
Abstract
In the present study, two alternative methods for identifying 13 salmon, trout and bream species were developed. Both of them are based on polymerase chain reaction (PCR) amplification of a cytochrome b gene fragment. Subsequently, different techniques were assayed to assign the PCR amplicons previously obtained to particular species. The first one is based on the restriction fragment length polymorphism (RFLP) and includes three endonucleases for generating species-specific restriction profiles, while the second one is based on the phylogenetic analysis of DNA sequences. The main novelty of this work lies in the applicability of the developed methods to all kinds of processed products, including those undergoing intensive processes of transformation, as for instance canned foods. Finally, the methods were applied to 25 commercial samples including some that had been subjected to intensive thermal treatment, allowing the detection of those incorrectly labeled (16%). Therefore, these methods are useful to check the fulfillment of labeling regulations for seafood products, verify the correct traceability in commercial trade, and for fisheries control.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The massive aquaculture production of salmon and trout has favored the presence of multiples species in the European markets. The main forms under which these species are marketed are whole, sliced, filleted, and as steaks, fresh or frozen. Among all the commercialized products it is worth pointing out smoked meat, but pate, cans, marinated, and other products are progressively taking more important place in the fishing industry.
The main species in salmonid production is the Atlantic salmon (Salmo salar), which has accounted for 54% of the total production in the year 2007. The second most important species is the rainbow trout (Onchorhynchus mykiss), with a share of 29% in this production. The third species in importance during the same period has been Coho salmon (Onchorhynchus kisutch) comprising 17% of the production [1]. The commercial value of salmonids depends on the species. For instance Salmo salar, Oncorhynchus tshawytscha, and O. nerka have greater commercial value than O. keta and O. gorbuscha. The bream species have a lower commercial value than salmon and trout, and due to this fact smoked bream slices are stained and commonly used as substitute of smoked salmon or trout.
In this sense, to guarantee the correct information to the consumers, the authorities of each region have established different regulations. For instance, in Europe the Regulation 104/2000 and 2065/2001 tries to protect the consumer’s rights [2]. The Codex Alimentarius Commission has also developed a standard outlining the main requirements for canned salmon [3]. These regulations establish the appropriate information about the commercial denomination and the scientific names of the species.
In order to enforce labeling regulations and prevent species substitution the necessity arises to develop analytical methods that can be used to determine the genuine species included in any product, independently of the degree of transformation which it has underwent. In this context, it is very important to have available analytical methods for the verification of the authenticity of raw materials included in fish and seafood products, since it allows controlling the correct labeling. Commercial kits exist as for example the Biofish salmon kit (Biotools) or the Biokits DNA Fish Speciation products (Tepnel) that permit the detection and the identification of salmonids, but all these can only be applied to two or three of the most important commercial species. In this sense the molecular methods are the best approach. As a proof the work of Rusell et al. might be cited in which ten salmon species have been identified using PCR–RFLP of a fragment of the cytochrome b (cyt b) gene [4]. Subsequently, the same authors validated the methodology mentioned above in food products [5]. Other studies centered on the genetic identification of salmon included too few individuals and did not cover the species present in the current international market [6–9]. The main disadvantage of all the previous studies is that these cannot be applied to highly processed products such as canned foods, because the amplified PCR product is longer than 200 bp. In this kind of products it is not possible to amplify DNA fragments of such a size [10–13]. Moreover, these methods do not include all the possible substitute species, and this fact can produce incorrect or not assignations.
For the reason, in the present work a molecular method was developed for authentication of salmon, trout, and bream species in any seafood product, including those that have undergone intensive thermal treatment. The proposed technique allows evaluating the correct labeling of products containing salmon, trout, and bream. This way this tool will assure the consumers of the accurate labeling and avoid unfair competition between fishing industries.
Materials and methods
Sample collection, storage
Samples of different salmon, trout, and bream species were collected from several locations around the world (Table 1). When it was possible, the individuals were identified according to morphological characters [14]. In other cases, fish tissues preserved in ethanol were provided by Universities and research centers located around the world. At least three individuals of each species were examined with the exception of Brama japonica, which was only available as two processed products. Once identified, the samples were labeled and preserved at −80 °C.
Moreover, 25 seafood products labeled as some salmon, trout and bream species were provided by import industries or purchased in supermarkets and shops from European countries and Chile, in order to apply the developed methodology to commercial samples.
DNA extraction
Genomic DNA was extracted from 30 mg of muscle in fresh and frozen samples, according to the method described by Roger and Bendich with slight modifications [15]. The obtained DNA was diluted in 100 μL of 1× Tris–EDTA (TE) buffer (Sigma). In the case of products used for the methodological validation and commercial samples, two methods were tested for DNA extraction. Both of them started from amounts of tissue comprising between 100 and 300 mg: (1) a method based on silica gel columns using the NucleoSpin Tissue kit (Macherey-Nagel), following the supplier’s protocol with minor changes (as to adapt the procedure to a high quantity of sample). (2) Extraction of DNA using the CTAB method previously described [15], and subsequent purification by means of the Nucleospin® Extract II kit (Macherey-Nagel) following the supplier’s protocol.
Extracted DNA was loaded on 1% agarose gels in 1× TBE buffer (Sigma) with 5 μg/mL of ethidium bromide (Sigma) using as standards the 100 to 1,500-bp DNA ladder (Dominion, MBL) and a 50-bp ladder (GE Healthcare). The electrophoresis was run at 70 V for 50 min and the gels were subsequently visualized using the Molecular Imager Gel Doc XR System transilluminator and the software Quantity One® v 4.5.2 (Bio-Rad).
The quality and quantity were determined by measuring the absorbance at 260 nm and the 260/280 nm and 234/260 ratios [16] using a NanoDropTM ND-1000 spectrophotometer (Thermo Scientific). DNA extractions were appropriately labeled and stored at −80 °C for subsequent tasks.
PCR amplification and DNA sequencing
A fragment of 1226 bp that include a partial region of the glutamic acid tRNA, the complete cytochrome b and partial region of the tRNA of threonine was amplified by PCR using the primers L14735 [17] and TRUCCYTB-R [18].
In all cases the PCR reactions were carried out in a total volume of 50 μL with the following composition: 100–300 ng of DNA template were added to a PCR mix consisting of 0.8 mM dNTP mix (Bioline), 5 μL 10× buffer, 2 mM MgCl2, 0.75 units of BioTaqTM DNA polymerase (Bioline), 0.8 μL of each primer (Sigma Genosys) and molecular biology grade water (Eppendorf) up to adjust to the final volume.
Polymerase chain reactions were carried out in a MyCyclerTM thermocycler (BIO-RAD). Conditions of cycling were as follows: a preheating step at 95 °C for 3 min, 35 cycles of amplification (95 °C for 30 s, 45 °C for 1 min, 72 °C for 1 min) and a final extension step of 72 °C for 3 min.
Polymerase chain reaction amplicons were visualized on 2% agarose gels (Sigma) in 1× TBE buffer (Sigma) as described above. Next, double-stranded DNA were purified using the Nucleospin© 96 Extract II (Macherey-Nagel) according to the manufacturer’s instructions. The concentration and purity were measured by means of the NanoDropTM ND-1000 spectrophotometer (Thermo Scientific) as described for DNA extraction. Subsequently, sequencing reactions of both DNA strands were carried out with the primers described previously in a final volume of 10 μL with the CEQ Dye Terminator cycle sequencing Quick Start kit (Beckman Coulter). Thermal cycle sequencing reaction and the subsequent sequencing products’ cleanup by ethanol precipitation were carried out in accordance with the manufacturer’s instructions (Beckman Coulter). The sequences were obtained on a CEQ 8800 Genetic Analysis System (Beckman Coulter). The sequences were analyzed with the Chromas 1.45 software [19] and aligned with Clustal W [20] available in the program BioEdit 7.0 [21]. The nucleotide sequences obtained were submitted to the GeneBank database of the National Centre for Biotechnology Information (NCBI).
The intra and inter-specific polymorphism analysis were performed from the resulting alignment using the DnaSP 4.0 software [22].
Primers design for species identification in heavily processed products
From the sequences of 1,226 bp two internal primers were designed: SALM-H (5′ ACG TCC TYC CRT GAG GAC A 3′) and SALM-L (5′ AAT CGT GTT AAR GTR GCG TTG T 3′).
PCR amplifications were carried out in the following conditions: a preheating step of 94 °C for 5 min followed by 35 cycles of amplification (94 °C for 20 s 52 °C for 20 s and 72 °C for 20 s) and a final extension step of 7 min at 72 °C.
Next, the PCR products obtained were visualized, cleaned, and sequenced as reported previously.
Development of an identification system based on the PCR–RFLP analysis
Restriction maps of the DNA sequences obtained were generated using the software Webcutter 2.0 [23]. The enzyme selection was determined by the ability of the restrictases to generate characteristic restriction profiles for each species with bands sizes easily distinguishable on agarose gels. 100 ng of PCR product purified on elution columns (Nucleospin© 96 Extract II, Macherey-Nagel) was digested with 2 units of each selected enzyme. Digestion reactions were incubated at 37 °C for 2 h. The results of the PCR–RFLP analysis were read as described above, except that the electrophoresis that was performed on 3% low-melting agarose gels (Pronadisa) at 70 V for 110 min. Sizes of fragments were estimated from the 50 bp DNA ladder (GE Healthcare).
Development of FINS methodology
Two phylogenetic analyses were carried out. The first of them was from sequences of the total cyt b gene and the second, from the sequences of the internal fragment herein designed.
These analyses were developed with Mega 4.0 [24] using the Tamura–Nei model [25] to calculate the genetic distances between sequences. The inference of the phylogenetic trees was carried out with the Neighbor-Joining method [26]. The reliability of the clades formed at the species level in the trees was evaluated by means of a bootstrap test with 2,000 replications.
Also, the MEGABLAST search available at NCBI was assessed to assign the sequences to a particular species [27].
Methodological validation
Individuals from different species were authenticated on the basis of their morphological traits and the main commercial treatments types were applied to them. The treatments included canning, smoking, marinating, preparation of carpaccio and pate, pickling, caking, and precooking for which different kinds of sauces and condiments were used.
The treatment applied to canned samples involved 121 °C of temperature and 1.2 bars of overpressure and the time varied depending on the size of the can. The smoking process combines two effects: on the one hand salting and drying steps and on the other the effect of temperature. The temperature corresponding to smoking of the fillets was raised to 121 °C until inside the product 60 °C were reached. The cooking time depended on the thickness of the fillets. The thermal treatment applied to pate was 121 °C for sterilized products and 90–100 °C for pasteurized products, and the time varied depending on the format size. All these treatments were carried out in the pilot plant of CECOPESCA (Spanish National Centre of Fish Processing Technology). Next, the products were analyzed with the methodology developed in the present work.
The coincidence percentage between the species identified on the basis of morphological traits and the genetic methodology developed was calculated to establish the specificity of the method.
Application to commercial products
After the validation of the methods developed in the present work, these were applied to 25 products labeled as some salmon, trout, and bream species. These products were acquired in supermarkets from Europe and Chile (the last country is the main salmon producer in the world). The purpose of these analyses was to evaluate the situation regarding the labeling of these products on the market.
Results and discussion
In the present work two methods for the genetic identification of salmon, trout, and bream species were designed. The first of them is based on the PCR–RFLP technique, and the second one on the phylogenetic analysis of a fragment of the cyt b gene.
DNA Extraction
Two methods were tried for DNA extraction from processed samples. The first of them was the NucleoSpin kit (Macherey-Nagel) and the second—the CTAB method followed by a purification step by means of the Nucleospin® Extract II kit (Machery-Nagel). The second one showed better results because higher quantity and better quality of DNA were obtained according to spectrophotometric measurements (data not shown). Therefore, the mentioned method was chosen for DNA extraction from processed samples.
Amplification and sequencing of PCR products
DNA amplification with the primers L14735/TRUCCYTB-R generated an amplicon of 1,224 bp in all the species included in the present study (Table 1), except in Brama spp and Salmo salar, in which the fragments were of 1,226 and 1,225 bp, respectively (accession numbers EU660676 to EU660716). The length polymorphism between the species was caused by the additional bases in the tRNA of the glutamic acid, so that there were no reading frame shifts for the cyt b gene.
In fresh or frozen fish it is possible to amplify a fragment of 1,226 bp. In the case of fishes that have undergone different treatments, it is not possible to amplify PCR products of this size, because the thermal treatment generates DNA fragmentation. This was the case of cans or smoked products, where fragments of little sizes were amplified. Quinteiro et al. [10] established a maximum fragment size in canned products of 176 bp to ensure the amplification. Other authors, under certain conditions, amplified fragments higher than 200 bp from canned products [28–31].
However, we consider that it is important to have at one’s disposal a method that can be used routinely, allowing the amplification of DNA easily and providing a reliable species identification. Herein the amplification of a small fragment is proposed that contains enough information to enable the differentiation of all the studied species. The primers applied amplified a fragment of 142 bp, being used to identify the species included in heavily processed products as canned or smoked.
The degradation of DNA in addition to the existence of PCR inhibitors in processed products as canned and smoked [28, 29] interfere in the PCR reaction. The presence of additives used in the alimentary industry as spices or sauces attenuates or inclusively inhibits the DNA amplification. Moreover, the different kinds of sauces added produce differences in the quantity and quality of the extracted DNA, this molecule being very sensitive to acid and alkaline agents. In this sense, it is worth highlighting the pickled products, in which the low pH produces higher DNA degradation [32]. For this reason, the strategy of amplification proposed facilitates the successful PCR amplification in any seafood product.
Development of an identification system based on the PCR–RFLP analysis
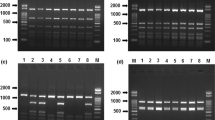
A genetic identification method based on the PCR–RFLP technique was developed. This technique has been widely used for several reasons. It the first place, it is simpler and faster than sequencing, and with a reduced cost. Moreover, it is especially useful for routine analysis when sequencing is not an affordable option. The studies based on the identification of marine species using PCR–RFLP analysis are numerous, both in invertebrate [12, 13] and vertebrate species [11, 33, 34]. Therefore, the RFLP represents a suitable technique to determine the identity of the salmonid species included in this work. Before digestion, the PCR products must be purified to remove the primer–dimer complexes in such a way that these will not interfere in the interpretation of the restriction profiles. The restrictases selected in this work were Afa I (Amersham pharmacia biotech), Mnl I, and Fau I (New England Biolabs). The DNA fragments generated in the digestion were compatible with the expected on the basis of the restriction map (Table 2, Fig. 1). These enzymes have a simple recognition sequence, reducing the risk of misidentification that other restrictases with more complex target can produce. The combined restriction profile generated by these enzymes allowed the identification of all the studied species in a univocal form. The exceptions were the species belonging to genus Brama, which had an identical restriction profile, being identified to the genus level.

RFLP analysis of PCR products obtained with the SALM primer set in all species included in this work. Lane PCR PCR products, lane 1 PCR products after digestion with Afa I, lane 2 PCR products after digestion with Mnl I, lane 3 PCR products after digestion with Fau I, lane L-50 molecular size marker (50 bp DNA ladder, GE Healthcare)
The DNA fragments with sizes of less than 50 bp generated in the restriction digestion were not used for the identification, because the established conditions for the agarose gels did not allow visualizing them.
The samples analyzed by the PCR–RFLP method did not show variations in the restriction profile, although the possible existence of variations in the recognition sites cannot be dismissed. However, the high number of samples that were taken into account during the development of the methodology assured high degree of reliability for the developed method.
Development of FINS methodology
The forensically informative nucleotide sequencing (FINS) technique described by Bartlett and Davidson [35] was used in the present study to develop an identification method for salmon, trout, and bream species. Sequences of unknown samples are compared with sequences belonging to pattern specimens on the basis of this technique.
The genetic distances between the cyt b gene sequences of all the studied species were estimated using the Tamura–Nei method [25]. The interspecific distances were calculated to know the degree of divergence between species and were in general two orders of magnitude higher than the intraspecific ones. The mean values for intraspecific and interspecific distances and their standard deviation were 0.003 ± 0.000 and 0.120 ± 0.007, respectively.
The phylogenetic analysis of the two fragments amplified (1,178 and 102 bp, fragments without primers) was carried out separately, allowing to establish the relationships among species by means of the construction of phylogenies using the two data sets. In the trees obtained, all the sequences belonging to individuals of the same species were grouped in the same cluster, allowing their identification. Three strongly supported clades were identified: Oncorhynchus, Salmo, and Brama, which corresponded to the three genera included in this work (Fig. 2). Moreover, bootstrap values of branches at level species were 100 for the matrix of 1,226 bp and higher than 70 for the 142 bp, reflecting the robustness of the phylogenetic tree (Figs. 2, 3). The internal fragment (142 bp) was used to identify the species included in foods that underwent thermal treatment (for instance, canned, smoked, and cooked products), where the DNA fragmentation impeded the amplification of higher amplicons.

Phylogenetic tree showing the relationships among the studied species, carried out from the alignment of 1,178 bp of the cyt b gene (fragment of 1,226 bp without primers)

Phylogenetic tree showing the relationships among the studied species, carried out from the alignment of 102 bp of the cyt b gene (fragment of 142 bp without primers)
Therefore, the two PCR products analyzed allowed the unequivocal identification of all the salmon, trout, and bream species included in this work.
BLAST analysis is a suitable technique to find regions of local similarity between sequences and can even be a suitable technique to identify species. This method is similar to the FINS since it uses DNA sequences and a database. Specifically, the MEGABLAST search available at NCBI was assessed to assign any salmon, trout, and bream DNA sequences to a particular species. The phylogenetic assignments generated by the proposed FINS technique were compared to the results obtained by BLAST, and the same species assignments were obtained (data not shown).
Therefore, these two techniques could be used to identify the salmon, trout, and bream species herein studied.
Methodological validation
The aim of the methodological validation was to check whether the manufacturing process which the processed food did undergo had no influence on the identification of these species.
Different products were prepared in the pilot plant of CECOPESCA simulating the conditions used in the food industry (Table 3). This approach is useful to assess the functioning and optimizing the conditions of the developed methodology. The standard individuals were subjected to several transformation processes, allowing to evaluate the influence of different variables on the genetic method herein proposed.
The species identified in these samples by means of the PCR–RFLP and FINS methods herein developed were in agreement with those based on morphological characters. Therefore, both of them show a specificity of 100%.
Application to commercial products
The methodologies developed were applied to 25 products labeled as some salmon, trout, and bream species, including different levels of transformation. This application allowed knowing the degree of fulfilment of the labeling regulations in these seafood products. In 16% of the products analyzed, the name of the species displayed in the label was not in agreement with the species contained, as determined by genetic analysis using the methodologies herein developed (Table 4). These groups subjected to mislabeling were salmon (75%) and trout (25%). The higher fraudulent labeling ratio can be due to the impossibility of carry out a species identification based on morphological traits in processed products. All the products that included some bream species were correctly labeled. Nevertheless, one of the samples analyzed, labeled as salmon, was identified as Brama (sample purchased in a restaurant).
Conclusions
In conclusion, in the present work, two DNA-based methods that allow the genetic identification of the most important commercialized species of salmon, trout, and bream were developed. The main advantage of the method developed compared to the ones published up to date is that besides including a high number of species, it is based on the use of a fragment with a size below 200 bp and can be applied to all kinds of processed products, including these undergoing intensive transformation processes. Moreover, the PCR–RFLP and FINS methods can be alternatively used depending on the equipment available in different laboratories. The possible applications of these methods are the following: normative control of raw and processed products, particularly the authenticity of imported species, the verification of the traceability of different fishing batches along the commercial chain, correct labeling, protection of the consumer’s rights, fair competence among fishing operators, and the fisheries’ control.
References
COUNCIL REGULATION (EC) No 104/2000 of 17 December 1999 on the Common Organisation of the Markets in Fishery and Aquaculture Products
Codex standard for canned salmon (CODEX STAN 3-1981)
Russell VJ, Hold GL, Pryde SE, Rehbein H, Quinteiro J, Rey-Mendez M, Sotelo CG, Perez-Martin RI, Santos AT, Rosa C (2000) Use of restriction fragment length polymorphism to distinguish between salmon species. J Agric Food Chem 48:2184–2188
Hold GL, Russell VJ, Pryde SE, Rehbein H, Quinteiro J, Rey-Mendez M, Sotelo CG, Perez-Martin RI, Santos AT, Rosa C (2001) Validation of a PCR–RFLP based method for the identification of salmon species in food products. Eur Food Res Technol 212:385–389
Carrera E, Garcia T, Cespedes A, Gonzalez I, Fernandez A, Hernandez PE, Martin R (1999) PCR–RFLP of the mitochondrial cytochrome oxidase gene: a simple method for discrimination between Atlantic salmon (Salmo salar) and rainbow trout (Oncorhynchus mykiss). J Sci Food Agric 79:1654–1658
Carrera E, Garcia T, Cespedes A, Gonzalez I, Fernandez A, Hernandez PE, Martin R (1999) Salmon and trout analysis by PCR–RFLP for identity authentication. J Food Sci 64:410–413
Carrera E, Garcia T, Cespedes A, Gonzalez I, Sanz B, Hernandez PE, Martin R (1998) Identification of Atlantic salmon (Salmo salar) and rainbow trout (Oncorhynchus mykiss) by using polymerase chain reaction amplification and restriction analysis of the mitochondrial cytochrome b gene. J Food Prot 61:482–486
Carrera E, García T, Céspedes A, González I, Fernández A, Asensio LM, Hernandez PE, Martin R (2000) Differentiation of smoked Salmo salar, Oncorhyncus mykiss and Brama raii using the nuclear marker 5S rRNA. Int J Food Sci Technol 35:401–406
Quinteiro J, Sotelo CG, Rehbein H, Pryde SE, Medina I, Perez-Martin RI, Rey-Mendez M, Mackie IM (1998) Use of mtDNA direct polymerase chain reaction (PCR) sequencing and PCR-restriction fragment length polymorphism methodologies in species identification of canned tuna. J Agric Food Chem 46:1662–1669
Santaclara FJ, Cabado AG, Vieites JM (2006) Development of a method for genetic identification of four species of anchovies: E. encrasicolus, E. anchoita, E. ringens and E. japonicus. Eur Food Res Technol 223:609–614
Santaclara FJ, Espiñeira M, Cabado AG, Aldasoro A, Gonzalez-Lavin N, Vieites JM (2006) Development of a method for the genetic identification of mussel species belonging to Mytilus, Perna, Aulacomya, and other genera. J Agric Food Chem 54:8461–8470
Santaclara FJ, Espiñeira M, Vieites JM (2007) Genetic identification of Squids (Families Ommastrephidae and Loliginidae) by PCR–RFLP and FINS methodologies. J Agric Food Chem 55:9913–9920
Mackie IM (1990) Identifying species of fish. Anal Proc 27:89–92
Roger SO, Bendich AJ (1988) Extraction of DNA from plant tissues, plant molecular biology manual, pp 1–10
Winfrey MR, Rot MA, Wortman AT (1997) UnraVeling DNA: molecular biology for the laboratory. Prentice Hall, New York
Burgener M (1997) Molecular species differentiation of fish and mammals. Ph.D. Thesis, University of Bern, Switzerland
Sevilla RG, Diez A, Noren M, Mouchel O, Jerome M, Verrez-Bagnis V, van Pelt H, Favre-Krey L, Krey G, Bautista JM (2007) Primers and polymerase chain reaction conditions for DNA barcoding teleost fish based on the mitochondrial cytochrome b and nuclear rhodopsin genes. Mol Ecol Notes 7:730–734
Mc Carthy C (1996) Chromas version 1.45, School of Health science. Griffifth University, Gold Coast Campus, Queensland, Australia
Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG (1997) The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res 25:4876–4882
Hall TA (1999) BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT, Nucleic Acids Symposium Series, pp 95–98
Rozas J, Sánchez-DelBarrio JC, Messeguer X, Rozas R (2003) DnaSP, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics 19:2496–2497
Heiman M (1997) Webcutter 2.0, http://www.firstmarket/cutter/cut2.html
Tamura K, Dudley J, Nei M, Kumar S (2007) MEGA4: molecular evolutionary genetics analysis (MEGA) software version 4.0. Mol Biol Evol 24:1596–1599
Tamura K, Nei M (1993) Estimation of the number of nucleotide substitutions in the control region of mitochondrial-DNA in humans and chimpanzees. Mol Biol Evol 10:512–526
Saitou N, Nei M (1987) The Neighbor-Joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4:406–425
Wheeler DL et al (2008) Database resources of the National Center for Biotechnology Information. Nucleic Acids Res 36(2):181–184
Pardo MA, Perez-Villareal B (2004) Identification of commercial canned tuna species by restriction site analysis of mitochondrial DNA products obtained by nested primer PCR. Food Chem 86:143–150
Ram JL, Ram ML, Baidoun FF (1996) Authentication of canned tuna and bonito by sequence and restriction site analysis of polymerase chain reaction products of mitochondrial DNA. J Agric Food Chem 44:2460–2467
Rehbein H, Kress G, Schmidt T (1997) Application of PCR-SSCP to species identification of fishery products. J Sci Food Agric 74:35–41
Unseld M, Beyermann B, Brandt P, Hiesel R (1995) Identification of the species origin of highly processed meat-products by mitochondrial-DNA sequences. PCR Methods Appl 4:241–243
Bauer T, Weller P, Hammes WP, Hertel C (2003) The effect of processing parameters on DNA degradation in food. Eur Food Res Technol 217:338–343
Espiñeira M, González.-Lavín N, Vieites JM, Santaclara FJ (2008) Authentication of anglerfish species (Lophius spp) by means of PCR-RFLP and FINS methodologies. J Agric Food Chem 56:10594–10599
Espiñeira M, González-Lavín N, Vieites JM, Santaclara FJ (2008) Development of a method for the genetic identification of flatfishes based on mitochondrial DNA sequence. J Agric Food Chem 56:8954–8961
Bartlett SE, Davidson WS (1992) FINS (forensically informative nucleotide sequencing): a procedure for identifying the animal origin of biological specimens. Biotechniques 12:408–411
Acknowledgments
We thank Miroslava Atanassova and Beatriz Herrero for their helpful assistance. We also thank Kristi Millar and Tobi Ming (Pacific Biological Station, Canada) for providing some of the samples included in this work.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Espiñeira, M., Vieites, J.M. & Santaclara, F.J. Development of a genetic method for the identification of salmon, trout, and bream in seafood products by means of PCR–RFLP and FINS methodologies. Eur Food Res Technol 229, 785–793 (2009). https://doi.org/10.1007/s00217-009-1107-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00217-009-1107-z