
Abstract
Elevated serum levels of the phosphate-regulating hormone fibroblast growth factor 23 (FGF23) are found in patients with phosphate wasting diseases and chronic kidney disease-mineral and bone disorder (CKD-MBD). These diseases are associated with rickets and renal osteodystrophy, respectively. FGF23 is secreted from osteoblastic cells and signals through FGFRs, membrane coreceptor alpha-Klotho (Klotho), and, possibly, a circulating form of Klotho. Despite the absence of detectable Klotho on osteoblastic cells, studies have suggested that forced FGF23 expression in osteoblasts inhibited mineralization. Thus, we examined the effects of exogenously applied FGF23 on osteoblastic MC3T3.E1 cell proliferation and differentiation, with and without soluble Klotho. MC3T3.E1 cells were cultured in osteoblast differentiation medium, supplemented with FGF23 (0.1–1,000 ng/mL), Klotho (50 ng/mL), the combination FGF23 + Klotho, and FGF2 (100 ng/mL) as a control. Neither FGF23 nor Klotho exposure affected proliferation of day 4 growth phase cells or mineralization of day 14 cultures. In contrast, FGF23 + Klotho resulted in inhibition of mineralization and osteoblast activity markers at day 14, and a slight, reproducible induction of proliferation. Inhibition of FGFR1, but not FGFR2 or FGFR3, completely restored FGF23 + Klotho-induced inhibition of alkaline phosphatase (ALP) activity at day 7. ALP activity was partially restored by the MAPK inhibitor U0126 but not inhibitors p38 and P13K. Thus, soluble Klotho enables FGF23 signaling in MC3T3.E1 cells, likely through FGFR 1(IIIc). Elevated FGF23 actions, in part, appear to parallel FGF2 with lower potency. In addition to affecting bone via indirect phosphate wasting pathways, supraphysiological FGF23 and soluble Klotho may directly impact bone in diseases with elevated FGF23 levels.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Fibroblast growth factor 23 (FGF23) is a phosphate-regulating hormone synthesized primarily in osteoblast lineage cells in bone and secreted into the circulation [1]. FGF23 acts by preventing the reabsorption of phosphate from the renal tubule, leading to increased phosphate excretion, and by limiting dietary absorption of phosphate through inhibition of renal 25-hydroxyvitamin D3, 1α-hydroxylase (1αOHase) [2–7]. In diseases associated with phosphate imbalance, serum levels of FGF23 are pathologically altered with elevations in phosphate wasting diseases, such as autosomal-dominant hypophosphatemic rickets (ADHR) [8], X-linked hypophosphatemic rickets (XLH) and tumor-induced osteomalacia (TIO) [9, 10], and autosomal-recessive hypophosphatemic rickets (ARHR) [11], as well as deficiencies in diseases of phosphate retention, such as tumoral calcinosis (TC) [12]. In addition, dialysis patients with secondary hyperparathyroidism (HPT) often have serum FGF23 levels several thousandfold higher than healthy individuals [3, 4, 7].
The elevated serum FGF23 levels in chronic kidney disease-mineral and bone disorder (CKD-MBD) patients may represent a compensatory response to increased serum phosphorus load due to kidney failure. However, in addition to sustained dietary phosphate load, FGF23 is upregulated by regulators of bone metabolism, including 1,25-dihydroxyvitamin D3 (1,25[OH]2D3) [13] and parathyroid hormone (PTH) [14], which are known to be altered in patients with CKD-MBD. It is well appreciated that sustained hormonal and mineral imbalances in patients with CKD contribute to the development of renal osteodystrophy (ROD), a severe bone disorder [15]. A significant amount of data indicates that alterations in PTH contribute to the disease process, whereas the role of FGF23 in the pathogenesis of CKD-MBD is much less clear. Others have demonstrated that FGF23 expression is increased in bone of a CKD-MBD rodent model [16]. Further, FGF23 in serum from patients with advanced CKD-MBD on dialysis was found to be intact and biologically active [17], suggesting that circulating FGF23 in CKD-MBD patients impacts tissues expressing the appropriate receptors.
FGF23 is a member of the endocrine FGF19 family, which, like other FGFs, signals through FGF receptors (FGFRs) [18, 19]. However, due to its atypical heparin binding domain, FGF23 binds FGFRs with low affinity [19], except in the presence of the coreceptor alpha-Klotho (Klotho), which confers high-affinity binding [20]. FGFRs are encoded by distinct genes—FGFRs 1, 2, 3, and 4—and each is characterized by a tyrosine kinase intracellular domain, a transmembrane domain, and three extracellular immunoglobulin domains (D1–D3). Several isoforms with different ligand specificities are generated by alternative splicing events. A splicing event in FGFRs 1, 2, and 3 excludes exon 8 or 9 in the third immunoglobulin domain (III) generating FGFR “c” and “b” forms, respectively. In cells transfected with different FGFRs and Klotho, FGF23 signaling occurred preferentially through the FGFR1(IIIc) isoform [21], suggesting that FGFR1(IIIc) is the predominant physiological FGF23 receptor.
Klotho is a single-pass transmembrane protein with a short intracellular domain found on FGF23 target tissues (such as kidney and parathyroid glands) [22, 23]. Klotho contains a short intracellular domain and two extracellular domains with similarity to glycosidases (KL1 and KL2). Two circulating forms of Klotho have been described in mice and humans. These two forms are generated by different processes: one involving an alternative RNA splicing event (to generate a 68-kDa, 549-aa KL1 domain containing protein) and the other involving a proteolytic cleavage of cell surface Klotho (to yield predominantly the entire 130-kDa extracellular KL1- and KL2-containing domain of Klotho) [24]. The soluble extracellular domain of Klotho is functional as a coreceptor for FGF23 in vitro, though it is less active than the membrane form [20].
Despite the absence of detectable Klotho mRNA expression in bone or bone marrow [23], forced FGF23 expression in rat calvarial cells suggested that supraphysiological FGF23 levels directly inhibit osteoblast mineralization [25]. Other studies using cell lines or forced FGFR expression demonstrated that supraphysiological levels of FGF23 and heparin/heparin sulfate glycosaminoglycans inhibit phosphate transport via the mitogen-activated protein kinase (MAPK) pathway in opossum kidney (OK) epithelial cells [26] or induced proliferation in FGFR-transfected murine bone marrow-derived pro-B (BaF3) cells [19]. These studies raise the possibility that FGF23 may have an autocrine/paracrine effect on osteoblastic cells in bone or other cells and signal in the absence of membrane Klotho.
To determine the impact of elevated FGF23 (as seen in patients with CKD-MBD) on MC3T3.E1 cells, which are derived from mouse calvarial osteoblasts, we examined the direct effects of FGF23 on MC3T3.E1 proliferation, differentiation, and cell signaling pathways in the presence and absence of soluble Klotho. In addition, we examined the effects of FGF23 on primary human proximal tubule epithelial cells (PTECs).
Methods
Cells and Reagents
Renal PTECs and culture medium were obtained from Lonza Bioscience (Walkersville, MD). Oversulfated heparin (OS-H, GT6020) was purchased from Neoparin (Alameda, CA). Antibodies for phospho-extracellular signal-regulated kinase (p-ERK1/2), phospho-p38 (p-p38) MAPK, and phosphorylated tyrosine (pTYR) were from Biosource/Invitrogen (Carlsbad, CA); ERK1/2 and p38 were from Upstate Biotech/Millipore (Temecula, CA); early growth response-1 protein (Egr-1) was from Santa Cruz Biotechnology (Santa Cruz, CA); and epidermal growth factor (EGF) was from R&D Systems (Minneapolis, MN). Human FGF23 wild-type (LO80106, stock concentration 1.7 mg/mL) and mutant R176Q (LO5316, stock 1.6 mg/mL) were made at Amgen (Thousand Oaks, CA). FGF2 (basic FGF) (lot BUE3108101, stock 100 μg/mL) was obtained from R&D Systems. The p38 inhibitor SB203580 (PHZ1253, lot 72547179A, stock 50 mg/mL, 132 mM) was from Invitrogen; the phosphoinositide 3-kinase (P13 K) inhibitor LY294002 (9901, lot 9, stock 15.3 mg/mL, 50 mM) was from Cell Signaling (Hayward, CA); FGFR2(IIIc) neutralizing antibody (MAB716, lot FSQ02, stock 5 mg/mL, 33 μM (ND50 = 0.67–2.7 nM) and FGFR3 neutralizing antibody (MAB710, lot FTD02, stock 5 mg/mL, 33 μM (ND50 = 20–80 nM) were from R&D Systems. The FGFR1 tyrosine kinase inhibitor SU 5402 and the MAPK pathway ERK1/2 inhibitor U0126 were from Signaling Technology (Beverly, MA).
Treatment of Cells
PTECs were cultured on 12- or 48-well plates at 1 to 2 × 106 cells/mL in 1 or 0.5 mL, respectively. Cells were typically ≤50% confluent at the time of treatments. Cells were exposed to 0–1,000 ng/mL human FGF23-wild-type or mutant (huFGF23R176Q) + OS-H (4 μg/mL) or Klotho (1 μg/mL).
Western Blot
Treated PTECs were harvested on ice in phosphate-buffered saline (PBS) and centrifuged. Cell pellets were resuspended in an appropriate volume of lysis buffer (50 mM Tris–HCl [pH 7.4], 50 mM NaCl, 5 mM EDTA, 1% Triton X-100, 0.05% SDS, 50 mM NaF, 10 mM β-glycerophosphate, 10 mM sodium pyrophosphate, 100 μM NaVO4, protease inhibitor cocktail; Roche Applied Science, Indianapolis, IN) and sonicated. After centrifugation at 4°C, the supernatant was removed and the protein concentration determined (BCA protein assay reagent; Pierce, Rockford, IL). An equal amount of total protein of each sample was resolved by SDS-PAGE and transferred onto nitrocellulose membrane (Invitrogen). Blots were blocked for 1 hour in Tris-buffered saline-Tween (TBST; 20 mM Tris–HCl [pH 7.6], 137 mM NaCl, 0.1% Tween-20) containing 5% milk (blocking buffer) before incubation with primary antibodies for 2 hours at room temperature. After three washes with TBST, blots were incubated with peroxidase-conjugated secondary antibodies (Amersham/GE Healthcare, Piscataway, NJ). Antibody binding was detected using enhanced chemiluminescence (Amersham/GE Healthcare).
p-ERK1/2 and ERK1/2 Detection
PTECs grown in 12-well plates for 36 hours were serum-deprived overnight and exposed to huFGF23 for 15 minutes, 1 hour, or 6 hours, depending on the experiment, followed by Western blot analysis. For p-ERK detection, blots were incubated with primary antibody (1:1,000 dilution) for 2 hours at room temperature, then secondary horseradish peroxidase-conjugated antibody (1:4,000). The signal was detected using enhanced chemiluminescence (Amersham/GE Healthcare) and BIOMAX AR film (Sigma-Aldrich, St. Louis, MO). To detect total ERK on the same blots, the blots were stripped with Western blot stripping buffer (Pierce) for 15 minutes with shaking. Filters were blocked for 1 hour, followed by incubation with primary antibody (1 hour) and secondary antibody (1 hour) and developed.
Egr-1 Detection
PTECs were grown in 12-well plates for 48 hours, pretreated with serum-free medium for 2 hours, and treated with huFGF23 overnight. Cell lysates were analyzed by Western blot.
Phosphotyrosine Detection
PTECs were grown and pretreated as for p-ERK1/2 and ERK1/2 and exposed to huFGF23 for 15 minutes, followed by Western blot analysis. Lysates were incubated with pTYR antibody (PY20).
Human FGF23R176Q
Both wild type FGF23 and huFGF23R176Q were expressed in a Chinese hamster ovary (CHO) cell line. Cells were transfected with Lipofectamine 2000 and selected in the absence of HT. For protein production, cells were placed in serum-free medium, and medium was harvested at 1 week. Wild type FGF23 and huFGF23R176Q were purified by cation exchange chromatography, followed by hydrophobic interaction chromatography (HIC). The purified protein was formulated in PBS.
Soluble Murine Klotho
Soluble murine Klotho (1-982) 6X-His, comprising the entire extracellular domain, was expressed in a CHO cell line. Top clones were selected, and production was performed under serum-free conditions for 7 days. Klotho was purified by immobilized metal ion affinity chromatography (IMAC), followed by preparative size exclusion chromatography (SEC) on S200 (GE Healthcare). Purified Klotho was formulated in PBS.
CHO-Klotho ELK-1 Luciferase Reporter Cells
Full-length murine Klotho was stably expressed in a CHO cell line. Klotho expression was enriched through FACS sorting with an anti-Klotho antibody (R&D Systems, AF1819). These cells were then transfected with the ELK-1 reporter luciferase reporter vector (Stratagene, Cedar Creek, TX), serum-deprived overnight, and treated for 4 hours with huFGF23R176Q. Luciferase reporter activity was assayed.
Cells and Treatment
MC3T3.E1 cells were cultured (2,500 cells/well in 96-well plate or 12,500 cells/well in 24-well plate) in differentiation medium—alpha-minimal essential medium (MEM)/10% fetal bovine serum (FBS)/ascorbic acid (50 μg/mL)/β-glycerolphosphate (BGP, 10 mM). Proliferation was performed on day 4 cultures; alkaline phosphatase (ALP) activity on day 7 cultures; and calcium accumulation, RNA isolation, von Kossa, and ALP staining on day 14 cultures. The day after plating, cells received fresh medium supplemented with huFGF23R176Q (0.1–1,000 ng/mL), murine Klotho (5, 50, and 500 ng/mL), combination of huFGF23R176Q and Klotho, or FGF2 (100 ng/mL) as a control. Culture medium with and without treatments was replaced every 48 hours. For effects of FGFR and MAPK pathway inhibitors for p38 (SB2035800), P13K (LY294002), FGFR2(IIIc) neutralizing antibody (MAB716), FGFR3 neutralizing antibody (MAB710), MC3T3.E1/BF cells were grown and treated as above, then exposed to the following: medium alone, medium/dimethysulfate (DMSO) vehicle control, Klotho (50 ng/mL), huFGF23R176Q (1,000 ng/mL), Klotho (50 ng/mL) + huFGF23R176Q (1,000 ng/mL) with and without different concentrations of inhibitors (five- or 10-fold dilutions) ranging from 0.7 nM to 7 μM for p38 inhibitor (SB203580), 0.014 to 140 μM for PI3-kinase inhibitor (LY294002), 0.05 to 500 nM for FGFR2(IIIc) neutralizing antibody (MAB 716), 0.2 nM to 2 μM for FGFR3 neutralizing antibody, 0.65 nM to 6.5 μM for FGFR1 tyrosine kinase inhibitor (SU5402), and 0.001 to 10 μM for ERK1/2 inhibitor (UO126).
Proliferation Assay for MC3T3.E1 Cells
Proliferation was determined on day 4 growth phase cells (FluoReporter Blue Fluorometric dsDNA Quantitation Kit, Invitrogen). Briefly, medium was replaced with distilled water (100 μL) and cells were incubated at 37°C for 1 hour and lysed by freeze-thawing the plate. Hoechst dye mix (100 μL) was added to each well, and contents were mixed thoroughly. Fluorescence was measured at 360 nm for excitation and at 460 nm for emission. Experiments were performed at least six times.
Calcium Accumulation
On day 14, calcium accumulation (Stanbio Laboratories, Boerne, TX) was determined by acid extraction and normalized to total protein (Coomassie Plus Reagent, Pierce) from the same well. Briefly, after washing cells (10 mM Tris–HCl, pH 7.4), 0.5 N HCl was added to wells, and the plate was freeze-thawed three times to lyse cells. Plates were shaken for 15 minutes to mix contents and then centrifuged at 2,000 rpm at 4°C to pellet debris. Calcium content was determined by adding an aliquot of supernatant to the o-Cresolphthalein Complexone-containing working solution. The calcium-Cresolphthalein Complexone Complex formed a fuchsia color, which was read at 575 nM. The experiment was performed at least six times.
von Kossa Staining, ALP Staining, and ALP Activity
Mineral deposition was visualized by the von Kossa staining method, which involved incubating cell layers in a solution of silver nitrate (3%) under UV light for 30 minutes. ALP staining was performed by incubating cells for 30 minutes at 37°C in Tris/glycine buffer (pH 8.4) containing disodium naphthol AS-MX phosphate (Sigma) and fast red TR salt (Sigma-Aldrich). After washing cell layers (10 mM Tris, HCl [pH 7–8]), cells were supplemented with glycine buffer (0.1 M glycine, 0.001 MgCl2 [pH 10.5]) and frozen and thawed twice to obtain cell lysates to determine ALP activity. Lysates were incubated with p-nitrophenyl phosphate disodium hexahydrate (PNPP, Sigma-Aldrich) solution for 30 minutes at 37°C. Para-nitrophenol was detected at 405 nM.
RNA Analysis
MC3T3.E1 cells were cultured as above in differentiation medium supplemented with FGF23 (1,000 ng/mL), Klotho (50 ng/mL), the combination huFGF23R176Q (1 or 1,000 ng/mL) + Klotho (50 ng/mL), or FGF2 (100 ng/mL) for 14 days. Total cellular RNA was isolated using STAT 60 (Teltest, Friendswood, TX). RNA integrity was confirmed on a Bioanalyzer (Agilent Technologies, Palo Alto, CA). B-DNA was performed on purified total cellular RNA using the Quantigene Screen Kit (Genospectra, Fremont, CA) following the manufacturer’s protocol (gene panel 1: FGFR 1b, 1c, 2b, 2c, 3c, and 4; gene panel 2: ALP, osteocalcin (OC), Runx2, type I collagen (a1) (COL1A1), bone sialoprotein (IBSP), osteopontin (SPP1). The experiment was performed two times.
Statistics
Statistical analyses were performed using GraphPad Prism (GraphPad, San Diego, CA). Significance was determined by one-way analysis of variance (ANOVA) followed by Dunnett’s post-test. All values represent mean ± standard error of mean (SEM).
Results
FGF23 and Soluble Klotho Can Elicit Signaling in Proximal Tubular Cells
To validate the functionality of human CHO-derived huFGF23R176Q and murine soluble Klotho, we examined the signaling response in a reporter cell, CHO-KL-ELK-1, and a model FGF23-target cell, normal human PTECs. A dose-dependent increase in luciferase activity in response to huFGF23R176Q (EC50 1.60 ng/mL) was demonstrated in CHO-KL-ELK-1 cells (Fig. 1a) but not CHO-KL cells or parental CHO cells (not shown). In PTECs, which were found to require exogenous OS-H or soluble Klotho for signaling, both wild-type and huFGF23R176Q dose-dependently increased p-ERK1/2 in the presence of OS-H (4 μg/mL). Wild type and huFGF23R176Q increased p-p38 and pTYR activity to a lesser extent (Fig. 1b, c). HuFGF23R176Q induced greater activity than wild-type FGF23, consistent with the increased stability of this mutation found in ADHR patients. In PTECs, increased p-ERK1/2 and pTYR activity were associated with upregulation of Egr-1, a signaling molecule downstream of FGF23 (Fig. 1d). In the presence of soluble Klotho (1 μg/mL) and huFGF23R176Q (100 ng/mL), PTECs exhibited enhanced p-ERK signaling compared to huFGF23R176Q + OS-H or Klotho alone (Fig. 2), confirming the functionality of murine soluble Klotho.

a FGF23 dose response in CHO-KL and ELK-1 luciferase reporter cells. Cells were serum-deprived overnight and exposed to huFGF23R176Q for 4 hours. Luciferase reporter activity was assayed. EC50 was 1.60 ng/mL. ***P < 0.001 compared with FGF23 (0.1 ng/mL). RLU, relative luminescent units. Values represent mean ± SEM. b FGF23 signals in PTECs. Supraphysiological levels of FGF23 and OS-H (4 μg/mL) increase p-ERK1/2 and p-p38 in human PTECs in vitro. PTECs were plated at 2 × 106 cells per 24-well plate and treated when <50% confluent with EGF (10 ng/mL), human wild type FGF23 or huFGF23R176Q (0–1,000 ng/mL) for 15 minutes, and Western blot was performed for total, p-ERK1/2, and p-p38 expression. c Supraphysiological levels of FGF23 and OS-H (4 μg/mL) increase tyrosine phosphorylation (pTYR) in human PTECs in vitro. PTECs were exposed to human wild-type FGF23 and huFGF23R176Q (1–1,000 ng/mL) for 15 minutes, followed by Western blot analysis with pTYR antibody (PY20). d Supraphysiological levels of huFGF23R176Q and OS-H (4 μg/mL) increase Egr-1 levels in human PTECs in vitro. PTECs were cultured in 12-well plates for 48 hours and serum-deprived for 2 hours, followed by exposure to huFGF23R176Q (0–1,000 ng/mL) in serum-free medium overnight. Cell lysates were analyzed by Western blot

Supraphysiological levels of FGF23 (100 ng/mL) and soluble Klotho (1 μg/mL) activate p-ERK1/2 in human PTECs in vitro. PTECs were cultured as above and exposed to control medium (PBS/bovine serum albumin), huFGF23R176Q (100 ng/mL) and soluble Klotho (1 μg/mL), huFGF23R176Q (100 ng/mL) + OS-H (4 μg/mL), PTH(1-34) (50 pM), and EGF (10 ng/mL) for 15 minutes, followed by Western blot analysis
FGF23 and Soluble Klotho Inhibit Mineralization in an Osteoblast Cell Line
To assess the effects of supraphysiological levels of serum FGF23, as seen in CKD-MBD patients, on proliferation and differentiation of osteoblastic cells, MC3T3.E1 cells were exposed to huFGF23R176Q, Klotho, or their combination. Abundant mineral was deposited over positive ALP-stained nodular areas in cultures exposed to lower concentrations (<100 ng/mL huFGF23R176Q + 50 ng/mL Klotho); however, exposure to higher concentrations (≥100 ng/mL huFGF23R176Q + 50 ng/mL Klotho) led to complete inhibition of osteoblastic differentiation, as demonstrated by absence of red ALP staining and associated black mineral deposits. Calcium quantification confirmed lack of mineral deposition (Fig. 3). Neither huFGF23R176Q nor Klotho exposure alone affected proliferation or calcium accumulation. Proliferation was stimulated 2- to 2.5-fold by FGF2 (Fig. 3a), which also inhibited mineral deposition (Fig. 3b–e), consistent with reports of FGF2 actions on osteoblastic cells in culture [27, 28]. Coadministration of huFGF23R176Q and Klotho, consistently and dose-dependently, induced a low level of proliferation (maximum of 1.6-fold at 100 and 1,000 ng/mL huFGF23R176Q + 50 ng/mL Klotho) associated with an inhibition of mineralization (Fig. 3). Consistent with the absence of mineralization, the expression of osteoblast differentiation markers ALP, OC, COL1A1, IBSP, and SPP1 was also downregulated in MC3T3.E1 cultures exposed to huFGF23R176Q + Klotho (Fig. 4). Similar results were found for FGF2. Cells exposed to huFGF23R176Q and OS-H or OS-H alone did not survive (data not shown), consistent with the activities of high levels of heparin on osteoblastic cells [29].

a FGF23 + Klotho, not FGF23 or Klotho, inhibited calcium accumulation in MC3T3.E1 cells. Cells were cultured in 96-well plates at 2,500/well in differentiation medium (alpha-MEM/10% FBS/ascorbic acid [50 μg/mL]/BGP [10 mM]) and supplemented with huFGF23R176Q (F), murine Klotho (KL), their combination, or FGF2 (basic FGF, bFGF) (100 ng/mL) for 14 days. Cell layers were solubilized with 0.5 N HCl, and calcium content was quantified, normalized to the total protein content in the same well, and expressed as micrograms per milligram protein. b Calcium accumulation per well (no normalization with protein). c Total protein per well. d HuFGF23R176Q + Klotho promoted a modest increase in MC3T3.E1 cell proliferation. Proliferation assays were performed on day 4 preconfluent cells. Bars represent mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001 compared with differentiation medium (Diff. Medium). e Dose-dependent inhibition of mineralization in the presence of the high concentrations of huFGF23R176Q (100 and 1,000 ng/mL) and Klotho (5, 50, and 500 μg/mL). Cells were grown as above on 24-well plates and stained for ALP or by the von Kossa method to detect black mineral deposits. These results are representative of six experiments

Osteoblast marker mRNA expression was inhibited in FGF23 + Klotho-treated MC3T3.E1 cells (B-DNA analysis). Cells were cultured in 12-well plates at 12,500/well in differentiation medium (alpha-MEM/10% FBS/ascorbic acid [50 μg/mL]/BGP [10 mM]) and supplemented with huFGF23R176Q (F) (1 and 1,000 ng/mL), murine Klotho (KL) (50 ng/mL), their combination, or FGF2 (bFGF) for 14 days. Gene panel ALP, OC, Runx2/Cbfa1, COL1A1, IBSP, and SPP1. Respective mRNA levels were normalized to cyclophilin mRNA levels. Bars represent mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001 compared with differentiation medium (Diff. Medium)
FGF23 and Soluble Klotho Signal Through FGFR1 and MAPK Pathway in MC3T3 Cells
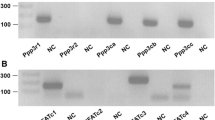
To identify potential underlying mechanisms, we sought to determine the FGFRs and signaling pathways involved in the FGF23- and Klotho-induced effects on proliferation and mineralization of MC3T3.E1 cells. MC3T3.E1 cells preferentially expressed FGFR1(IIIc), FGFR2(IIIc), and FGFR3(IIIc) isoforms, whereas the FGFR(IIIb) isoform and FGFR4 were undetectable (Fig. 5a). HuFGF23R176Q + Klotho treatment upregulated FGFR1(IIIc) and FGFR3(IIIc) and downregulated FGFR2(IIIc), whereas FGF2 upregulated FGFR1(IIIc) and downregulated both FGFR2(IIIc) and FGFR3(IIIc) (Fig. 5b). Consistent with previous reports of the absence of Klotho in bone cells [21], we did not detect Klotho transcripts in rat bone sections by in situ hybridization; however, Klotho transcripts were readily visible on kidney distal tubules with antisense, but not sense, probes (not shown). Further, we did not detect significant Klotho expression on the surface of MC3T3.E1 cells by FACS analysis with anti-Klotho antibody (data not shown).

FGFRs were expressed and modulated by FGF23 + Klotho, but not Klotho or FGF23 alone (B-DNA analysis). Cells were cultured in 12-well plates in differentiation medium (alpha-MEM/10% FBS/ascorbic acid [50 μg/mL]/BGP [10 mM]) and supplemented with huFGF23R176Q (F) (1 and 1,000 ng/mL), murine Klotho (KL) (50 ng/mL), their combination, or FGF2 (bFGF) for 14 days. a FGFR “IIIc” forms are preferentially expressed in MC3T3.E1 cells exposed to differentiation medium alone. b FGFR1(IIIc). c FGFR2(IIIc). d FGFR3(IIIc). Gene panel FGFR1(IIIb), FGFR1(IIIc), FGFR2(IIIb), FGFR2(IIIc), FGFR3(IIIc) and FGFR4. Bars represent mean ± SEM. *P < 0.05, **P < 0.01 compared with differentiation medium (Diff. Medium)
The huFGF23R176Q + Klotho-induced suppression of MC3T3.E1 cell differentiation, as measured by ALP activity, was fully blocked by the FGFR1 inhibitor SU5402 (Fig. 6a) and partially blocked by the MAPK pathway ERK1/2 inhibitor U0126 (Fig. 6b). In contrast, the p38 inhibitor SB203580, the PI3K inhibitor LY294002, and FGFR2(IIIc) and FGFR3 neutralizing antibodies were without effect on the huFGF23R176Q + Klotho-induced suppression of MC3T3.E1 cell differentiation (data not shown).

ALP activity was variably restored in the presence of FGFR and MAPK inhibitors. Cells were grown in 24-well plates and exposed to huFGF23R176Q (FGF23, F) (1,000 ng/mL), murine Klotho (KL) (50 ng/mL), huFGF23R176Q + Klotho, or FGF2 (100 ng/mL) in the presence or absence of FGFR inhibitors or MAPK pathway inhibitors for 7 days. Inhibitor alone and FGF2 (bFGF) (100 ng/mL) served as controls. a ALP activity was completely restored in the presence of FGFR1 inhibitor SU5402. b ALP activity was partially restored in the presence of the MAPK, ERK1/2 inhibitor UO126. All values represent mean ± SEM. *P < 0.05, ***P < 0.001 compared with huFGF23R176Q + Klotho (KL + F) or differentiation medium (Diff. Medium) as designated
Discussion
Supraphysiological levels of FGF23 in the presence of soluble Klotho were associated with increased MC3T3.E1 cell proliferation and concomitant inhibition of mineralization and bone cell marker gene expression in vitro. These results support the hypothesis that elevated FGF23 found in CKD-MBD patients may contribute to bone pathologies by directly acting on bone cells, possibly in an autocrine/paracrine manner.
Initially, we examined the activity of purified FGF23 on signaling via the MAPK pathway in normal human PTECs. As reported for FGF23 and heparin in OK PTECs [26], FGF23 led to phosphorylation of ERK and p38 in PTECs. This phosphorylation was only observed in the presence of OS-H, suggesting that high levels of FGF23 can signal in PTECs in the absence of Klotho. Further supporting heparin-mediated signaling, a previous report demonstrated that BaF3 cells expressing FGFRs can respond to FGF23 with increased proliferation and decreased sodium phosphate transporter expression in the presence of OS-H [19]. The necessity for soluble Klotho and/or heparin in FGF23 signaling in PTECs was further demonstrated in our study by the FGF23-induced ERK phosphorylation in the presence, but not the absence, of soluble Klotho. Although recent studies have identified Klotho in proximal tubule fluid, urine, and PTECs [30], suggesting that FGF23 may directly target PTEC membrane Klotho, we did not find any evidence to support the role of membrane Klotho. However, the level of membrane Klotho in our PTEC cultures may have been below that needed for signaling, requiring the addition of soluble Klotho. This would support a model in which renal distal tubules act as the primary source of secreted Klotho that targets PTECs [22].
Recently, FGF23 in serum of CKD-MBD dialysis patients was found to be intact and active [17]. Thus, we examined whether chronically elevated levels of FGF23 as seen in CKD-MBD patients (100- to 10,000-fold above normal values) could signal in extrarenal tissues such as bone cells. We examined the direct effects of FGF23 on osteoblastic MC3T3.E1 cell proliferation and differentiation in vitro because studies in vivo may be hampered by coexisting hormonal (e.g., PTH, vitamin D) and mineral abnormalities that each have proven effects on bone cells. Serum Klotho concentrations have been reported to be approximately two- to threefold higher in healthy children than adults (adult approximate mean 600 pg/mL) [31, 32]. Levels of serum Klotho in XLH children and adults were no different from those in respective healthy individuals [31]. In CKD patients, circulating serum Klotho levels are yet to be reported, but acute kidney injury in humans and rodent models was associated with decreased Klotho protein in blood, kidneys, and urine [33].
In mouse osteoblastic MC3T3.E1 cells soluble Klotho appeared to confer specificity to endogenous osteoblastic FGFRs for FGF23, as reported for other cells [21]. Signaling in the presence of elevated FGF23 + Klotho increased proliferation slightly and inhibited mineralization and bone marker gene expression. The “IIIc” forms of FGFR1, FGFR2, and FGFR3 were preferentially expressed in MC3T3.E1 cells; FGF23 + Klotho upregulated FGFR1(IIIc) and FGFR3(IIIc) and downregulated FGFR2(IIIc). The expression profile of FGFRs in MC3T3.E1 cells parallels that found for rat calvarial cultures and developing fetal mouse calvaria in that “IIIb” forms and FGFR4 were not expressed [25, 34]. To address the molecular mechanisms associated with FGF23 action, we examined canonical FGFR signal-transduction pathways as possible mediators of FGF23 action. The complete rescue of ALP levels in the presence of FGFR1 inhibitor SU5402 indicated that FGFR1 was the dominant receptor through which FGF23 signals in MC3T3.E1 cells. This was underscored by the lack of effect by FGFR2(IIIc) and FGFR3 neutralizing antibodies on ALP levels. ALP activity was partially restored by the MAPK pathway ERK1/2 inhibitor U0126, indicating that additional pathways were likely involved. However, inhibitors for p38 or PI3K could not rescue ALP inhibition induced by FGF23. The results with FGFR1 inhibitor SU5402 support those in rat calvarial osteoblast cultures overexpressing FGF23 by adenoviral delivery [25] and provide further support for FGFR1 as the predominant FGF23 receptor in osteoblastic cells. In the latter study, overexpression of FGF23 alone inhibited calvarial osteoblast mineralization [25], whereas here soluble Klotho was essential for FGF23-induced inhibition of mineralization of MC3T3.E1 cells. The reason for this is unknown but could be stage-specific, differential expression of proteoglycans/heparins and/or related to different cell sources and culture methodologies [35]. In the present study, elevated FGF23 actions appeared, in part, to parallel FGF2 actions with lower potency. FGF2 is an important regulator of osteoblast function and bone formation. FGF2 stimulated proliferation, inhibited mineralization, and increased FGFR1(IIIc) expression, while decreasing FGFR2(IIIc) and FGFR3(IIIc) expression. FGF2 has been reported to induce osteoblast proliferation and differentiation in vitro, but the effects are stage-specific [36].
Although exposure of MC3T3.E1 cells to soluble heparin led to cell death in this study, FGF23 signaling in bone cells may be stabilized by specific locally produced heparins, residing in the extracellular matrix, in the presence or absence of soluble Klotho.
The concentrations of FGF23 used in this study include those present in CKD-MBD patient serum. Normal serum concentrations of FGF23 range 5–50 pg/mL, whereas in CKD-MBD patients levels have been reported to range from 100- to 10,000-fold normal [3, 4, 7]. Thus, 1 ng/mL is 20- to 200-fold normal levels, and 100 ng/mL is 2,000- to 20,000-fold normal levels.
Klotho-deficient mice have elevated FGF23 levels (2,000-fold levels found in wild-type mice) [21] but the same phenotype as FGF23-deficient mice [5], a finding that does not support a pathological role for supraphysiological FGF23 levels on nontarget tissues. However, Klotho-deficient animals survive for about 3 months [23], while CKD-MBD patients are exposed to sustained high FGF23 levels for years. In addition, Klotho-deficient mice lack soluble Klotho, which might be present in CKD-MBD patient serum derived from tissues other than the kidneys.
In summary, FGF23/Klotho inhibited osteoblastic cell mineralization and osteoblast marker gene expression, likely via FGFR1 signaling. In addition, FGF23/Klotho increased FGFR1(IIIc) and FGFR3(IIIc) expression in MC3T3.E1 cells. Thus, in addition to affecting bone via indirect phosphate wasting pathways, supraphysiological FGF23 (and soluble Klotho) may directly impact bone in diseases with sustained elevated FGF23 levels.
References
Riminucci M, Collins MT, Fedarko NS, Cherman N, Corsi A, White KE, Waguespack S, Gupta A, Hannon T, Econs MJ, Bianco P, Gehron Robey P (2003) Fgf-23 in fibrous dysplasia of bone and its relationship to renal phosphate wasting. J Clin Invest 112:683–692
Bai X, Miao D, Li J, Goltzman D, Karaplis AC (2004) Transgenic mice overexpressing human fibroblast growth factor 23 (r176q) delineate a putative role for parathyroid hormone in renal phosphate wasting disorders. Endocrinology 145:5269–5279
Kazama JJ, Sato F, Omori K, Hama H, Yamamoto S, Maruyama H, Narita I, Gejyo F, Yamashita T, Fukumoto S, Fukagawa M (2005) Pretreatment serum fgf-23 levels predict the efficacy of calcitriol therapy in dialysis patients. Kidney Int 67:1120–1125
Larsson T, Nisbeth U, Ljunggren O, Juppner H, Jonsson KB (2003) Circulating concentration of fgf-23 increases as renal function declines in patients with chronic kidney disease, but does not change in response to variation in phosphate intake in healthy volunteers. Kidney Int 64:2272–2279
Shimada T, Kakitani M, Yamazaki Y, Hasegawa H, Takeuchi Y, Fujita T, Fukumoto S, Tomizuka K, Yamashita T (2004) Targeted ablation of fgf23 demonstrates an essential physiological role of fgf23 in phosphate and vitamin d metabolism. J Clin Invest 113:561–568
Shimada T, Urakawa I, Yamazaki Y, Hasegawa H, Hino R, Yoneya T, Takeuchi Y, Fujita T, Fukumoto S, Yamashita T (2004) Fgf-23 transgenic mice demonstrate hypophosphatemic rickets with reduced expression of sodium phosphate cotransporter type IIa. Biochem Biophys Res Commun 314:409–414
Weber TJ, Liu S, Indridason OS, Quarles LD (2003) Serum fgf23 levels in normal and disordered phosphorus homeostasis. J Bone Miner Res 18:1227–1234
White KE, Evans WE, O’Riordan JLH, Speer MC, Econs MJ, Lorenz-Depiereux B, Grabowski M, Meitinger T, Strom TM (2000) Autosomal dominant hypophosphataemic rickets is associated with mutations in FGF23. Nat Genet 26:345–348
Jonsson KB, Zahradnik R, Larsson T, White KE, Sugimoto T, Imanishi Y, Yamamoto T, Hampson G, Koshiyama H, Ljunggren O, Oba K, Yang IM, Miyauchi A, Econs MJ, Lavigne J, Juppner H (2003) Fibroblast growth factor 23 in oncogenic osteomalacia and X-linked hypophosphatemia. N Engl J Med 348:1656–1663
Shimada T, Mizutani S, Muto T, Yoneya T, Hino R, Takeda S, Takeuchi Y, Fujita T, Fukumoto S, Yamashita T (2001) Cloning and characterization of fgf23 as a causative factor of tumor-induced osteomalacia. Proc Natl Acad Sci USA 98:6500–6505
Feng JQ, Ward LM, Liu S, Lu Y, Xie Y, Yuan B, Yu X, Rauch F, Davis SI, Zhang S, Rios H, Drezner MK, Quarles LD, Bonewald LF, White KE (2006) Loss of dmp1 causes rickets and osteomalacia and identifies a role for osteocytes in mineral metabolism. Nat Genet 38:1310–1315
Benet-Pages A, Orlik P, Strom TM, Lorenz-Depiereux B (2005) An fgf23 missense mutation causes familial tumoral calcinosis with hyperphosphatemia. Hum Mol Genet 14:385–390
Shimada T, Hasegawa H, Yamazaki Y, Muto T, Hino R, Takeuchi Y, Fujita T, Nakahara K, Fukumoto S, Yamashita T (2004) Fgf-23 is a potent regulator of vitamin D metabolism and phosphate homeostasis. J Bone Miner Res 19:429–435
Lavi-Moshayoff V, Wasserman G, Meir T, Silver J, Naveh-Many T (2010) PTH increases FGF23 gene expression and mediates the high-FGF23 levels of experimental kidney failure: a bone parathyroid feedback loop. Am J Physiol Renal Physiol 299:F882–F889
Moe SM (2006) Vascular calcification and renal osteodystrophy relationship in chronic kidney disease. Eur J Clin Invest 36(Suppl 2):51–62
Saji F, Shiizaki K, Shimada S, Okada T, Kunimoto K, Sakaguchi T, Hatamura I, Shigematsu T (2009) Regulation of fibroblast growth factor 23 production in bone in uremic rats. Nephron Physiol 111:p59–p66
Shimada T, Urakawa I, Isakova T, Yamazaki Y, Epstein M, Wesseling-Perry K, Wolf M, Salusky IB, Juppner H (2010) Circulating fibroblast growth factor 23 in patients with end-stage renal disease treated by peritoneal dialysis is intact and biologically active. J Clin Endocrinol Metab 95:578–585
Itoh N (2010) Hormone-like (endocrine) Fgfs: their evolutionary history and roles in development, metabolism, and disease. Cell Tissue Res 342:1–11
Yu X, Ibrahimi OA, Goetz R, Zhang F, Davis SI, Garringer HJ, Linhardt RJ, Ornitz DM, Mohammadi M, White KE (2005) Analysis of the biochemical mechanisms for the endocrine actions of fibroblast growth factor-23. Endocrinology 146:4647–4656
Kurosu H, Ogawa Y, Miyoshi M, Yamamoto M, Nandi A, Rosenblatt KP, Baum MG, Schiavi S, Hu MC, Moe OW, Kuro-o M (2006) Regulation of fibroblast growth factor-23 signaling by klotho. J Biol Chem 281:6120–6123
Urakawa I, Yamazaki Y, Shimada T, Iijima K, Hasegawa H, Okawa K, Fujita T, Fukumoto S, Yamashita T (2006) Klotho converts canonical fgf receptor into a specific receptor for fgf23. Nature 444:770–774
Farrow EG, Davis SI, Summers LJ, White KE (2009) Initial fgf23-mediated signaling occurs in the distal convoluted tubule. J Am Soc Nephrol 20:955–960
Kuro-o M, Matsumura Y, Aizawa H, Kawaguchi H, Suga T, Utsugi T, Ohyama Y, Kurabayashi M, Kaname T, Kume E, Iwasaki H, Iida A, Shiraki-Iida T, Nishikawa S, Nagai R, Nabeshima YI (1997) Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature 390:45–51
Huang CL (2010) Regulation of ion channels by secreted klotho: mechanisms and implications. Kidney Int 77:855–860
Wang H, Yoshiko Y, Yamamoto R, Minamizaki T, Kozai K, Tanne K, Aubin JE, Maeda N (2008) Overexpression of fibroblast growth factor 23 suppresses osteoblast differentiation and matrix mineralization in vitro. J Bone Miner Res 23:939–948
Yamashita T, Konishi M, Miyake A, Inui K, Itoh N (2002) Fibroblast growth factor (fgf)-23 inhibits renal phosphate reabsorption by activation of the mitogen-activated protein kinase pathway. J Biol Chem 277:28265–28270
Kasperk CH, Wergedal JE, Mohan S, Long DL, Lau KH, Baylink DJ (1990) Interactions of growth factors present in bone matrix with bone cells: effects on DNA synthesis and alkaline phosphatase. Growth Factors 3:147–158
Rodan SB, Wesolowski G, Thomas KA, Yoon K, Rodan GA (1989) Effects of acidic and basic fibroblast growth factors on osteoblastic cells. Connect Tissue Res 20:283–288
Hausser HJ, Brenner RE (2004) Low doses and high doses of heparin have different effects on osteoblast-like saos-2 cells in vitro. J Cell Biochem 91:1062–1073
Hu MC, Shi M, Zhang J, Pastor J, Nakatani T, Lanske B, Razzaque MS, Rosenblatt KP, Baum MG, Kuro-o M, Moe OW (2010) Klotho: a novel phosphaturic substance acting as an autocrine enzyme in the renal proximal tubule. FASEB J 24:3438–3450
Carpenter TO, Insogna KL, Zhang JH, Ellis B, Nieman S, Simpson C, Olear E, Gundberg CM (2010) Circulating levels of soluble klotho and fgf23 in X-linked hypophosphatemia: circadian variance, effects of treatment, and relationship to parathyroid status. J Clin Endocrinol Metab 95:E352–E357
Yamazaki Y, Imura A, Urakawa I, Shimada T, Murakami J, Aono Y, Hasegawa H, Yamashita T, Nakatani K, Saito Y, Okamoto N, Kurumatani N, Namba N, Kitaoka T, Ozono K, Sakai T, Hataya H, Ichikawa S, Imel EA, Econs MJ, Nabeshima Y (2010) Establishment of sandwich elisa for soluble alpha-klotho measurement: age-dependent change of soluble alpha-klotho levels in healthy subjects. Biochem Biophys Res Commun 398:513–518
Hu MC, Shi M, Zhang J, Quinones H, Kuro-o M, Moe OW (2010) Klotho deficiency is an early biomarker of renal ischemia-reperfusion injury and its replacement is protective. Kidney Int 78:1240–1251
Rice DP, Aberg T, Chan Y, Tang Z, Kettunen PJ, Pakarinen L, Maxson RE, Thesleff I (2000) Integration of fgf and twist in calvarial bone and suture development. Development 127:1845–1855
Jackson RA, Murali S, van Wijnen AJ, Stein GS, Nurcombe V, Cool SM (2007) Heparan sulfate regulates the anabolic activity of mc3t3-e1 preosteoblast cells by induction of runx2. J Cell Physiol 210:38–50
Debiais F, Hott M, Graulet AM, Marie PJ (1998) The effects of fibroblast growth factor-2 on human neonatal calvaria osteoblastic cells are differentiation stage specific. J Bone Miner Res 13:645–654
Acknowledgement
The authors thank Jon Nilsen, PhD (Amgen, Inc.), for editorial assistance.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
All authors are or were (N.H.) full time employees of Amgen and own stock in Amgen.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Shalhoub, V., Ward, S.C., Sun, B. et al. Fibroblast Growth Factor 23 (FGF23) and Alpha-Klotho Stimulate Osteoblastic MC3T3.E1 Cell Proliferation and Inhibit Mineralization. Calcif Tissue Int 89, 140–150 (2011). https://doi.org/10.1007/s00223-011-9501-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00223-011-9501-5