
Abstract
Several rare inherited disorders have been described that show phenotypic overlap with Paget’s disease of bone (PDB) and in which PDB is a component of a multisystem disorder affecting muscle and the central nervous system. These conditions are the subject of this review article. Insertion mutations within exon 1 of the TNFRSF11A gene, encoding the receptor activator of nuclear factor kappa B (RANK), cause severe PDB-like disorders including familial expansile osteolysis, early-onset familial PDB and expansile skeletal hyperphosphatasia. The mutations interfere with normal processing of RANK and cause osteoclast activation through activation of nuclear factor kappa B (NFκB) independent of RANK ligand stimulation. Recessive, loss-of-function mutations in the TNFRSF11B gene, which encodes osteoprotegerin, cause juvenile PDB and here the bone disease is due to unopposed activation of RANK by RANKL. Multisystem proteinopathy is a disorder characterised by myopathy and neurodegeneration in which PDB is often an integral component. It may be caused by mutations in several genes including VCP, HNRNPA1, HNRNPA2B1, SQSTM1, MATR3, and TIA1, some of which are involved in classical PDB. The mechanisms of osteoclast activation in these conditions are less clear but may involve NFκB activation through sequestration of IκB. The evidence base for management of these disorders is somewhat limited due to the fact they are extremely rare. Bisphosphonates have been successfully used to gain control of elevated bone remodelling but as yet, no effective treatment exists for the treatment of the muscle and neurological manifestations of MSP syndromes.



Similar content being viewed by others

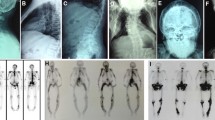

Change history
19 April 2021
A Correction to this paper has been published: https://doi.org/10.1007/s00223-021-00850-3
References
Osterberg PH, Wallace RG, Adams DA, Crone RS, Dickson GR, Kanis JA, Mollan RA, Nevin NC, Sloan J, Toner PG (1988) Familial expansile osteolysis. A new dysplasia. J Bone Joint Surg Br 70:255–260
Whyte MP, Mills BG, Reinus WR, Podgornik MN, Roodman GD, Gannon FH, Eddy MC, McAlister WH (2000) Expansile skeletal hyperphosphatasia: a new familial metabolic bone disease. J Bone Miner Res 15:2330–2344
Nakatsuka K, Nishizawa Y, Ralston SH (2003) Phenotypic characterization of early onset Paget’s disease of bone caused by a 27-bp duplication in the TNFRSF11A gene. J Bone Miner Res 18:1381–1385
Bakwin H, Eiger MS (1956) Fragile bones and macrocranium. J Pediatr 49:558–564
Schafer AL, Mumm S, El-Sayed I, McAlister WH, Horvai AE, Tom AM, Hsiao EC, Schaefer FV, Collins MT, Anderson MS, Whyte MP, Shoback DM (2014) Panostotic expansile bone disease with massive jaw tumor formation and a novel mutation in the signal peptide of RANK. J Bone Miner Res 29:911–921
Kim HJ, Kim NC, Wang YD, Scarborough EA, Moore J, Diaz Z, MacLea KS, Freibaum B, Li S, Molliex A, Kanagaraj AP, Carter R, Boylan KB, Wojtas AM, Rademakers R, Pinkus JL, Greenberg SA, Trojanowski JQ, Traynor BJ, Smith BN, Topp S, Gkazi AS, Miller J, Shaw CE, Kottlors M, Kirschner J, Pestronk A, Li YR, Ford AF, Gitler AD, Benatar M, King OD, Kimonis VE, Ross ED, Weihl CC, Shorter J, Taylor JP (2013) Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature 495:467–473
Gennari L, Rendina D, Falchetti A, Merlotti D (2019) Paget’s disease of bone. Calcif Tissue Int. https://doi.org/10.1007/s00223-019-00522-3
Enderle A, Willert HG (1979) Osteolytic-expansive type of familial Paget’s disease (osteitis deformans). Pathol Res Pract 166:131–139
Palenzuela L, Vives-Bauza C, Fernandez-Cadenas I, Meseguer A, Font N, Sarret E, Schwartz S, Andreu AL (2002) Familial expansile osteolysis in a large Spanish kindred resulting from an insertion mutation in the TNFRSF11A gene. J Med Genet 39:E67
Johnson-Pais TL, Singer FR, Bone HG, McMurray CT, Hansen MF, Leach RJ (2003) Identification of a novel tandem duplication in exon 1 of the TNFRSF11A gene in two unrelated patients with familial expansile osteolysis. J Bone Miner Res 18:376–380
Whyte MP, Reinus WR, Podgornik MN, Mills BG (2002) Familial expansile osteolysis (excessive RANK effect) in a 5-generation American kindred. Medicine (Baltimore) 81:101–121
Hughes AE, Shearman AM, Weber JL, Barr RJ, Wallace RG, Osterberg PH, Nevin NC, Mollan RA (1994) Genetic linkage of familial expansile osteolysis to chromosome 18q. Hum Mol Genet 3:359–361
Hughes AE, Ralston SH, Marken J, Bell C, MacPherson H, Wallace RG, Van Hul W, Whyte MP, Nakatsuka K, Hovy L, Anderson DM (2000) Mutations in TNFRSF11A, affecting the signal peptide of RANK, cause familial expansile osteolysis. Nat Genet 24:45–48
Anderson DM, Maraskovsky E, Billingsley WL, Dougall WC, Tometsko ME, Roux ER, Teepe MC, DuBose RF, Cosman D, Galibert L (1997) A homologue of the TNF receptor and its ligand enhance T-cell growth and dendritic-cell function. Nature 390:175–179
Simonet WS, Lacey DL, Dunstan CR, Kelley M, Chang MS, Luthy R, Nguyen HQ, Wooden S, Bennett L, Boone T, Shimamoto G, DeRose M, Elliott R, Colombero A, Tan HL, Trail G, Sullivan J, Davy E, Bucay N, Renshaw-Gegg L, Hughes TM, Hill D, Pattison W, Campbell P, Boyle WJ (1997) Osteoprotegerin: a novel secreted protein involved in the regulation of bone density. Cell 89:309–319 (see comments)
Crockett JC, Mellis DJ, Shennan KI, Duthie A, Greenhorn J, Scott DI, Ralston SH, Helfrich MH, Rogers MJ (2011) Signal peptide mutations in rank prevent downstream activation of NFkappaB. J Bone Miner Res 26:1926–1938
Albagha OME, Rojas J, van’t Hof RJ, Dorin J, Ralston SH (2007) A mouse model of early onset Paget’s disease of bone caused by an insertion mutation affecting the RANK signal peptide. Calcif Tissue Int 80:S34–S34
Pahl HL, Baeuerle PA (1997) The ER-overload response: activation of NF-kappa B. Trends Biochem Sci 22:63–67
Hughes AE, Barr RJ (1996) Familial expansile osteolysis: a genetic model of Paget’s disease. In: Sharpe PT (ed) The molecular biology of Paget’s disease. R.G.Landes Company, London, pp 179–199
Ke YH, Yue H, He JW, Liu YJ, Zhang ZL (2009) Early onset Paget’s disease of bone caused by a novel mutation (78dup27) of the TNFRSF11A gene in a Chinese family. Acta Pharmacol Sin 30:1204–1210
Whyte MP, Tau C, McAlister WH, Zhang X, Novack DV, Preliasco V, Santini-Araujo E, Mumm S (2014) Juvenile Paget’s disease with heterozygous duplication within TNFRSF11A encoding RANK. Bone 68:153–161
Chong B, Hegde M, Fawkner M, Simonet S, Cassinelli H, Coker M, Kanis J, Seidel J, Tau C, Tuysuz B, Yuksel B, Love D, Cundy T (2003) Idiopathic hyperphosphatasia and TNFRSF11B mutations: relationships between phenotype and genotype. J Bone Miner Res 18:2095–2104
Allen CA, Hart BL, Taylor CL, Clericuzio CL (2008) Bilateral cavernous internal carotid aneurysms in a child with juvenile paget disease and osteoprotegerin deficiency. AJNR Am J Neuroradiol 29:7–8
Polyzos SA, Cundy T, Mantzoros CS (2018) Juvenile Paget disease. Metabolism 80:15–26
Patnaik R, Baidya DK, Maitra S (2015) Unanticipated difficult intubation in a patient with juvenile Paget disease. J Clin Anesth 27:427–428
Gottesman GS, Madson KL, McAlister WH, Nenninger A, Wenkert D, Mumm S, Whyte MP (2016) Auricular ossification: a newly recognized feature of osteoprotegerin-deficiency juvenile Paget disease. Am J Med Genet A 170A:978–985
Whyte MP, Singhellakis PN, Petersen MB, Davies M, Totty WG, Mumm S (2007) Juvenile Paget’s disease: the second reported, oldest patient is homozygous for the TNFRSF11B “Balkan” mutation (966_969delTGACinsCTT), which elevates circulating immunoreactive osteoprotegerin levels. J Bone Miner Res 22:938–946
Whyte MP, Obrecht SE, Finnegan PM, Jones JL, Podgornik MN, McAlister WH, Mumm S (2002) Osteoprotegerin deficiency and juvenile Paget’s disease. N Engl J Med 347:175–184
Cundy T, Hegde M, Naot D, Chong B, King A, Wallace R, Mulley J, Love DR, Seidel J, Fawkner M, Banovic T, Callon KE, Grey AB, Reid IR, Middleton-Hardie CA, Cornish J (2002) A mutation in the gene TNFRSF11B encoding osteoprotegerin causes an idiopathic hyperphosphatasia phenotype. Hum Mol Genet 11:2119–2127
Grasemann C, Schundeln MM, Hovel M, Schweiger B, Bergmann C, Herrmann R, Wieczorek D, Zabel B, Wieland R, Hauffa BP (2013) Effects of RANK-ligand antibody (denosumab) treatment on bone turnover markers in a girl with juvenile Paget’s disease. J Clin Endocrinol Metab 98:3121–3126
Grasemann C, Unger N, Hovel M, Arweiler-Harbeck D, Herrmann R, Schundeln MM, Muller O, Schweiger B, Lausch E, Meissner T, Kiewert C, Hauffa BP, Shaw NJ (2017) Loss of functional osteoprotegerin: more than a skeletal problem. J Clin Endocrinol Metab 102:210–219
Naot D, Choi A, Musson DS, Simsek Kiper PO, Utine GE, Boduroglu K, Peacock M, DiMeglio LA, Cundy T (2014) Novel homozygous mutations in the osteoprotegerin gene TNFRSF11B in two unrelated patients with juvenile Paget’s disease. Bone 68:6–10
Saki F, Karamizadeh Z, Nasirabadi S, Mumm S, McAlister WH, Whyte MP (2013) Juvenile paget’s disease in an Iranian kindred with vitamin D deficiency and novel homozygous TNFRSF11B mutation. J Bone Miner Res 28:1501–1508
Janssens K, de Vernejoul MC, de Freitas F, Vanhoenacker F, Van Hul W (2005) An intermediate form of juvenile Paget’s disease caused by a truncating TNFRSF11B mutation. Bone 36:542–548
Middleton-Hardie C, Zhu Q, Cundy H, Lin JM, Callon K, Tong PC, Xu J, Grey A, Cornish J, Naot D (2006) Deletion of aspartate 182 in OPG causes juvenile Paget’s disease by impairing both protein secretion and binding to RANKL. J Bone Miner Res 21:438–445
Doyle FH, Woodhouse NJ, Glen AC, Joplin GF, MacIntyre I (1974) Healing of the bones in juvenile Paget’s disease treated by human calcitonin. Br J Radiol 47:9–15
Horwith M, Nunez EA, Krook L, Viteri F, Torun B, Mena E, Suh SM, Eisenberg E, MacIntyre I, Whalen JP (1976) Hereditary bone dysplasia with hyperphosphatasaemia: response to synthetic human calcitonin. Clin Endocrinol (Oxford) 5(Suppl):341S–352S
Woodhouse NJ, Fisher MT, Sigurdsson G, Joplin GF, MacIntyre I (1972) Paget’s disease in a 5-year-old: acute response to human calcitonin. Br Med J 4:267–269
Kraszeski JL, Avramides A, Wallach S, Hussain MN (1981) Three adult cases resembling hereditary bone dysplasia. Metab Bone Dis Relat Res 3:9–16
Demir E, Bereket A, Ozkan B, Topcu M (2000) Effect of alendronate treatment on the clinical picture and bone turnover markers in chronic idiopathic hyperphosphatasia. J Pediatr Endocrinol Metab 13:217–221
Spindler A, Berman A, Mautalen C, Ubios J, Santini AE (1992) Chronic idiopathic hyperphosphatasia. Report of a case treated with pamidronate and a review of the literature. J Rheumatol 19:642–645
Cassinelli HR, Mautalen CA, Heinrich JJ, Miglietta A, Bergada C (1992) Familial idiopathic hyperphosphatasia (FIH): response to long-term treatment with pamidronate (APD). Bone Miner 19:175–184
Cundy T, Wheadon L, King A (2004) Treatment of idiopathic hyperphosphatasia with intensive bisphosphonate therapy. J Bone Miner Res 19:703–711
Polyzos SA, Anastasilakis AD, Litsas I, Efstathiadou Z, Kita M, Arsos G, Moralidis E, Papatheodorou A, Terpos E (2010) Profound hypocalcemia following effective response to zoledronic acid treatment in a patient with juvenile Paget’s disease. J Bone Miner Metab 28:706–712
Polyzos SA, Singhellakis PN, Naot D, Adamidou F, Malandrinou FC, Anastasilakis AD, Polymerou V, Kita M (2014) Denosumab treatment for juvenile Paget’s disease: results from two adult patients with osteoprotegerin deficiency (“Balkan” mutation in the TNFRSF11B gene). J Clin Endocrinol Metab 99:703–707
Cundy T, Davidson J, Rutland MD, Stewart C, DePaoli AM (2005) Recombinant osteoprotegerin for juvenile Paget’s disease. N Engl J Med 353:918–923
McBride TI (1966) Paget’s disease and muscular dystrophy. Report of an unusual association in one family. Scott Med J 11:238–243
Tucker WS Jr, Hubbard WH, Stryker TD, Morgan SW, Evans OB, Freemon FR, Theil GB (1982) A new familial disorder of combined lower motor neuron degeneration and skeletal disorganization. Trans Assoc Am Physicians 95:126–134
Kimonis VE, Kovach MJ, Waggoner B, Leal S, Salam A, Rimer L, Davis K, Khardori R, Gelber D (2000) Clinical and molecular studies in a unique family with autosomal dominant limb-girdle muscular dystrophy and Paget disease of bone. Genet Med 2:232–241
Kovach MJ, Waggoner B, Leal SM, Gelber D, Khardori R, Levenstien MA, Shanks CA, Gregg G, Al Lozi MT, Miller T, Rakowicz W, Lopate G, Florence J, Glosser G, Simmons Z, Morris JC, Whyte MP, Pestronk A, Kimonis VE (2001) Clinical delineation and localization to chromosome 9p13.3-p12 of a unique dominant disorder in four families: hereditary inclusion body myopathy, Paget disease of bone, and frontotemporal dementia. Mol Genet Metab 74:458–475
Watts GD, Wymer J, Kovach MJ, Mehta SG, Mumm S, Darvish D, Pestronk A, Whyte MP, Kimonis VE (2004) Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin-containing protein. Nat Genet 36:377–381
Waggoner B, Kovach MJ, Winkelman M, Cai D, Khardori R, Gelber D, Kimonis VE (2002) Heterogeneity in familial dominant Paget disease of bone and muscular dystrophy. Am J Med Genet 108:187–191
Laurin N, Brown JP, Morissette J, Raymond V (2002) Recurrent mutation of the gene encoding sequestosome 1 (SQSTM1/p62) in Paget disease of bone. Am J Hum Genet 70:1582–1588
Hocking LJ, Lucas GJA, Daroszewska A, Mangion J, Olavesen M, Nicholson GC, Ward L, Bennett ST, Wuyts W, Van Hul W, Ralston SH (2002) Domain specific mutations in sequestosome 1 (SQSTM1) cause familial and sporadic Paget’s disease. Hum Mol Genet 11:2735–2739
Obaid R, Wani SE, Azfer A, Hurd T, Jones R, Cohen P, Ralston SH, Albagha OM (2015) Optineurin negatively regulates osteoclast differentiation by modulating NF-kappaB and interferon signaling: implications for Paget’s disease. Cell Rep 13:1096–1102
Taylor JP (2015) Multisystem proteinopathy: intersecting genetics in muscle, bone, and brain degeneration. Neurology 85:658–660
Benatar M, Wuu J, Fernandez C, Weihl CC, Katzen H, Steele J, Oskarsson B, Taylor JP (2013) Motor neuron involvement in multisystem proteinopathy: implications for ALS. Neurology 80:1874–1880
Al-Tahan S, Al-Obeidi E, Yoshioka H, Lakatos A, Weiss L, Grafe M, Palmio J, Wicklund M, Harati Y, Omizo M, Udd B, Kimonis V (2018) Novel valosin-containing protein mutations associated with multisystem proteinopathy. Neuromuscul Disord 28:491–501
Le Ber I, Van Bortel I, Nicolas G, Bouya-Ahmed K, Camuzat A, Wallon D, De Septenville A, Latouche M, Lattante S, Kabashi E, Jornea L, Hannequin D, Brice A, French research Network on FF-A (2014) hnRNPA2B1 and hnRNPA1 mutations are rare in patients with “multisystem proteinopathy” and frontotemporal lobar degeneration phenotypes. Neurobiol Aging 35:934 e935–e936
van den Boom J, Meyer H (2018) VCP/p97-Mediated Unfolding as a principle in protein homeostasis and signaling. Mol Cell 69:182–194
Rabinovich E, Kerem A, Frohlich KU, Diamant N, Bar-Nun S (2002) AAA-ATPase p97/Cdc48p, a cytosolic chaperone required for endoplasmic reticulum-associated protein degradation. Mol Cell Biol 22:626–634
Tresse E, Salomons FA, Vesa J, Bott LC, Kimonis V, Yao TP, Dantuma NP, Taylor JP (2010) VCP/p97 is essential for maturation of ubiquitin-containing autophagosomes and this function is impaired by mutations that cause IBMPFD. Autophagy 6:217–227
Brandman O, Stewart-Ornstein J, Wong D, Larson A, Williams CC, Li GW, Zhou S, King D, Shen PS, Weibezahn J, Dunn JG, Rouskin S, Inada T, Frost A, Weissman JS (2012) A ribosome-bound quality control complex triggers degradation of nascent peptides and signals translation stress. Cell 151:1042–1054
Kim NC, Tresse E, Kolaitis RM, Molliex A, Thomas RE, Alami NH, Wang B, Joshi A, Smith RB, Ritson GP, Winborn BJ, Moore J, Lee JY, Yao TP, Pallanck L, Kundu M, Taylor JP (2013) VCP is essential for mitochondrial quality control by PINK1/Parkin and this function is impaired by VCP mutations. Neuron 78:65–80
Meerang M, Ritz D, Paliwal S, Garajova Z, Bosshard M, Mailand N, Janscak P, Hubscher U, Meyer H, Ramadan K (2011) The ubiquitin-selective segregase VCP/p97 orchestrates the response to DNA double-strand breaks. Nat Cell Biol 13:1376–1382
Dai RM, Li CC (2001) Valosin-containing protein is a multi-ubiquitin chain-targeting factor required in ubiquitin-proteasome degradation. Nat Cell Biol 3:740–744
Buchan JR, Kolaitis RM, Taylor JP, Parker R (2013) Eukaryotic stress granules are cleared by autophagy and Cdc48/VCP function. Cell 153:1461–1474
Custer SK, Neumann M, Lu H, Wright AC, Taylor JP (2010) Transgenic mice expressing mutant forms VCP/p97 recapitulate the full spectrum of IBMPFD including degeneration in muscle, brain and bone. Hum Mol Genet 19:1741–1755
Weihl CC, Miller SE, Hanson PI, Pestronk A (2007) Transgenic expression of inclusion body myopathy associated mutant p97/VCP causes weakness and ubiquitinated protein inclusions in mice. Hum Mol Genet 16:919–928
Badadani M, Nalbandian A, Watts GD, Vesa J, Kitazawa M, Su H, Tanaja J, Dec E, Wallace DC, Mukherjee J, Caiozzo V, Warman M, Kimonis VE (2010) VCP associated inclusion body myopathy and paget disease of bone knock-in mouse model exhibits tissue pathology typical of human disease. PLoS ONE. https://doi.org/10.1371/journal.pone.0013183
Weihl CC, Dalal S, Pestronk A, Hanson PI (2006) Inclusion body myopathy-associated mutations in p97/VCP impair endoplasmic reticulum-associated degradation. Hum Mol Genet 15:189–199
Ju JS, Fuentealba RA, Miller SE, Jackson E, Piwnica-Worms D, Baloh RH, Weihl CC (2009) Valosin-containing protein (VCP) is required for autophagy and is disrupted in VCP disease. J Cell Biol 187:875–888
Ritson GP, Custer SK, Freibaum BD, Guinto JB, Geffel D, Moore J, Tang W, Winton MJ, Neumann M, Trojanowski JQ, Lee VM, Forman MS, Taylor JP (2010) TDP-43 mediates degeneration in a novel Drosophila model of disease caused by mutations in VCP/p97. J Neurosci 30:7729–7739
Mahboubi H, Stochaj U (2017) Cytoplasmic stress granules: dynamic modulators of cell signaling and disease. Biochim Biophys Acta Mol Basis Dis 1863:884–895
Buchberger A, Schindelin H, Hanzelmann P (2015) Control of p97 function by cofactor binding. FEBS Lett 589:2578–2589
Salajegheh M, Pinkus JL, Taylor JP, Amato AA, Nazareno R, Baloh RH, Greenberg SA (2009) Sarcoplasmic redistribution of nuclear TDP-43 in inclusion body myositis. Muscle Nerve 40:19–31
Bucelli RC, Arhzaouy K, Pestronk A, Pittman SK, Rojas L, Sue CM, Evila A, Hackman P, Udd B, Harms MB, Weihl CC (2015) SQSTM1 splice site mutation in distal myopathy with rimmed vacuoles. Neurology 85:665–674
Purice MD, Taylor JP (2018) Linking hnRNP function to ALS and FTD pathology. Front Neurosci 12:326
Ditlev JA, Case LB, Rosen MK (2018) Who’s in and who’s out-compositional control of biomolecular condensates. J Mol Biol 430:4666–4684
Fecto F, Siddique T (2012) UBQLN2/P62 cellular recycling pathways in amyotrophic lateral sclerosis and frontotemporal dementia. Muscle Nerve 45:157–162
Le Ber I, Camuzat A, Guerreiro R, Bouya-Ahmed K, Bras J, Nicolas G, Gabelle A, Didic M, De Septenville A, Millecamps S, Lenglet T, Latouche M, Kabashi E, Campion D, Hannequin D, Hardy J, Brice A, French C, Genetic Research Network on FF-A (2013) SQSTM1 mutations in French patients with frontotemporal dementia or frontotemporal dementia with amyotrophic lateral sclerosis. JAMA Neurol 70:1403–1410
Teyssou E, Takeda T, Lebon V, Boillee S, Doukoure B, Bataillon G, Sazdovitch V, Cazeneuve C, Meininger V, LeGuern E, Salachas F, Seilhean D, Millecamps S (2013) Mutations in SQSTM1 encoding p62 in amyotrophic lateral sclerosis: genetics and neuropathology. Acta Neuropathol 125:511–522
Lee Y, Jonson PH, Sarparanta J, Palmio J, Sarkar M, Vihola A, Evila A, Suominen T, Penttila S, Savarese M, Johari M, Minot MC, Hilton-Jones D, Maddison P, Chinnery P, Reimann J, Kornblum C, Kraya T, Zierz S, Sue C, Goebel H, Azfer A, Ralston SH, Hackman P, Bucelli RC, Taylor JP, Weihl CC, Udd B (2018) TIA1 variant drives myodegeneration in multisystem proteinopathy with SQSTM1 mutations. J Clin Invest 128:1164–1177
Mehta SG, Watts GD, Adamson JL, Hutton M, Umberger G, Xiong S, Ramdeen S, Lovell MA, Kimonis VE, Smith CD (2007) APOE is a potential modifier gene in an autosomal dominant form of frontotemporal dementia (IBMPFD). Genet Med 9:9–13
Albagha OM, Visconti MR, Alonso N, Langston AL, Cundy T, Dargie R, Dunlop MG, Fraser WD, Hooper MJ, Isaia G, Nicholson GC, Del Pino MJ, Gonzalez-Sarmiento R, Di SM, Tenesa A, Walsh JP, Ralston SH (2010) Genome-wide association study identifies variants at CSF1, OPTN and TNFRSF11A as genetic risk factors for Paget’s disease of bone. Nat Genet 42:520–524
Maruyama H, Morino H, Ito H, Izumi Y, Kato H, Watanabe Y, Kinoshita Y, Kamada M, Nodera H, Suzuki H, Komure O, Matsuura S, Kobatake K, Morimoto N, Abe K, Suzuki N, Aoki M, Kawata A, Hirai T, Kato T, Ogasawara K, Hirano A, Takumi T, Kusaka H, Hagiwara K, Kaji R, Kawakami H (2010) Mutations of optineurin in amyotrophic lateral sclerosis. Nature 465:223–226
Rezaie T, Child A, Hitchings R, Brice G, Miller L, Coca-Prados M, Heon E, Krupin T, Ritch R, Kreutzer D, Crick RP, Sarfarazi M (2002) Adult-onset primary open-angle glaucoma caused by mutations in optineurin. Science 295:1077–1079
Funding
This work was is supported in part by an Advanced Investigator Award from the European Commission to SHR (787270) and by grants from the National Institutes of Health (R35 NS097974) and the Howard Hughes Medical Institute to JPT.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Prof. Stuart H. Ralston reported receiving grant funding to his institution from Abbvie, Amgen, Eli Lilly, and Pfizer, and reported receiving consultancy funding to his institution from Novartis and Merck. Dr. J. Paul Taylor has no interests to declare.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Ralston, S.H., Taylor, J.P. Rare Inherited forms of Paget’s Disease and Related Syndromes. Calcif Tissue Int 104, 501–516 (2019). https://doi.org/10.1007/s00223-019-00520-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00223-019-00520-5