the 150-kDa protein recognized by claudin 1 antibodies in NPins of MDCK I (not shown).
Phosphorylation State of Claudins
Claudin 1 and claudin 4 immunoprecipitates of cell fractions were analyzed by immunoblotting with antibodies to phosphothreonine and phosphotyrosine and the corresponding claudins. The amount of precipitated claudins of 18–20 kDa from each fraction was changed by PMA treatment in the same manner as for nonprecipitated claudins.
MDCK I
In control cells, threonine- and tyrosine-phosphorylated claudin 1 of 18–20 kDa was detected in NPins but only tyrosine-phosphorylated claudin 1 in NPsol (Fig. 3a). In NPins, both threonine and tyrosine phosphorylations were reduced by 2-h incubation with PMA but recovered after 18 h with PMA. In NPsol, the amount of tyrosine-phosphorylated claudin 1 was decreased after 18-h incubation in parallel with a decrease in the amount of the claudin protein.

NPsol and NPins fractions of PMA-activated MDCK I and HT29 cells were immunoprecipitated (IP) with claudins 1 and 4, respectively, and immunoblotted (WB) for phosphothreonine (p-thre), and phosphotyrosine (p-tyr). In MDCK I cells, a claudin 1 in NPins was dephosphorylated after 2-h incubation and recovered after 18 h and c the tyrosine phosphorylation of claudin 4 decreased. b, d In HT29 cells, the phosphorylation of claudins 1 and 4 was unchanged. The approximate estimated molecular mass of the claudins was 16–18 kDa. c = control cells
Claudin 4 displayed phosphorylation on threonine and tyrosine in both fractions (Fig. 3c). PMA reduced the tyrosine phosphorylation of claudin 4 in NPins and yielded a smaller apparent molecular mass of the molecule, whereas in NPsol a parallel decrease was detected in tyrosine phosphorylation and the amount of the protein. A slight increase in threonine phosphorylation could be detected in NPsol.
HT29
Threonine and tyrosine phosphorylations of both claudins 1 and 4 were detected in the NPsol, but not NPins, of control cells (Fig. 3b, d). PMA did not alter the phosphorylation of the claudins.
Discussion
General
In this study, we compared the cellular partitioning and the state of phosphorylation of claudins in two epithelial cell lines, i.e., the canine kidney cell line MDCK I and the human adenocarcinoma cell line HT29; the latter was stimulated to form a differentiated small intestine-like epithelium by growth in glucose-free medium (Pinto et al. 1982; Zweibaum et al. 1985; Neutra and Louvard 1989). The two cell lines display distinct barrier characteristics; i.e., MDCK I cells form a very tight monolayer with high TER (up to 5,000 Ω × cm2) and low permeability to uncharged molecules, while HT29 cells form a leaky barrier with low TER (<200 Ω × cm2) and high permeability (Sjo et al. 2003).
Localization of Different Claudins
When assessing the localization of claudins 1–5 in MDCK I and HT29 cells by immunostaining and confocal imaging, first, expression of claudins 1, 3, 4 and 5 was observed in both cell lines and, additionally, claudin 2 in HT29 cells (Fig. 1a, b). The presence of claudin 2 is thought to confer a leaky phenotype to epithelial TJs (Escaffit et al. 2005); this is in accordance with the detection of claudin 2 in HT29 cells but not in MDCK I cells, confirming an earlier report (Furuse et al. 1999). In the MDCK I cell monolayer, representing a tight epithelium, claudins 1, 3, 4 and 5 were all localized apically at the cell–cell contacts (Fig. 1a, x-y sections). We presume that this localization represents the TJ, where colocalization of claudin 1 and the strictly apically localized TJ protein occludin has been demonstrated (Furuse et al. 1993; Sjo et al. 2003). In HT29 cells, claudins 1–5 were seen as diffuse bands at apical cell–cell contacts (Fig. 1b, x-y sections). The absence of distinct bands could indicate that the complete copolymerization required for formation of TJs was not achieved, which resulted in a weak barrier function of HT29 cell layers (Sjo et al. 2003). Moreover, claudins 1, 3, 4 and 5 were found also along the lateral interfaces of adjacent cells in both HT29 and MDCK I (Fig. 1a, b; z sections). In MDCK cells, claudin 4, but not claudins 1, 3 and 5, appeared apically as distinct dots in the z sections, suggesting a concentration of claudin 4 at the TJ. The lateral localizations of claudins, different from the strictly apical localization of the integral TJ protein occludin (Furuse et al. 1993; Sjo et al. 2003), have also been seen in rat intestine and epididymis and in human intestine and airway epithelium (Gregory et al. 2001; Rahner et al. 2001; Coyne et al. 2003; Escaffit et al. 2005). This pattern could reflect a pool of proteins that can be recruited into the junction to rapidly alter paracellular transport properties in response to a physiological stimulus. It has also been suggested that claudins contribute to cell adhesion (Kuhn et al. 2007). In HT29 cells, the claudins were also to some extent seen along the entire membrane.
Detergent Solubility
The claudins were recovered in both the NPsol and NPins fractions of cell lysates (Fig. 2a, b). Since the TJ strands are assumed to be resistant to detergent extraction (Stevenson et al. 1988), these findings speak for the existence of claudins both as a junctional complex and as a more soluble pool of proteins. Our findings explain the lateral localization demonstrated by immunofluorescence. The distribution of claudins in two distinct subcellular pools has also been described earlier (de Oliveira et al. 2005).
In addition to the claudins of 16–18 kDa, we found immunoreactive protein bands of higher molecular weight (45/47, 56, 150 kDa) in lysates of both MDCK I and HT29, after immunoblotting with claudin 1 and 4 antibodies. The bands are recognized by monoclonal as well as polyclonal antibodies. Some of the bands had a density close to that of the putatively monomeric claudin bands of 16–18 kDa. The recognition by both monoclonal and polyclonal antibodies suggests a specific reaction and could represent either oligomers of claudins (Mitic et al. 2003) or complexes of claudins with other TJ proteins, not solubilized with a standard SDS concentration. Although native gel electrophoresis preferably should be used to detect oligomers, partial retention of claudin oligomers can be achieved by SDS-PAGE and reduced conditions (Coyne et al. 2003).
PKC Activation
In a previous study, we demonstrated that PKC activation promotes the barrier function and increases the apical expression of the TJ proteins occludin and claudin 1 in HT29 cells. By contrast, in MDCK I cells it reduced the barrier and caused the redistribution of occludin and claudin 1 from the apical part of the cells (Sjo et al. 2003, 2005). The present investigation delineated a PKC-induced increase in claudin 1 but also in claudins 3, 4 and 5 and possibly claudin 2 at the apical cell–cell contacts of HT29 cells (Fig. 1b). In parallel, the cells changed from an irregular form to a honeycomb structure, which we also reported in the earlier study (Sjo et al. 2003). The claudins formed, however, only partially continuous bands. This suggests that they were not completely incorporated into the TJ structure, resulting in an improved but not fully developed barrier (Sjo et al. 2003). By contrast, in MDCK I cells PKC activation caused a decrease in and fragmentation of claudins 1, 3, 4 and 5 at the apical interface between cells after 18 h preceded by an increase in claudins 4 and 5 after 2 h (Fig. 1a). Claudin 2 could not, however, be detected either before or after PKC activation.
The reduction of the epithelial barrier and loss of proteins from the TJ in PKC-activated MDCK I cells imply a decreased complexity of the TJ. Hence, one would expect a redistribution of claudins from NPins to NPsol. Our results revealed, however, a decreased amount of claudins 1, 3, 4 and 5 in NPsol and a parallel increase in NPins of the cell lysates (Fig. 2a), suggesting a reorganization of the claudins into more insoluble complexes. The changes may reflect altered interactions between junctional proteins and the perijunctional F-actin cytoskeleton or altered association of claudins with membrane lipid raft microdomains (Nusrat et al. 2000). This could be a protective mechanism during the cellular stress induced by PMA (Kachar and Pinto da Silva 1981; Lynch et al. 1995). The claudins could thus respond by reorganizing into less soluble complexes recovered not only from the TJ but also from other parts of the cell, e.g., the adherens junctions where they may have scaffolding functions. Accordingly, by immunofluorescence there was loss of claudins from the TJ region after 18 h. The decreased solubility of claudins after 2 h paralleled the temporarily increased but less distinct apical staining of claudins 4 and 5, possibly representing claudin-containing complexes in close vicinity of the TJ.
Recently, it has been proposed that claudins can be regulated by PKC-mediated phosphorylation, contributing to increased or decreased barrier function in the cells. Thus, in Caco-2 cells, inactivation of PKC-θ reduced phosphorylation of claudins 1 and 4 and decreased the membrane/cytoskeletal distribution of the claudins (Banan et al. 2005). In ovarian cancer cells, phorbol ester-mediated PKC activation induced phosphorylation of claudin 4 and decreased barrier function (D’Souza et al. 2007); and in human epidermal keratinocytes, formation of TJ was suggested to be regulated by a PKC-induced phosphorylation of claudin 4 (Aono and Hirai 2008). In our study, activation of PKC by phorbol ester reduced claudin 1 phosphorylation in NPins of MDCK I cells after 2 h, although the amount of claudin 1 was increased. Likewise, in earlier studies, it has been shown that occludin is dephosphorylated by PMA after 2 h (Clarke et al. 2000; Sjo et al. 2003). Since PKC is a serine/threonine kinase, dephosphorylation of proteins could be the result of a downregulation or a secondary effect of PKC. Interestingly, claudin 1 phosphorylation returned after 18 h incubation, but at that time the distribution of claudin 1 was totally fragmented at the apical level (Fig. 1a) and the function of the barrier remained impaired (Sjo et al. 2003). Consequently, phosphorylated claudin 1 may not be localized exclusively at the TJ but also at lateral membranes. PKC activation of MDCK I cells resulted in a small reduction of claudin 4 phosphorylation in the insoluble fraction but without the dual effects seen for claudin 1. The decrease in the apparent molecular mass of claudin 4 after 2 h also indicates dephosphorylation of the claudin. Still, the apical distribution of claudin 4 was not reduced but rather enhanced after 2 h with PMA.
HT29 cells normally form a leaky barrier with low TER and high permeability. Unlike in MDCK I cells, it was strengthened by 2- and 18-h PKC activations (Sjo et al. 2003), which could possibly be attributed to an apical accumulation of claudins 1, 3, 4 and 5 (Fig. 1b). The amounts of claudins in either fraction of HT29 cells showed only small increases after 2 h but claudins 1, 3 and 5 increased after 18 h, as demonstrated by immunoblotting (Fig. 2a). This could suggest a rapid redistribution of claudins from a cellular pool to the TJ, as demonstrated by immunofluorescence, combined with a slower PKC-induced differentiation process including synthesis of specific claudins, i.e., claudins 1, 3 and 5. Such a proposal is supported by a previous suggestion that claudins are developmentally regulated (Turksen and Troy 2004). In nonfractionated cell lysates, increases in the expression of claudins were not detectable after 18-h incubation.
In MDCK I control cells, phosphorylated claudins 1 and 4 were found in both fractions, whereas in HT29 control cells these were recovered mainly in NPsol and the phosphorylation of claudins 1 and 4 was not changed by PKC activation. However, since PKC induced only a minor increase in the apical distribution of the claudins and a small improvement in barrier function (Sjo et al. 2003), a change in the phosphorylation state might be limited and below detection.
Taken together, our findings demonstrate that TJ regulation is associated with specific alterations in the expression and destination of individual claudins, in cells with very tight (MDCK I) and leaky (HT29) TJs, respectively. We also conclude that PKC-induced claudin reorganization is accompanied by distinct changes in detergent solubility and the phosphorylation state of the claudin isomers.
References
Amasheh S, Meiri N, Gitter AH, Schoneberg T, Mankertz J, Schulzke JD, Fromm M (2002) Claudin-2 expression induces cation-selective channels in tight junctions of epithelial cells. J Cell Sci 115:4969–4976
Anderson JM, Van Itallie CM (1995) Tight junctions and the molecular basis for regulation of paracellular permeability. Am J Physiol Gastrointest Liver Physiol 269:G467–G475
Ando-Akatsuka Y, Saitou M, Hirase T, Kishi M, Sakakibara A, Itoh M, Yonemura S, Furuse M, Tsukita S (1996) Interspecies diversity of the occludin sequence: cDNA cloning of human, mouse, dog, and rat-kangaroo homologues. J Cell Biol 133:43–47
Andreeva AY, Krause E, Muller EC, Blasig IE, Utepbergenov DI (2001) Protein kinase C regulates the phosphorylation and cellular localization of occludin. J Biol Chem 276:38480–38486
Aono S, Hirai Y (2008) Phosphorylation of claudin-4 is required for tight junction formation in a human keratinocyte cell line. Exp Cell Res 314:3326–3339
Balda MS, Matter K (1998) Tight junctions. J Cell Sci 111(Pt 5):541–547
Banan A, Zhang LJ, Shaikh M, Fields JZ, Choudhary S, Forsyth CB, Farhadi A, Keshavarzian A (2005) Theta isoform of protein kinase C alters barrier function in intestinal epithelium through modulation of distinct claudin isotypes: a novel mechanism for regulation of permeability. J Pharmacol Exp Ther 313:962–982
Chiba H, Osanai M, Murata M, Kojima T, Norimasa S (2008) Transmembrane proteins of tight junctions. Biochim Biophys Acta 1778:588–600
Clarke H, Soler AP, Mullin JM (2000) Protein kinase C activation leads to dephosphorylation of occludin and tight junction permeability increase in LLC-PK1 epithelial cell sheets. J Cell Sci 113(Pt 18):3187–3196
Coyne CB, Gambling TM, Boucher RC, Carson L, Johnson LG (2003) Role of claudin interactions in airway tight junctional permeability. Am J Physiol Lung Cell Mol Physiol 285:L1166–L1178
D’Souza T, Indig FE, Morin PJ (2007) Phosphorylation of claudin-4 by PKCepsilon regulates tight junction barrier function in ovarian cancer cells. Exp Cell Res 313:3364–3375
de Oliveira SS, de Oliveira IM, De Souza W, Morgado-Diaz JA (2005) Claudins upregulation in human colorectal cancer. FEBS Lett 579:6179–6185
Escaffit F, Boudreau F, Beaulieu JF (2005) Differential expression of claudin-2 along the human intestine: implication of GATA-4 in the maintenance of claudin-2 in differentiating cells. J Cell Physiol 203:15–26
Fogh J, Trempe G (1975) New tumor cell lines. Plenum, New York
Fujibe M, Chiba H, Kojima T, Soma T, Wada T, Yamashita T, Sawada N (2004) Thr203 of claudin-1, a putative phosphorylation site for MAP kinase, is required to promote the barrier function of tight junctions. Exp Cell Res 295:36–47
Furuse M, Hirase T, Itoh M, Nagafuchi A, Yonemura S, Tsukita S (1993) Occludin: a novel integral membrane protein localizing at tight junctions. J Cell Biol 123:1777–1788
Furuse M, Fujita K, Hiiragi T, Fujimoto K, Tsukita S (1998) Claudin-1 and -2: novel integral membrane proteins localizing at tight junctions with no sequence similarity to occludin. J Cell Biol 141:1539–1550
Furuse M, Sasaki H, Tsukita S (1999) Manner of interaction of heterogeneous claudin species within and between tight junction strands. J Cell Biol 147:891–903
Furuse M, Furuse K, Sasaki H, Tsukita S (2001) Conversion of zonulae occludentes from tight to leaky strand type by introducing claudin-2 into Madin-Darby canine kidney I cells. J Cell Biol 153:263–272
Gonzalez-Mariscal L, Betanzos A, Nava P, Jaramillo BE (2003) Tight junction proteins. Prog Biophys Mol Biol 81:1–44
Gregory M, Dufresne J, Hermo L, Cyr D (2001) Claudin-1 is not restricted to tight junctions in the rat epididymis. Endocrinology 142:854–863
Guillemot L, Paschoud S, Pulimeno P, Foglia A, Citi S (2008) The cytoplasmic plaque of tight junctions: a scaffolding and signalling center. Biochim Biophys Acta 1778:601–613
Gumbiner B (1987) Structure, biochemistry, and assembly of epithelial tight junctions. Am J Physiol Cell Physiol 253:C749–C758
Ikenouchi J, Furuse M, Furuse K, Sasaki H, Tsukita S (2005) Tricellulin constitutes a novel barrier at tricellular contacts of epithelial cells. J Cell Biol 171:939–945
Kachar B, Pinto da Silva P (1981) Rapid massive assembly of tight junction strands. Science 213:541–544
Krause G, Winkler L, Mueller SL, Haseloff RF, Piontek J, Blasig IE (2008) Structure and function of claudins. Biochim Biophys Acta 1778:631–645
Kuhn S, Koch M, Nubel T, Ladwein M, Antolovic D, Klingbeil P, Hildebrand D, Moldenhauer G, Langbein L, Franke WW, Weitz J, Zoller M (2007) A complex of EpCAM, claudin-7, CD44 variant isoforms, and tetraspanins promotes colorectal cancer progression. Mol Cancer Res 5:553–567
Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685
Lynch RD, Tkachuk-Ross LJ, McCormack JM, McCarthy KM, Rogers RA, Schneeberger EE (1995) Basolateral but not apical application of protease results in a rapid rise of transepithelial electrical resistance and formation of aberrant tight junction strands in MDCK cells. Eur J Cell Biol 66:257–267
Martin-Padura I, Lostaglio S, Schneemann M, Williams L, Romano M, Fruscella P, Panzeri C, Stoppacciaro A, Ruco L, Villa A, Simmons D, Dejana E (1998) Junctional adhesion molecule, a novel member of the immunoglobulin superfamily that distributes at intercellular junctions and modulates monocyte transmigration. J Cell Biol 142:117–127
Matter K, Balda M (2003) Signalling to and from tight junctions. Nat Rev Mol Cell Biol 4:225–236
Mitic LL, Unger VM, Anderson JM (2003) Expression, solubilization, and biochemical characterization of the tight junction transmembrane protein claudin-4. Protein Sci 12:218–227
Morita K, Sasaki H, Furuse M, Tsukita S (1999) Endothelial claudin: claudin-5/TMVCF constitutes tight junction strands in endothelial cells. J Cell Biol 147:185–194
Neutra M, Louvard D (1989) Differentiation of intestinal cells in vitro. Alan R. Liss, New York
Nusrat A, Parkos CA, Verkade P, Fole CS, Liang TW, Innis-Whitehouse W, Eastburn KK, Madara JL (2000) Tight junctions are membrane microdomains. J Cell Sci 113(Pt 10):1771–1781
Pinto M, Appay M-D, Simon-Assmann P, Chevalier G, Dracopoli N, Fogh J, Zweibaum A (1982) Enterocytic differentiation of cultured human colon cancer cells by replacement of glucose by galactose in the medium. Biol Cell 44:193–196
Powell DW (1981) Barrier function of epithelia. Am J Physiol Gastrointest Liver Physiol 241:G275–G288
Rahner C, Mitic LL, Anderson JM (2001) Heterogeneity in expression and subcellular localization of claudins 2, 3, 4, and 5 in the rat liver, pancreas, and gut. Gastroenterology 120:411–422
Reuss L, Cheung LY, Grady TP (1981) Mechanisms of cation permeation across apical cell membrane of Necturus gallbladder: effects of luminal pH and divalent cations on K+ and Na+ permeability. J Membr Biol 59:211–224
Sakakibara A, Furuse M, Saitou M, Ando-Akatsuka Y, Tsukita S (1997) Possible involvement of phosphorylation of occludin in tight junction formation. Cancer Biochem Biophys 137:1393–1401
Sasaki H, Matsui C, Furuse K, Mimori-Kiyosue Y, Furuse M, Tsukita S (2003) Dynamic behavior of paired claudin strands within apposing plasma membranes. Proc Natl Acad Sci USA 100:3971–3976
Schneeberger EE, Lynch RD (1992) Structure, function, and regulation of cellular tight junctions. Am J Physiol Lung Cell Mol Physiol 262:L647–L6461
Simon DB, Lu Y, Choate KA, Velazquez H, Al-Sabban E, Praga M, Casari G, Bettinelli A, Colussi G, Rodriguez-Soriano J, McCredie D, Milford D, Sanjad S, Lifton RP (1999) Paracellin-1, a renal tight junction protein required for paracellular Mg2+ resorption. Science 285:103–106
Sjo A, Magnusson KE, Peterson KH (2003) Distinct effects of protein kinase C on the barrier function at different developmental stages. Biosci Rep 23:87–102
Sjo A, Magnusson KE, Peterson KH (2005) Association of alpha-dystrobrevin with reorganizing tight junctions. J Membr Biol 203:21–30
Stevenson BR, Siliciano JD, Mooseker MS, Goodenough DA (1986) Identification of ZO-1: a high molecular weight polypeptide associated with the tight junction (zonula occludens) in a variety of epithelia. J Cell Biol 103:755–766
Stevenson BR, Anderson JM, Bullivant S (1988) The epithelial tight junction: structure, function and preliminary biochemical characterization. Mol Cell Biochem 83:129–145
Sundqvist T, Magnusson KE (1985) Modelling of intestinal permeability in man to polyethylene glycols (PEG 400 and PEG 1000). Acta Physiol Scand 125:289–296
Tamagawa H, Takahashi I, Furuse M, Yoshitake-Kitano Y, Tsukita S, Ito T, Matsuda H, Kiyono H (2003) Characteristics of claudin expression in follicle-associated epithelium of Peyer’s patches: preferential localization of claudin-4 at the apex of the dome region. Lab Invest 83:1045–1053
Towbin H, Staehelin T, Gordon J (1979) Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci USA 76:4350–4354
Tsukamoto T, Nigam SK (1999) Role of tyrosine phosphorylation in the reassembly of occludin and other tight junction proteins. Am J Physiol Renal Physiol 276:F737–F750
Tsukita S, Furuse M, Itoh M (2001) Multifunctional strands in tight junctions. Nat Rev Mol Cell Biol 2:285–293
Turksen K, Troy TC (2001) Claudin-6: a novel tight junction molecule is developmentally regulated in mouse embryonic epithelium. Dev Dyn 222:292–300
Turksen K, Troy TC (2004) Barriers built on claudins. J Cell Sci 117:2435–2447
Van Itallie CM, Anderson JM (2004) The molecular physiology of tight junction pores. Physiology (Bethesda) 19:331–338
Van Itallie CM, Anderson JM (2006) Claudins and epithelial paracellular transport. Annu Rev Physiol 68:403–429
Van Itallie CM, Fanning AS, Anderson JM (2003) Reversal of charge selectivity in cation or anion-selective epithelial lines by expression of different claudins. Am J Physiol Renal Physiol 285:F1078–F1084
Van Itallie CM, Holmes J, Bridges A, Gookin JL, Coccaro MR, Proctor W, Colegio OR, Anderson JM (2008) The density of small tight junction pores varies among cell types and is increased by expression of claudin-2. J Cell Sci 121:298–305
Zweibaum A, Pinto M, Chevalier G, Dussaulx E, Triadou N, Lacroix B, Haffe K, Brun JL, Rousset M (1985) Enterocytic differentiation of a subpopulation of the human colon tumor cell line HT-29 selected for growth in sugar-free medium and its inhibition by glucose. J Cell Physiol 122:21–29
Acknowledgements
This study was supported by the King Gustaf Vth 80-Year Foundation, the Swedish Research Council (VRM) and the Lion Club Research Foundation for Young Investigators.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Sjö, A., Magnusson, KE. & Peterson, K.H. Protein Kinase C Activation Has Distinct Effects on the Localization, Phosphorylation and Detergent Solubility of the Claudin Protein Family in Tight and Leaky Epithelial Cells. J Membrane Biol 236, 181–189 (2010). https://doi.org/10.1007/s00232-010-9289-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00232-010-9289-7