
Abstract
Pacific lion-paw scallops were collected from natural aggregations in Laguna Ojo de Liebre (Pacific Ocean), the Gulf of California, and from aquaculture facilities for genetic diversity analyses. Mitochondrial DNA sequencing uncovered two highly supported clades separated by 2.5% divergence. Data from ten microsatellite markers suggest individuals from these mitogroups are introgressed, raising questions about the mitotype origin. Some evidence suggests gene flow between La Paz and Ojo de Liebre; otherwise the Gulf of California and Ojo de Liebre are acting as two distinct populations. It is unclear whether translocations between sites have influenced the observed genetic structure or whether gene flow has been facilitated by past geologic events. Finally, scallops spawned for aquaculture are unique from the wild and have significantly less diversity. These results warrant the attention of managers and producers who should work to monitor and conserve genetic diversity in both wild and aquaculture populations.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Genetic diversity of a species can lend information about population connectivity, adaptive potential, and allow insight into past events. In addition, measures of diversity are important in management—especially for species that inhabit disturbed areas or are manipulated for culture and/or harvest. The Pacific lion-paw scallop (Nodipecten subnodosus) is such a species, which exhibits a relatively unique geographic distribution, inhabiting waters of both the Pacific and Gulf of California (Sea of Cortez) coasts of the Baja California Peninsula, Mexico.
Over the past 5 million years, the Baja California Peninsula was bisected by two seaways connecting the Pacific Ocean with the Gulf of California. Approximately 3 million years ago (Ma), a seaway was found across the southern portion of the Peninsula at the Isthmus of La Paz while the second, mid-Peninsular seaway is estimated to have occurred 1 Ma connecting the Pacific to the Gulf of California near what is now Santa Rosalia (Upton and Murphy 1997; Terry et al. 2000; Stepien et al. 2001; Bernardi et al. 2003) (Fig. 1). Evidence of the separation of the land masses by one or both of these seaways has been demonstrated by divergence on either side of the proposed seaway(s) in terrestrial taxa (Upton and Murphy 1997; Blair et al. 2009; Riddle et al. 2000; Riginos 2005; Lindell et al. 2005, 2006). However, the extent of connectivity between marine habitats afforded by the seaway(s) and the resulting influence on the evolution of the Pacific lion-paw scallop is unknown and difficult to predict, as prior studies of fish distributed similarly to the Pacific lion-paw scallop have shown varying patterns of interspecific diversity around the Baja California Peninsula. Some species have been shown to have fixed differences between coastal populations (Stepien et al. 2001; Bernardi et al. 2003), while others appear to be panmicticly distributed (Bernardi et al. 2003). Fished and the focus of aquaculture, the Pacific lion-paw scallop is only recently the subject of genetic investigations at the population level.

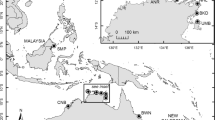
Sampling locations around the Baja California Peninsula, Mexico. Dotted lines indicate approximate sites of hypothesized historical seaways connecting the Pacific Ocean and Gulf of California. Inset (lower left): Lagoons Ojo de Liebre and Guerrero Negro. Aggregations sampled in Ojo de Liebre are noted: Canal Barcazas (OLCB), El Datil (OLD), Banco la Concha (OLBC), El Conchalito (OLC), La Ventana (OLV), El Alambre (OLA), El Mariscal (OLM)
The largest known population of the Pacific lion-paw scallop around the Baja California Peninsula is in the Lagoon Ojo de Liebre, within the Vizcaino Biosphere Reserve on the Pacific coast (Fig. 1). In the Reserve, harvest is restricted to members of local communities in order to maintain a self-sustaining population (Masso-Rojas et al. 2000). Population estimates for scallops in the Lagoon Ojo de Liebre have ranged from 8 million individuals in 1996 to 18.2 million in 2004 (German Ponce-Diaz, Personal Communication). Total biomass fluctuates within and between sites (Masso-Rojas et al. 2000; Ponce-Diaz, Personal Communication); however, from 1996 to 2004, biomass was consistently estimated to be greatest in La Ventana, followed by El Alambre. On the other hand, outside the Reserve, in the Lagoon Guerrero Negro immediately to the north of Ojo de Liebre, aquaculture ventures have been established and fishermen report that few to no wild lion-paw scallops remain. The reason for fluctuations in population size and the lack of other large populations outside of the Lagoon Ojo de Liebre are largely unknown although generally assumed to be caused by stochastic environmental conditions, alteration of habitat, and fishing pressure. Across the Baja California Peninsula in the Gulf of California, scallop aggregations range from Bahia de Los Angeles in the north to Bahia de La Paz in the south (Fig. 1). Aggregations in the Gulf of California are sparsely distributed and smaller in biomass relative to those in Ojo de Liebre.
As noted, aquaculture ventures for the Pacific lion-paw scallop have been established along the Pacific coast of the Peninsula in the Lagoon Guerrero Negro and also in Bahia Tortugas. Spawners for use in aquaculture production are collected from wild populations, most often the Lagoon Ojo de Liebre; however, the origin of aquaculture stock is not well documented. Furthermore, there are many examples of aquaculture procedures resulting in a loss of genetic diversity in culture stock relative to wild populations, and this phenomenon may be occurring in cultured Pacific lion-paw scallops as well (Petersen et al. 2008 and references cited therein; Dixon et al. 2008; Lind et al. 2009; Yuan et al. 2009). An understanding of the relationships between and within collections and how diversity of aquaculture populations relates to the wild are critical for the management and sustainability of both wild and cultured scallops.
Reproductive characteristics are also important in understanding potential population relationships. In the lion-paw scallop, the timing of gonad maturation and reproduction is related to water temperature and potentially also influenced by the abundance of phytoplankton (Arellano-Martinez et al. 2004a, b). Though the foremost spawning effort occurs annually from September to November in the Lagoon Ojo de Liebre (Arellano-Martinez et al. 2004a) and approximately May–June in the Gulf of California (as observed in La Paz; JL Ramirez, Personal Communication), some scallops have been observed to remature and spawn a second time during the year depending upon environmental conditions (Arellano-Martinez et al. 2004a). Mature at 1 year of age, Pacific lion-paw scallops are highly fecund. Each scallop can produce 20 million eggs per spawning event (Maldonado-Amparo et al. 2004) and as functional hermaphrodites can self-fertilize. It is believed that self-fertilization is not common (Petersen et al. 2008); though, this has not been extensively studied in the wild. Finally, broadcast spawning produces planktonic larvae that disperse in the water column for approximately 3 weeks before settlement onto the substrate.
The purpose of this study was to survey the genetic variation between and within natural, wild populations of the Pacific lion-paw scallop as well as those from aquaculture ventures using nuclear DNA (microsatellite) and mitochondrial DNA (mtDNA) data. Collections from both the Pacific Ocean and Gulf of California allowed inference of both modern and historical connectivity across the Peninsula. This information was collected with the goal of better understanding the relationships among aggregations. Samples from aquaculture companies also allow for an important comparison of diversity in aquaculture populations to that in the wild.
Materials and methods
Sample collection
Gulf of California
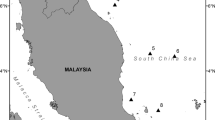
Samples were obtained from four locations including Bahia de La Paz (GCBL), Loreto (GCLG), Bahia de Los Angeles (GCLA), and Isla Tiburón (GCIT) (Fig. 1; Table 1). Collections from Loreto were obtained at three different time points, from January to June, 2006. Two sampling efforts, in February and October of 2006, make up the Bahia de Los Angeles collection, while the Bahia de La Paz and Isla Tiburón samples were each obtained in a single effort, in May 2006 and August 2008, respectively.
Pacific coast
Samples from the Lagoon Ojo de Liebre were collected at three time points (Fig. 1; Table 1). The first group of samples was taken in November 2004 from four locations: El Conchalito (OLC1), Canal de las Barcazas (OLCB), Banco la Concha (OLBC1), and El Datil (OLD). In May 2005, the second set of samples was obtained from fishermen and represented El Alambre (OLA) and a second collection from El Conchalito (OLC2). The final sample was also obtained from fishermen in December 2005 and consisted of scallops from El Mariscal (OLM), La Ventana (OLV), and a second sample from Banco la Concha (OLBC2). Scallops obtained from fishermen were minimally 15 cm in shell height per harvest regulations. For all other collections, individuals with shell height 2–5 cm were classified as juveniles, while those greater than 5 cm were classified as adults.
Aquaculture
Two companies provided samples from their aquaculture ventures, which are located in the Lagoon Guerrero Negro. Those include one sample from Pacific Big Clam (AqPBC) and two from the aquaculture cooperative Sociedad Cooperativa Complejo Lagunar de Guerrero Negro (AqSC). A fourth sample was obtained from Sol Azul in Bahia Tortugas (AqBT), also in the Pacific but southwest of Lagoons Guerrero Negro and Ojo de Liebre (Fig. 1). All aquaculture samples were collected in December 2007 with the exception of the sample from Bahia Tortugas, which was collected in September 2008 (Table 1). The samples from AqSC represent two different spawns and time points of development, juvenile (3 month, AqSCJ) and preadult (6 month, AqSCP). Scallops that were collected as spat from wild aggregations in the Lagoon Ojo de Liebre were also obtained from Sol Azul. This sample (AqWS) represented individuals that would be grown and used to spawn future generations for aquaculture. All samples, with the exception of those from fishermen and the aquaculture companies, were obtained by diving.
Microsatellite data collection and analyses
Genomic DNA was extracted from all samples using the Promega Wizard SV96 extraction kit. Additionally, genomic DNA was provided from a single individual classified as N. subnodosus and collected in Panama for a prior study (Puslednik and Serb 2008). Polymerase chain reaction (PCR) of a suite of 14 microsatellite DNA markers (Ibarra et al. 2006; Petersen et al. 2009) was employed to genotype each individual. One marker (Nsub2A1A06) was amplified and scored individually, while the other 13 loci were amplified in 4 multiplex reactions (Multiplex A: NsubA005, NsubA208, NsubC205, NsubA245; B: NsubA249, NsubA1F03, NsubA004; C: NsubA1H09, NsubA2H05, NsubA235; D: NsubC261, NsubA1G09, NsubA1H12). Marker specific primer conditions are found in the previous literature and can also be obtained from the authors. The general PCR protocol was as follows: 5–10 ng template DNA, 1X Promega buffer, 1.6 mM each dNTP, 0.375 units of Promega Go Taq Flexi polymerase, MgCl2 specific to each locus, primers ranging from 0.03 to 0.08 μM, depending upon their relative fluorescence in the multiplex, plus water resulting in a final volume of 10 μl. Thermal cycler profiles consisted of 94°C for 3 min, followed by 30 cycles of 94°C for 30 s, marker-dependant annealing temperature (Ta) for 30 s and 72°C for 30 s, and a final extension period of 10 min. PCR product was diluted with 70 μl of nanopure water. LIZ600 (ABI) size standard (0.20 μl) and 8.8 μl of Hi–Di formamide (Gel Company) were added to 1 μl of each diluted sample and denatured for 3 min at 95°C. Electrophoresis took place on an ABI 3130xl with all loci labeled with 5′ 6-FAM, VIC, NED, or PET dyes for visualization.
Polymorphism Information Content (PIC) values were calculated in Cervus 3.0 (Marshall et al. 1998; Kalinowski et al. 2007). GDA (Lewis and Zaykin 2001) was used to calculate observed and expected heterozygosities as well as to perform exact tests (Guo and Thompson 1992) for Hardy–Weinberg Equilibrium (HWE) and linkage disequilibrium (LD) with 10,000 permutations to determine significance. Allelic richness was calculated for each population using FSTAT (Goudet 2001), which measures the average number of alleles per locus for each collection, accounting for unequal sample size through rarefaction. Expanding on private allele analysis, the program ADZE (Szpiech et al. 2008) was used to examine alleles private to combinations of populations via a rarefaction approach. This method differs from that of Kalinowski (2004) by considering alleles found in all collections within a group, therefore assisting in identifying shared variation that is unique relative to all other samples.
Exact tests of genic differentiation between temporal samples in Loreto and Los Angeles were performed in GenePop (Raymond and Rousset 1995). With all collections from Loreto and Los Angeles pooled by respective sampling location, pairwise values of theta (θ) an F ST estimator that corrects for sample size (Weir and Cockerham 1984) were calculated in FSTAT as was the inbreeding coefficient, F IS. The significance of F IS was determined by 2,400 randomizations of the data. G′ST (Hedrick 2005), a standardized measure of F ST, was calculated to report the proportion of the maximum level of differentiation observed as determined by the homozygosity of the collections. The program RecodeData 0.1 (Meirmans 2006) was used to transform the data, allowing for the G′ST calculation. The proportion of genetic variance explained by differences among and within collections was evaluated using a hierarchical AMOVA in Arlequin 3.1 (Excoffier et al. 2005).
Mean population pairwise relatedness was calculated using the estimator of Queller and Goodnight (1989) implemented in GenAlEx 6 (Peakall and Smouse 2006). Relatedness was calculated using the baseline allele frequencies of the major genetic cluster to which each collection was assigned. The allele frequencies of wild, Ojo de Liebre samples were used in the calculations of relatedness within the samples spawned for aquaculture. An estimate of the number of breeders (N b) and 95% confidence intervals (CI) in each collection was obtained using the heterozygosity method of Zhdanova and Pudovkin (2008) implemented in Nb_HetEx with 10,000 bootstrap iterations.
Clustering within and among samples was tested using the Bayesian model implemented in STRUCTURE 2.2 (Pritchard et al. 2000). The admixture model with correlated allele frequencies was used for runs with 100,000 burn-in and 100,000 MCMC repetitions with three different sets of samples: (1) all wild samples, (2) Gulf of California samples, and (3) Ojo de Liebre wild samples. The number of clusters (K) was determined by averaging log Pr(X|K) over five repetitions for various values of K and choosing the value of K considering those with the maximum average likelihood and low variance across runs that also made biological sense. The graphical output was visualized using Distruct (Rosenberg 2004).
Mitochondrial DNA sequencing and analyses
Randomly selected individuals were sequenced at the mitochondrial 12S and 16S rRNA subunits using the primers and protocol in Petersen et al. (2008) to survey mtDNA diversity within each sampling location. A total of 95 individuals (ten from each Gulf location, three from each Pacific aggregation, and at least five from each culture sample) were sequenced along with two N. nodosus collected from the Atlantic off the coast of Venezuela. Additionally, 12S and 16S sequence reported in Puslednik and Serb (2008) from the N. subnodosus specimen collected in Panama was added from GenBank (accession no. EU379427 and EU379481). All sequences were visualized and aligned in Sequencher ver4.8 (Gene Codes Corp, Ann Arbor, MI). Pairwise distances between mitotypes were computed using the Kimura 2-parameter model in MEGA ver 4 (Tamura et al. 2007).
Homogeneity between mitochondrial regions of all N. subnodosus mitotypes was assessed using a partition homogeneity test (PHT) with 1,000 replications in PAUP4.0b10 (Swofford 2003). 12S and 16S sequence from each individual were concatenated in Sequencher. The phylogeny was estimated using a Bayesian framework in MrBayes v3.1 (Ronquist and Huelsenbeck 2003) assuming the most likely model of nucleotide evolution as determined using the Akaike information criterion (AIC) in MrModeltest (Nylander 2004) and N. nodosus as an outgroup. Two simultaneous runs of 3,000,000 generations were performed with a random starting tree and sampling every 100 generations. The initial 3,000 trees were discarded as burn-in samples, and the 50% majority rule consensus tree was visualized with FigTree v1.2.2 (Rambaut 2009). Mitotypes were also visualized via a median-joining haplotype network constructed using Network 4.5.1.6 (http://www.fluxus-engineering.com; Bandelt et al. 1999).
Assignment of all wild collected individuals to one of the two resulting mtDNA clades was performed using the 12S rRNA PCR product and restriction enzyme BauI (Fermentas), which recognizes a restriction site unique to individuals with the Clade II mitotype. PCR product (5 μl) amplified as in Petersen et al. (2008) was digested with 3 units of enzyme, 2 μl of tango buffer, and water to a total volume of 10 μl and digested overnight at 37°C. 3 μl of the digest was electrophoresed on a 2% agarose gel for 30 min at 125 volts. Previously sequenced samples of each mitotype were included in each digest as scoring/digestion controls.
Results
mtDNA analyses
A 314-bp fragment of the 12S locus and a 389-bp fragment of the 16S locus were obtained in the N. subnodosus alignment. Pairwise differences between Baja California mitotypes ranged from a single insertion to 13-bp differences (pairwise distance = 4.23%) within the 12S region and from a single base pair change to nine changes (pairwise distance = 1.84%) in the 16S region. A PHT including both N. nodosus and N. subnodosus found no significant heterogeneity between gene regions (P = 1.0); therefore, these fragments were concatenated for further analyses. Divergence between concatenated N. subnodosus mitotypes ranged from the single insertion to 21 single nucleotide polymorphisms (SNPs) (Kimura pairwise distance = 2.78%), while N. subnodosus samples from the Baja California Peninsula, Mexico, and N. nodosus differed by an average pairwise value of 6.17% (Table 2).
The most common mitotype, (a), was found in 42 of 95 individuals sequenced; that and three other mitotypes were shared between coasts (Table 3). Sequence data from the sample collected in Panama placed it into Clade I. The resulting Bayesian tree (Fig. 2) of wild caught samples further illustrates the divergence between clades, as does the mitotype network (Supplementary Figure 1). Restriction digest of the wild samples revealed a 26.6% incidence of the Clade I mitotype in Ojo de Liebre. In the Gulf of California, a total of six individuals were assigned by RFLP to Clade I including two from La Paz, three from Loreto, and one from Isla Tiburón (Fig. 3); the overall incidence of the Clade I mitotype in the Gulf of California was 3.3%.

Majority rule consensus tree created by Bayesian analysis of 703 bp of 12S and 16S rRNA mtDNA sequence. N. subnodosus mitotypes are labeled Nsub. Two distinct clades are observed in the N. subnodosus samples (Clade I and II). Posterior probabilities greater than 0.5 are shown

Incidence of the Clade I mtDNA mitotype in each wild collection as determined by PCR–RFLP
Microsatellite analyses: wild collections
Pooling of samples
Exact tests of genic differentiation showed that the temporal samples from Los Angeles were not significantly different from one another (P = 0.13); on the other hand, before correction for multiple testing, the third sample from Loreto (June 2006) differed from the two other samples from that location (P = 0.03). Due to the small size of that sample (N = 8), the lack of significance after Bonferroni correction, and the fact that no population level results change with or without the inclusion of these samples into the data set, all three samples from Loreto and the two from Los Angeles were pooled based upon sampling location for all further analyses. Similarly, no two individual collections in Ojo de Liebre were statistically divergent from another collected during the same sampling period; therefore, samples from Ojo de Liebre were pooled according to the date of collection for all analyses (referred to as OL1-3).
Marker diversity
A preliminary linkage map (JLP, unpublished data) show loci NsubA1H09, NsubA2H05, NsubA1H12, and Nsub2A1A06 to be in linkage groups with other loci in this study. These four loci were removed from all analyses. No other pairs of loci showed consistent LD across samples although LD was present in both wild and cultured samples. In the Gulf of California, LD was present in five pairs of loci, but none remained significant after sequential Bonferroni correction (Holm 1979; Rice 1989) within each sample (total of 45 tests per sample). In Ojo de Liebre, 70 total pairs of loci showed significant LD before correction, 7 in OL1, 32 in OL2, and 31 in OL3. Sixteen pairs (24.4%) remained significant in OL2 after correction for multiple tests and 11 (13.3%) within OL3. PIC values for the microsatellites ranged from 0.47 (NsubA208) to 0.86 (NsubA1G09), averaging 0.71 across loci.
Significant departure from HWE was found in 16 tests (α = 0.05) before correction for multiple testing. After applying a sequential Bonferroni correction, only one locus remained significantly different from the expectations of HWE, NsubC261 in OL2, showing heterozygote deficiency. F IS was significantly different from zero in GCIT. Overall expected and observed heterozygosities from the wild were similar to one another and similar to AqWS (Table 1).
Relatedness & N b
All wild samples in Ojo de Liebre had mean pairwise relatedness values within the bounds of the CI expected under the null hypothesis of random mating. The Gulf of California samples, examined separately, showed high relatedness in the Loreto sample, and significantly lower relatedness than the expected value in the sample from La Paz (Fig. 4). Confidence intervals (95%) of the estimates of N b in the Gulf and Ojo de Liebre populations and the wild spat collected by producers had a median and upper bound of infinity (Table 1).

Mean pairwise relatedness ±standard error for each of the wild (left) and aquaculture (right) collections based upon microsatellite genotypes. Circles designate the upper and lower bounds of the 95% confidence interval for the null hypothesis (random mating)
Population divergence
Comparing the collections parsed by sampling date in Ojo de Liebre resulted in one significant θ value between OL1 and OL2. No significance was found within the comparisons among samples in the Gulf (Table 4). Significant differences were found in all comparisons between wild samples in Ojo de Liebre and those in the Gulf of California. The pairwise θ values comparing collections between coasts were generally an order of magnitude larger than comparisons within either the Gulf of California or Ojo de Liebre (Table 4). Standardized G′ST values also show that on average less than 1% of the maximum possible differentiation is found in comparisons within the Gulf of California and within Ojo de Liebre (Table 4). Between coasts, 14–24% of the possible differentiation is observed. Hierarchical AMOVA showed that most variation within either coast is captured within collections, while less than 1% is found among collections. A low but significant proportion of the variation (3.8%) is explained by the coastal difference (Table 5). Allele frequencies for all samples can be found in the Supplemental Material.
Allelic richness
Allelic richness adjusted for sample size was similar across all wild collections and within the sample of spat (AqWS) collected from Ojo de Liebre. Private alleles were found in all samples but were most prevalent in the Gulf of California sites (Table 1; Fig. 5a, e), even given the discrepancy in sample sizes. Parsing the samples into two populations (Gulf of California and Pacific coast), the most frequent private allele was found in the Gulf of California with an occurrence of 1.6%. Microsatellite genotyping of the sample from Panama revealed a total of eight private alleles at the eight loci amplified. Because it was a single sample, it was not included in any other microsatellite analyses.

The mean number of private alleles per locus (microsatellite) for: a all collections, b Gulf of California collections compared only to themselves, c Gulf of California collections compared to all wild collections including Ojo de Liebre, d Ojo de Liebre samples paired with each of the Gulf samples, relative to all other collections, and e all Gulf of California and all Ojo de Liebre samples. Note the scale of the axes differ among graphs
Private allele analysis using rarefaction in ADZE further illustrated the trend of more prevalent private alleles in the Gulf of California (Fig. 5a, e). The samples from the three northernmost regions of the Gulf of California were similar in the mean number of private alleles per locus as were the three collections from Ojo de Liebre. On examining the number of private alleles per locus in only the Gulf of California samples (neglecting the data from the Pacific), Loreto is least unique within the collections, while La Paz appears most unique (Fig. 5b). When the information from the Pacific is added, La Paz shows a decrease in the mean number of private alleles per locus compared to the other Gulf samples (Fig. 5c). Pairing each sample in the Gulf of California with Ojo de Liebre one at a time, and comparing the pair to the rest of the wild samples, shows scallops in La Paz share more private alleles with the Pacific than those in the other Gulf of California locations (Fig. 5d).
STRUCTURE
With only the wild samples considered, the most likely value of K was 2 (log Pr(X|K) = −22,782.9), distinguishing the Ojo de Liebre samples from those taken from the Gulf of California (Fig. 6). The mean value of log Pr(X|K) for K = 3 was greater (−22,708.3) than that of K = 2, but had higher variance across runs, and the resulting plots showing assignment of each individual within the collections did not clarify any relationships between individuals or collections. Assuming K = 2 in the wild samples, the average q-value (assignment probability) for self-assignment of Gulf of California samples to their own cluster was 0.90, and 0.87 in Ojo de Liebre. Twelve Gulf of California individuals (6.8%) and 43 from Ojo de Liebre (10.8%) did not assign to the cluster representing their geographic region. No structure was found considering only the samples collected in the Gulf of California or those within Ojo de Liebre (K = 1; log Pr(X|K) = −15,384 and −15,407.1, respectively).

STRUCTURE output for all wild collections (K = 2) based upon microsatellite data. Each vertical line represents one individual, and each color represents the proportion of assignment to either of the two clusters
Microsatellite analyses: aquaculture
In AqBT, 29 (53.5%) pairs of loci were in LD while in all other aquaculture collections, a total of 7 pairs had significant values (1 in AqSCJ, 5 in AqSCP, 1 in AqPBC) after Bonferroni correction. Nineteen tests for HWE were significant at α = 0.05. Twelve remained significant after correction including four loci each in AqSCJ and AqPBC (NsubA005, NsubC205, NsubC261, and NsubA1G09), and four in AqBT (NsubC205, NsubA249, NsubA004, and NsubC261). No departure from HWE was observed in AqSCP. Unlike the wild samples, the departure from HWE in all aquaculture samples was in the direction of excess heterozygosity. Aquaculture samples also had less diversity as measured by expected heterozygosity and allelic richness (Table 1). The mean pairwise relatedness values for all samples spawned in aquaculture were significantly higher than that expected at random and at levels suggesting a high proportion of full (0.5) and/or half (0.25) siblings while the sample of wild spat being grown by producers was not significant (Fig. 4). For N b in samples spawned for aquaculture, the upper bound for AqSCP included infinity; however, all samples had a median estimate less than 26 and in some cases approximately 2 (AqSCJ and AqPBC).
With the exception of the sample of wild spat collected for future aquacultural production (AqWS), samples provided by the aquaculture companies were significantly different than all collections from the wild (Table 4). Additionally, all aquaculture samples differed from one another with the exception of AqSCP and AqPBC. G′ST values, when comparing all samples spawned in aquaculture to those in the wild, show 21–69% of the possible variation is observed (Table 4). Aquaculture samples had decreased allelic richness relative to the wild (Table 1). For both AqSC samples and AqPBC, no locus had more than four alleles, contrary to the wild where no sample had fewer than four alleles at any one locus (and up to 23 alleles at NsubA1G09 in the GCLA sample). If the assumption is made that only two spawners contributed to these samples, chi-square tests for allele frequency assuming Mendelian inheritance, the observed genotype identity and ratios of the progeny, similar deviations from HWE between collections, and the presence of only two mitotypes in AqSCJ and AqPBC, suggest that these samples were derived from the same two spawners. Following this assumption, these data allowed for putative parental genotypes of these collections to be constructed using the allele segregation patterns. Working from the inferred parental genotypes, two individuals from AqPBC appear to have resulted from self-fertilization while one progeny had a single allele that was not shared by any other individuals in the sample. In the AqSCP sample, although no more than four alleles were observed at any locus, three mitotypes were found in the five sequenced individuals, and genotype combinations suggest the progeny were derived from more than two spawners. Though the AqBT sample showed only two mitotypes in eight individuals sequenced, the observation of ten alleles at one locus supports the conclusion that at least five spawners contributed to this sample.
Discussion
mtDNA diversity
While this study was originally designed to examine among population differentiation, the unexpected divergence in mtDNA overshadows the evidence of population structure revealed by the microsatellite data. Comparing prior studies of the 16S locus, the level of divergence between clades of the Pacific lion-paw scallop is comparable to that which Saavedra and Peña (2004) found between true species (Pecten fumatus and P. jacobaeus), although the divergence between N. nodosus and N. subnodosus in this study is almost 2.5 times that between the two mitochondrial clades of N. subnodosus. In the aforementioned study and others, the rate of mtDNA evolution for mollusks has been estimated to range from 0.04 to 0.54% per million years (Ó Foighil et al. 1995; Rawson and Hilbish 1995; Jazefowicz and Ó Foighil 1998). High fecundity in marine mollusks is often linked to high prevalence of null alleles and speculated accelerated sequence evolution (Williams 1975; Hedgecock et al. 2004); therefore unusually slow rates of mtDNA evolution would be surprising. However, even assuming a “high” rate of evolution given the prior estimates in related species suggests divergence between the two mtDNA clades dates back approximately 5 million years (assuming a rate of 0.5%/MY). While this is only a rough estimate of divergence, geologic evidence suggests the Gulf of California was formed about this time (Riddle et al. 2000; reviewed in Murphy and Aguirre-Leon 2002). One could speculate that populations became isolated as the Gulf of California was formed, leading to genetic drift and the divergence observed. Then, a further hypothesis could assert that secondary contact via the most recent (1 Ma) mid-Peninsular seaway, and/or modern translocation (see “Discussion” below) led to introgression of the populations as seen by the lack of nuclear differentiation between clades. While these hypotheses are untested, support for recent connectivity is corroborated by the sharing of mitotypes between Ojo de Liebre and the Gulf of California, including the most common mitotype (a). Within Ojo de Liebre, microsatellites cannot distinguish members of the mtDNA clades from one another (P = 0.29). In the Gulf of California, there is a significant difference between members of Clade I and Clade II (P = 0.28). However, with only six samples from Clade I (of 173 total) and high allelic richness, this may not be an accurate comparison of these groups. Finally, q-values of assignment in STRUCTURE do not differ between members of the clades, nor are the individuals of each clade distinguished in cluster analyses. With all evidence considered, it seems that there has been complete introgression between members of the two mitochondrial clades.
Regardless of the origin of the mtDNA divergence, these results introduce new questions regarding the taxonomic status of the Pacific lion-paw scallop and genus Nodipecten, which has historically been based upon differences in rib count and arrangement, the node scheme of the left valve of the shell, and geographic location (Smith 1991). Shell morphology was not documented for the scallops in this study as the question of phylogenetic identity was not anticipated. Even so, morphological variation is present within the species and accounts of species distribution along the Pacific coast of North and Central America differ, or at most are unclear. For example, the reported range of N. subnodosus varies from Keen (1971), who reports it ranges as far south as Peru, to Smith (1991), who reports the southern boundary of N. subnodosus lies at the tip of the Baja California Peninsula while a third species, N. arthriticus, is present southward. Recent phylogenetic work on Pectinids include that by Saavedra and Peña (2006) and Puslednik and Serb (2008), which show N. subnodosus and N. nodosus to be monophyletic; however, neither report N. arthriticus. Furthermore, with the exception of the single sample from Panama, which falls into Clade I, no mtDNA sequence is available for Nodipecten spp. south of the Baja California Peninsula.
Microsatellite analyses: wild collections
Marker utility
For analysis of among- and within-population relationships, the ten microsatellite loci used in this investigation had high levels of diversity, which revealed significant population structure between coasts and dissimilarity between wild and cultured samples. Observed and expected heterozygosities were remarkably similar across samples from the wild, and also in the sample of wild spat collected for aquaculture; the average H e (0.77) is comparable to that reported in other wild marine mollusks (Kenchington et al. 2006; Lind et al. 2009). Little deviation from HWE was observed. In the wild sample where it was present, deviation was in the direction of heterozygote deficit and may be due to null alleles, which have been documented in several marine mollusks (McGoldrick et al. 2000; Launey et al. 2002; Hedgecock et al. 2004; Reece et al. 2004; Kenchington et al. 2006; Galindo-Sanchez et al. 2008, among others) as well as in controlled spawns of this species (Petersen et al. 2008; JLP, unpublished data). However, heterozygote deficit was not observed across samples as would be expected with null alleles, and therefore all 10 loci were kept in the analyses.
Ojo de Liebre
After pooling the Ojo de Liebre samples based upon collection date, the significant pairwise comparison between OL1 and OL2 was surprising giving the lack of structure found between individual collections. Also no structure within Ojo de Liebre was detected using Bayesian clustering methods. Compared to the other wild collections, OL2 and OL3 did have a high incidence of LD. While no relatedness values in Ojo de Liebre were significant, OL2 and OL3 differ from the other wild samples in that they were obtained from fisherman. Harvest is limited by a minimum size requirement, so it is possible that during sample collection a biased sample was obtained based upon size, selection of closely related individuals, and/or those that have a similar genetic advantage for growth and fitness. In addition to affecting measures of LD, the sampling scheme could also have contributed to the variation observed between OL2 and OL1 (not collected from fishermen). On the other hand, if this were the case, similar significance would be expected comparing OL1 and OL3, which was not evident. While temporal variation for these or other reasons (i.e., selective mortality) would be of note for the management of Ojo de Liebre scallops, the lack of significant structure between all other pairwise comparisons of Ojo de Liebre collections and between these samples and the wild spat collected for culture in 2007 suggests that the minor temporal variation in the Lagoon is not a driving factor in the overall genetic structure observed. Dispersal of gametes and/or larvae, possibly facilitated by a complex system of connected channels, appears to have produced a population that is near panmixia in Ojo de Liebre. These data, together with Benzie and Smith-Keune (2006) who showed no variance between age classes in Pinctada maxima, added confidence to our ability to make comparisons between the coasts and across the years sampled.
Gulf of California
Despite the geographic distance between samples in the Gulf of California, no significant population structure was found based on indices of θ or STRUCTURE analyses. Relatedness calculations, however, show significant, increased relatedness in GCLG and significant, decreased relatedness in GCBL. In GCLG, the relatedness observed could be due to the relatively high proportion of juveniles in the sample. Based upon the size criteria used to distinguish age classes, juveniles represent a single year class while adults likely represent multiple generations. Conversely, this logic does not fit the low value of relatedness in the smaller GCBL sample, which consisted entirely of juveniles. Low relatedness in GCBL, relative to the entire Gulf of California population, may be evidence of gene flow from Ojo de Liebre (discussed below). When calculating relatedness using each collection as its own baseline, no significant deviation from expected values is observed (data not shown).
Significant F IS was found in GCIT, though not accompanied by elevated pairwise relatedness. No signs of a significant reduction in population size were detected through the estimated number of breeders (N b) in the Gulf of California population as a whole. Not shown here, estimates of N e as calculated by the linkage disequilibrium method of Waples and Do (2008) showed similar patterns within the wild collections with intervals bounded by infinity. While estimates of N b may be good for relative comparisons, many assumptions are included in the process. In the method of Zhdanova and Pudovkin (2008) used here, the assumptions include that the samples are from a closed population and that there are nonoverlapping generations. While these assumptions are generally true in aquaculture settings, they are not often the case in the wild. Furthermore, this method is most capable of estimating the number of breeders when the true value is less than 30, which is not the case in many wild situations, and is reported to have the most power with 200 or more progeny sampled (not the case in any individual collection here) (Zhdanova and Pudovkin 2008). That said, all N b estimates should be considered with those caveats in mind and are best used to examine relative measures among samples.
Structure between coasts
Connectivity between marine mollusk habitats is often explained by the potential for high dispersal of their mass-spawned gametes and drift of the planktonic larvae. Population structure has previously been found in oysters (Benzie and Smith-Keune 2006; Rose et al. 2006; Galindo-Sanchez et al. 2008) and scallops (Zhan et al. 2009), largely attributed to the action of ocean currents. In the case of the sea scallop, Tremblay et al. (1994) and Kenchington et al. (2006) have used particle tracking software to examine recruitment of larvae. The former study found that significant biological and environmental effects determine to where the larvae will disperse, while the latter showed that dispersal closely followed current patterns. In the Gulf of California, complex seasonal upwellings and various gyres have been shown to disperse particles (and likely gametes and larvae) both north and southward (Marinone 2003; Gutierrez et al. 2004; Marinone et al. 2009). As directionality of the currents change from a general southward flow in April to northward in June (Marinone et al. 2009), the path that larvae in the Gulf of California take likely varies based upon the timing of the spawn and the directionality of the currents. Furthermore, the apparent connectivity between sample locations may be enhanced by gene flow among unsampled aggregations intermediate to those studied.
While overall indices of θ were similar in the Gulf of California and Ojo de Liebre (0.002 and 0.001, respectively), the θ value comparing all Gulf of California and Pacific samples (0.04) differs by an order of magnitude suggesting there is not sufficient gene flow between coasts to result in panmixia. The proportion of variation explained between coasts is less than 4%, but the divergence is significant and not surprising due to the geographic barrier imposed by the Peninsula, following prior studies of Stepien et al. (2001) and Bernardi et al. (2003). The distinction between the Gulf of California and Pacific samples is also supported by cluster analyses in STRUCTURE.
One of the main purposes of this investigation was to provide baseline data to allow for monitoring of the genetic diversity in the populations and the detection of changes in population structure due to natural or anthropogenic events. In this system, it is acknowledged (documented and hearsay) that movement of scallops across the Peninsula has taken place for the purpose of aquaculture and for research (from Guerrero Negro to La Paz (Barrios-Ruiz et al. 2003); from Ojo de Liebre to Norte de Sinaloa (Diarte-Plata 2007); undocumented translocation from Ojo de Liebre to Loreto). In many of these instances, it is reported that the translocated individuals perform poorly and often die (Diarte-Plata 2007; Martha Castro-Poloni, Personal Communication), as seen in experimental translocation of catarina scallops across the Baja California Peninsula (Cruz et al. 1998). However, Barrios-Ruiz et al. (2003) reported gametogenesis and a spawn in lion-paw scallops from Guerrero Negro that were moved to La Paz. Examining the populations for signs of translocation, assignment of individuals in STRUCTURE showed that several individuals assigned (q > 0.5) to the opposite cluster from which they were sampled. While these individuals could reflect translocations, as noted, the large majority of known translocations are individuals moved from the Pacific to the Gulf of California. STRUCTURE results would suggest the opposite trend is most common. La Paz is a known recipient of scallops that originated in the Pacific (Barrios-Ruiz et al. 2003); however, misassignment of individuals from La Paz is not disproportionately more frequent than in other collections. For more precise monitoring purposes, it would be valuable to develop forensic markers able to resolve the population of origin for these scallops. Private alleles, which could potentially serve this purpose, were common in every collection but most were rare and do not offer much value as diagnostic markers. Future work will focus on microsatellite sequencing that has shown substantial SNP variation in microsatellite flanking regions that may allow for more accurate population assignment in the future (JLP, unpublished data).
While in this instance private alleles are not feasible for forensic purposes, analyses of shared alleles private to combinations of populations in ADZE did uncover evidence of potential gene flow between Ojo de Liebre and La Paz. As Fig. 5b illustrates, when the diversity of Ojo de Liebre is not considered, La Paz appears to be more unique (more private alleles/locus) than the other Gulf of California samples. But when alleles from Ojo de Liebre are considered (Fig. 5c), the uniqueness of La Paz drops, suggesting it shares more alleles with Ojo de Liebre than the other Gulf of California collections, further supported by Fig. 5d. It is uncertain whether these data reflect recent translocations, although with La Paz having the highest incidence of the Clade I mitotype in the Gulf of California, the possibility of past translocations successfully reproducing in La Paz seems feasible. If this were the case, the influence of genetic material from the Pacific may also help to explain the lower than expected values of relatedness in the La Paz collection. However, if the presence of the Clade I mitotype in the Gulf of California was due to translocation, the scallops themselves were not—STRUCTURE analyses assign each to the population from which it was collected.
Diversity of aquaculture populations
As aquaculture develops for this species, it is important to understand the diversity of the cultured populations and how it relates to that of the wild. The levels of diversity in the wild were not reflected in the samples spawned for aquaculture as shown by decreased measures of heterozygosity, allelic richness, and estimates of N b, as well as high pairwise relatedness. Unlike heterozygote deficiency in wild samples, deviations from HWE in the samples spawned in aquaculture were due to heterozygote excess, which could be a consequence of genetic drift in these small populations. Additionally, Bierne et al. (1998) and Launey and Hedgecock (2001) have characterized a high incidence of genetic load in mollusks, which could also contribute to the observed excess heterozygosity.
Notably, the sample of wild spat collected from the producers did not show reduced genetic diversity, indicating that diversity is lost somewhere in the process of spawning or in selection for aquaculture conditions. The use of few spawners to create aquaculture cohorts was supported by estimates of N b and prior studies of this species (Petersen et al. 2008) and oysters (Lind et al. 2009) that show the familial make-up of progeny created by mass spawns can be significantly skewed from null expectations. Linkage disequilibrium prevalent in the AqBT sample could be explained by family structure, which was suggested by clustering analysis and both allelic and genotypic frequencies (data not shown). Although the AqBT sample had decreased diversity relative to the wild, it contained the most diversity of all the samples from aquaculture facilities, illustrating how the success of multiple spawners in production can improve the diversity of these spawns. In AqSCJ and AqPBC, chi-square tests and genotypic combinations support the assertion that the same two spawners were used in the creation of both samples. As all scallops sold to producers in 2007 were provided by one individual (Marcelo Armenta, Personal Communication), this is a reasonable conclusion.
Three mitotypes were observed in the five AqSCP samples sequenced, and N b was estimated to be 25. However, this sample had the highest mean relatedness values observed (0.54) in any sample and no more than four alleles per locus. When sampled, these scallops were noted to have shell abnormalities consistent with those observed in catarina scallops produced in the laboratory by the most severe form of inbreeding, self-fertilization (AMI, unpublished data). Unlike the other aquaculture samples, it appears that the spawners used to create this population were highly related, which is likely as this sample was produced from spawners that were themselves bred in captivity (unlike the other aquaculture samples). The evidence of highly related spawners and evidence of potential inbreeding along with the “high” estimate of N b illustrates again that this estimate is useful only as an approximation of the effective breeding size.
With many examples of a loss of genetic diversity in cultured populations (Gaffney et al. 1992; Hedgecock et al. 1992; Blake et al. 1997; Li et al. 2004; Dixon et al. 2008; Lind et al. 2009; Yuan et al. 2009), acknowledging that this is taking place in Pacific lion-paw scallop culture is especially important as grow-out occurs in open waters adjacent to natural populations. Populations of lion-paw scallop artificially spawned for production sexually mature before harvest and therefore have the potential to introgress with and thus alter the make-up of the native populations (Allendorf and Ryman 1987; Bekkevold et al. 2006). The introgression of individuals from culture into the wild, whether intentional or not, can affect the diversity in the wild. Following Ryman and Laikre (1991), consider the case if cultured samples from AqSCJ (N e ~2) were to inhabit and introgress with a wild aggregation (N e ~1,000). Even if the cultured scallops make up only 5% of the total population, the N e of the aggregation could be reduced by more than 50%. Illustrations of this phenomenon are nicely presented in Gaffney (1996) along with further discussion of the importance of maintaining N e and diversity in the wild. From the standpoint of the producers, the maintenance of diversity in aquaculture may benefit production as inbreeding has been shown to have negative effects on growth of abalone (Deng et al. 2005) as well as growth and fitness of oysters (Evans et al. 2004) and scallop (Ibarra et al. 1995; Zheng et al. 2008). Loss of natural diversity may also eliminate natural genetic variability that could be drawn upon in selective efforts to improve the quality of the aquaculture populations.
Summary
In summary, mitochondrial analyses showed two divergent groups, both represented in Ojo de Liebre and the Gulf of California although at differing frequencies. As microsatellite data show, scallops from both mitogroups appear to be introgressed, raising questions pertaining to the origin of the mitotypes found in this species. Microsatellite data has shown that the Pacific lion-paw scallop aggregations in the Gulf of California and those in the Lagoon Ojo de Liebre are acting as two genetically distinct populations. Some evidence suggests gene flow between the Pacific population studied and that in La Paz. It is uncertain whether translocations have influenced the genetic structure observed, although future translocations are warned against due to the significant population structure found here. Samples of scallops being grown in aquaculture are unlike any wild populations, clearly less diverse, and also should be prevented from contributing to the diversity of the wild. Besides serving as a baseline for future monitoring of the species, these results warrant the attention of managers and producers of scallops who should work to ensure genetic diversity is sustained in wild and cultured populations. Finally, a complete survey of the genus Nodipecten would be helpful in elucidating the relationship between scallops around the Baja California Peninsula with other aggregations in the Pacific.
References
Allendorf FW, Ryman N (1987) Genetic management of hatchery stocks. In: Ryman M, Utter F (eds) Population genetics and fishery management. Univ of Washington Press, Seattle, pp 141–159
Arellano-Martinez M, Racotta IS, Ceballos-Vazquez BP, Elorduy-Garay JF (2004a) Biochemical composition, reproductive activity, and food availability of the lion’s paw scallop Nodipecten subnodosus in the Laguna Ojo de Liebre, B.C.S., Mexico. J Shellfish Res 23:15–23
Arellano-Martinez M, Ceballos-Vazquez BP, Villalejo-Fuerte M, Garcia-Dominguez F, Elorduy-Garay JF, Esliman-Salgado A, Racotta IS (2004b) Reproduction of the lion’s paw scallop Nodipecten subnodosus Sowerby, 1935 (bivalvia: pectinidae) from Laguna Ojo de Liebre, B.C.S., Mexico. J Shellfish Res 23:723–730
Bandelt HJ, Forster P, Röhl A (1999) Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol 16:37–48
Barrios-Ruiz D, Chavez-Villalba J, Caceres-Martinez C (2003) Growth of Nodipecten subnodosus (Bivalvia: Pectinidae) in La Paz Bay, Mexico. Aqua Res 34:633–639
Bekkevold D, Hansen MM, Nielsen EE (2006) Genetic impact of gadoid culture on wild fish populations: predictions, lessons from salmonids, and possibilities for minimizing adverse effects. J Mar Sci 63:198–208
Benzie JAH, Smith-Keune C (2006) Microsatellite variation in Australian and Indonesian pearl oyster Pincatada maxima populations. Mar Ecol Prog Ser 214:197–211
Bernardi G, Findley L, Rocha-Olivares A (2003) Vicariance and dispersal across Baja California in disjunct marine fish populations. Evolution 57:1599–1609
Bierne N, Launey S, Naciri-Graven Y, Bonhomme F (1998) Early effect of inbreeding as revealed by microsatellite analyses on Ostrea edulis larvae. Genetics 148:1893–1906
Blair C, Mendez de la Cruz FR, Ngo A, Lindell J, Lathrop A, Murphy RW (2009) Molecular phylogenetics and taxonomy of leaf-toed geckos (Phyllodactylidae: Phyllodactulus) inhabiting the peninsula of Baja California. Zootaxa 2027:28–42
Blake SG, Blake NJ, Oeterling MJ, Graves JE (1997) Genetic divergence and loss of diversity in two cultured populations of the bay scallop, Argopecten irradians. J Shellfish Res 16:55–58
Cruz P, Ramírez JL, Garcia GA, Ibarra AM (1998) Genetic differences between two populations of catarina scallop (Argopecten ventricosus) for adaptations for growth and survival in a stressful environment. Aquaculture 166:321–335
Deng Y, Liu X, Zhang G, Guo X (2005) Inbreeding depression and maternal effects on early performance of Pacific abalone. N Am J Aquacult 67:231–236
Diarte-Plata G (2007) Cultivo experimental de la almeja mano de león Nodipecten subnodosus (Sowerby, 1835) en la coasta Norte de Sinaloa, México. Master thesis, Instituto Politecnico Nacional, Guasave, Sinaloa, México
Dixon TJ, Coman GJ, Arnold SJ, Sellars MJ, Lyons RE, Dierens L, Preston NP, Li Y (2008) Shifts in genetic diversity during domestication of black tiger shrimp, Penaeus monodon, monitored using two muliplexed microsatellite systems. Aquaculture 283:1–6
Evans F, Matson S, Brake J, Langdon C (2004) The effects of inbreeding on performance traits of adult Pacific oysters (Crassostrea gigas). Aquaculture 230:89–98
Excoffier L, Laval G, Schneider S (2005) ARLEQUIN version 3.1: an integrated software package for population genetics data analysis. Evol Bioinform 1:47–50
Gaffney PM (1996) The role of genetics in shellfish restoration. Aquat Living Resour 19:277–282
Gaffney PM, Davis CV, Hawes RO (1992) Assessment of drift and selection in hatchery populations of oysters (Crassostrea virginica). Aquaculture 105:1–20
Galindo-Sanchez CE, Gaffney PM, Perez-Rostro CI, de la Rosa-Velez J, Candela J, Cruz P (2008) Assessment of genetic diversity of the eastern oyster Crassostrea virginica in Veracruz, Mexico using microsatellite markers. J Shellfish Res 27:721–727
Goudet J (2001) FSTAT, a program to estimate and test gene diversities and fixation indices (version 2.9.3). Updated from Goudet (1995). Available at http://www2.unil.ch/popgen/softwares/fstat.htm
Guo S, Thompson EA (1992) Performing the exact test of Hardy-Weinberg proportion for multiple alleles. Biometrics 48:361–372
Gutierrez OQ, Marinone SG, Pares-Sierra A (2004) Lagrangian surface circulation in the Gulf of California from a 3D numerical model. Deep Sea Res II 51:659–672
Hedgecock D, Chow V, Waples RS (1992) Effective population numbers of shellfish broodstocks estimated from temporal variance in allele frequencies. Aquaculture 108:215–232
Hedgecock D, Li G, Hubert S, Bucklin K, Ribes V (2004) Widespread null alleles and poor cross-species amplification of microsatellite DNA loci cloned from the Pacific oyster, Crassostrea gigas. J Shellfish Res 23:379–385
Hedrick PW (2005) A standardized genetic differentiation measure. Evolution 59:1633–1638
Holm S (1979) A simple sequentially rejective multiple test procedure. Scand J Statist 6:65–70
Ibarra AM, Cruz P, Romero BA (1995) Effects of inbreeding on growth and survival of self-fertilized catarina scallop larvae, Argopecten circularis. Aquaculture 134:37–47
Ibarra AM, Petersen JL, Famula TR, May B (2006) Characterization of 35 microsatellite loci in the Pacific lion-paw scallop (Nodipecten subnodosus) and their cross-species amplification in four other scallops of the Pectinidae family. Mol Ecol Notes 6:153–156
Jozefowicz CJ, Ó Foighil D (1998) Phylogenetic analysis of southern hemisphere flat oysters based on partial mitochondrial 16S rDNA gene sequences. Mol Phylogenet Evol 10:426–435
Kalinowski ST (2004) Counting alleles with rarefaction: private alleles and hierarchical sampling designs. Conserv Genet 5:539–543
Kalinowski ST, Taper ML, Marshall TC (2007) Revising how the computer program CERVUS accommodates genotyping error increases success in paternity assignment. Mol Ecol 16:1099–1106
Keen MA (1971) Sea shells of tropical west America, 2nd edn. Stanford Univ Press, Stanford
Kenchington EL, Patwary MU, Zouros E, Bird CJ (2006) Genetic differentiation in relation to marine landscape in a broadcast-spawning bivalve mollusk (Placopecten magellanicus). Mol Ecol 15:1781–1796
Launey S, Hedgecock D (2001) High genetic load in the Pacific Oyster Crassostrea gigas. Genetics 159:255–265
Launey S, Ledu C, Boudry P, Bonhomme B, Naciri-Graven Y (2002) Geographic structure in the European flat oyster (Ostrea edulis L.) as revealed by microsatellite polymorphism. J Hered 93:331–338
Lewis PO, Zaykin D (2001) Genetic data analysis: computer program for the analysis of allelic data. Available at http://lewis.eeb.uconn.edu/lewishome/software.html
Li Q, Park C, Endo T, Kijima A (2004) Loss of genetic variation at microsatellite loci in hatchery strains of the Pacific abalone (Haliotis discus hannai). Aquaculture 235:207–222
Lind CE, Evans BS, Knauer J, Taylor JJU, Jerry DR (2009) Decreased genetic diversity and a reduced effective population size in cultured silver-lipped pearl oysters (Pinctada maxima). Aquaculture 286:12–19
Lindell J, Mendez de la Cruz FR, Murphy RW (2005) Deep genealogical history without population differentiation: discordance between mtDNA and allozyme divergence in the zebra-tailed lizard (Callisaurus draconoides). Mol Phylogenet Evol 36:682–694
Lindell J, Ngo A, Murphy RW (2006) Deep genealogies and the mid-peninsular seaway of Baja California. J Biogeogr 33:1327–1331
Maldonado-Amparo R, Ramirez JL, Avila S, Ibarra AM (2004) Triploid lion-paw scallop (Nodipecten subnodosus); growth, gametogenesis, and gametic cell frequencies when grown at a high food availability site. Aquaculture 235:185–205
Marinone SG (2003) A three-dimensional model of the mean and seasonal circulation of the Gulf of California. J Geophys Res 108:3325
Marinone SG, Gonzalez JI, Figueroa JM (2009) Prediction of currents and sea surface elevation in the Gulf of California from tidal to season scales. Environ Modell Softw 24:140–143
Marshall TC, Slate J, Kruuk LEB, Pemberton JM (1998) Statistical confidence for likelihood-based paternity inference in natural populations. Mol Ecol 7:639–655
Masso-Rojas A, Morales-Bojorquez E, Talavera-Mayer J, Fajardo-Leon M, Hernandez-Valenzuela R (2000) La Pesqueria de Almeja Mano de Leon, Baja California. In: Cisneros-Mata MA, Belendez-Moreno LF, Zarate-Becerra E, Gaspar-Dillanes MT, Lopez-Gonzales L, Saucedo-Ruiz C, Tovar-Avila J (eds) Sustentabilidada y pesca responsable en Mexico, evaluacion y manejo. Instituto Nacional de Pesca, Mexico, pp 349–366
McGoldrick DJ, Hedgecock D, English LJ, Baoprasertkul P, Ward RD (2000) The transmission of microsatellite alleles in Australian and North American stocks of the Pacific oyster (Crassostrea gigas): selection and null alleles. J Shellfish Res 19:779–788
Meirmans PG (2006) Using the AMOVA framework to estimate a standardized genetic differentiation measure. Evolution 60:2399–2402
Murphy RW, Aguirre-Leon G (2002) The non-avian reptiles: origins and evolution. In: Case TJ, Cody ML, Ezcurra E (eds) A new island biogeography of the Sea of Cortés. Oxford, New York, pp 181–220
Nylander JAA (2004) MrModeltest v2. Distributed by the author. Evolutionary Biology Centre, Uppsala Univ, Sweden
Ó Foighil D, Gaffney PM, Hilbish TJ (1995) Differences in mitochondrial 16S ribosomal gene sequences allow discrimination among American [Crassostrea virginica (Gmelin)] and Asian [C. gigas (Thunberg) C. ariakensis Wakiya] oyster species
Peakall R, Smouse PE (2006) GENALEX 6: genetic analysis in Excel. Population genetic software for teaching and research. Mol Ecol Notes 6:288–295
Petersen JL, Ibarra AM, Ramirez JL, May B (2008) An induced mass spawn of the hermaphroditic lion-paw scallop, Nodipecten subnodosus: genetic assignment of maternal and paternal parentage. J Hered 99:337–348
Petersen JL, Ibarra AM, May B (2009) Thirty-seven additional microsatellite loci in the Pacific lion-paw scallop (Nodipecten subnososus) and cross-amplification in other Pectinids. Conserv Genet Resour 1:101–105
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Puslednik L, Serb JM (2008) Molecular phylogenetics of the Pectinidae (Mollusca: Bivalvia) and effect of increased taxon sampling and outgroup selection on tree topology. Mol Phylogenet Evol 48:1178–1188
Queller DC, Goodnight KF (1989) Estimating relatedness using genetic markers. Evolution 43:258–275
Rambaut A (2009) Figtree v1.2.2. Institute of Evolution Biology, University of Edinburgh. Available at http://tree.bio.ed.ac.uk/software/figree
Rawson PD, Hilbish TJ (1995) Evolutionary relationships among the male and female mitochondrial DNA lineages in the Mytilus edulis species complex. Mol Biol Evol 12:893–901
Raymond M, Rousset F (1995) GENEPOP: population genetics software for exact tests and ecumenicism. J Hered 86:248–249
Reece KS, Ribeiro WL, Gaffney PM, Carnegie RB, Allen SK Jr (2004) Microsatellite marker development and analysis in the Eastern oyster (Crassostrea virginica): confirmation of null alleles and non-Mendelian segregation ratios. J Hered 95:346–352
Rice WR (1989) Analysing tables of statistical tests. Evolution 43:223–225
Riddle BR, Hafner DJ, Alexander LF, Jaeger JR (2000) Cryptic vicariance in the historical assembly of a Baja California Peninsular desert biota. PNAS 97:14438–14443
Riginos C (2005) Cryptic vicariance in Gulf of California fishes parallels vicariant patterns found in Baja California mammals and reptiles. Evolution 59:2678–2690
Ronquist F, Huelsenbeck JP (2003) MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19:1572–1574
Rose CG, Paynter KT, Hare MP (2006) Isolation by distance in the Eastern oyster, Crassostrea virginica, in Chesapeake Bay. J Hered 97:158–170
Rosenberg NA (2004) Distruct: a program for the graphical display of Structure results. Mol Ecol Notes 4:137–138
Ryman N, Laikre L (1991) Effects of supportive breeding on the genetically effective population size. Conserv Biol 5:325–329
Saavedra C, Peña JB (2004) Phylogenetic relationships of commercial European and Australasian king scallops (Pecten spp.) based on partial 16S ribosomal RNA gene sequences. Aquaculture 235:153–166
Saavedra C, Peña JB (2006) Phylogenetics of American scallops (Bivalvia: Pectinidae) based on partial 16S and 12S ribosomal RNA gene sequences. Mar Biol 150:111–119
Smith JT (1991) Cenozoic giant Pectinids from California and Tertiary Caribbean Provinces: Lyropecten, Macrochlamys, Vertipecten, and Nodipecten species. USGS, Washington DC, p 166
Stepien CA, Rosenblatt RH, Bargmeyer BA (2001) Phylogeography of the spotted sand bass, Paralabrax maculatofasciatus: divergence of Gulf of California and Pacific coast populations. Evolution 55:1852–1862
Swofford DL (2003) PAUP*: phylogenetic analysis using parsimony (* and other methods). Sinauer Associates, Inc., Sunderland
Szpiech ZA, Jakobsson M, Rosenberg NA (2008) ADZE: a rarefaction approach for counting alleles private to combinations of populations. Bioinformatics 24:2498–2504
Tamura K, Dudley J, Nei M, Kumar S (2007) MEGA4: molecular evolutionary genetics analysis (MEGA) software version 4.0. Mol Biol Evol 24:1596–1599
Terry A, Bucciarelli G, Bernardi G (2000) Restricted gene flow and incipient speciation in disjunct Pacific Ocean and Sea of Cortez populations of a reef fish species, Girella nigricans. Evolution 54:652–659
Tremblay MJ, Loder JW, Werner FE, Naimie CE, Page FH, Sinclair MM (1994) Drift of sea scallop larvae Placopecten magellanicus on Georges Bank: a model study of the roles of mean advection, larval behavior and larval origin. Deep Sea Res II 41:7–49
Upton DE, Murphy RW (1997) Phylogeny of the side-blotched lizards (Phrynosomatidae: Uta) based on mtDNA sequences: support for a midpeninsular seaway in Baja California. Mol Phylogenet Evol 8:104–113
Waples RS, Do C (2008) LDNE: a program for estimating effective population size from data on linkage disequilibrium. Mol Ecol Resour 8:753–756
Weir BS, Cockerham CC (1984) Estimating F-statistics for the analysis of population structure. Evolution 38:1358–1370
Williams GC (1975) Sex and evolution. Princeton University Press, Princeton
Yuan T, He M, Huang L (2009) Intraspecific genetic variation in mitochondrial 16S rRNA and COI genes in domestic and wild populations of Huaguizhikong scallop Chlamys nobilis Reeve. Aquaculture 289:19–25
Zhan A, Hu J, Hu X, Zhou Z, Hui M, Wang S, Peng W, Wang M, Bao Z (2009) Fine-scale population genetic structure of Zhikong scallop (Chlamys farreri): do local marine currents drive geographical differentiation? Mar Biotechnol 11:223–235
Zhdanova OL, Pudovkin AI (2008) Nb_HetEx: a program to estimate the effective number of breeders. J Hered 99:694–695
Zheng H, Zhang G, Guo X, Liu X (2008) Inbreeding depression for various traits in two cultured populations of the American bay scallop, Argopecten irradians irradians Lamark (1819) introduced into China. J Exp Mar Biol Ecol 364:42–47
Acknowledgments
UCMEXUS dissertation research fellowship, UC Davis Jastro-Shields, Hart-Cole-Goss Summer Fellowship, and UCD and Humanity Awards to J. L. P.; CONACYT-SEMARNAT S0010-2006-1, grant 23397 to A. M. I.; J. L. Ramirez (CIBNOR) for sample collection; J. Serb (Iowa State Univ) for providing DNA of N. subnodosus from Panama; Cesar Lodeiros for N. nodosus from Venezuela; B. Mahardja and E. Ringelman for laboratory assistance; A. M. Schreier and R. S. Schwartz and two anonymous reviewers provided helpful comments on earlier versions of the manuscript.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by C. Riginos.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Petersen, J.L., Ibarra, A.M. & May, B. Nuclear and mtDNA lineage diversity in wild and cultured Pacific lion-paw scallop, Nodipecten subnodosus (Baja California Peninsula, Mexico). Mar Biol 157, 2751–2767 (2010). https://doi.org/10.1007/s00227-010-1534-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00227-010-1534-1