
Abstract
In this study, we compare combustion ion chromatography (CIC) and high resolution-continuum source-graphite furnace molecular absorption spectrometry (HR-CS-GFMAS) with respect to their applicability for determining organically bound fluorine sum parameters. Extractable (EOF) and adsorbable (AOF) organically bound fluorine as well as total fluorine (TF) were measured in samples from river Spree in Berlin, Germany, to reveal the advantages and disadvantages of the two techniques used as well as the two established fluorine sum parameters AOF and EOF. TF concentrations determined via HR-CS-GFMAS and CIC were comparable between 148 and 270 μg/L. On average, AOF concentrations were higher than EOF concentrations, with AOF making up 0.14–0.81% of TF (determined using CIC) and EOF 0.04–0.28% of TF (determined using HR-CS-GFMAS). The results obtained by the two independent methods were in good agreement. It turned out that HR-CS-GFMAS is a more sensitive and precise method for fluorine analysis compared to CIC. EOF and AOF are comparable tools in risk evaluation for the emerging pollutants per- and polyfluorinated alkyl substances; however, EOF is much faster to conduct.

Graphical abstract
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Substitution of hydrogen with fluorine in organic molecules affects their chemical and physical properties, e.g., increased chemical and thermal stability and/or oleophobic as well as hydrophobic properties [1, 2]. These molecules are described according to Buck et al. as per- and polyfluorinated alkyl substances (PFASs) [3]. PFASs are organic compounds in which all the hydrogen atoms on at least one carbon atom are replaced by fluorine [3]. Highly fluorinated organic substances are used as surfactants in technical applications, e.g., as water and grease protection agents in carpets [4], clothing [5], and food packaging [6], and as fire-extinguishing agents [7]. During production, use, and disposal of these industrial products, PFASs enter the environment. Because of their extreme persistence, PFASs accumulate in the abiotic environment, e.g., ground [8] and surface water [9], soil [10], and air [11], as well as in the biotic environment [12, 13]. Overall, there are large amounts of human exposure pathways so that PFASs can be detected in human serum [14, 15] as well as in human breast milk [16]. The concerning notice that PFASs can be found ubiquitously, even in the arctic environment [17], and that there are potential negative effects on the environment and human health [18] lead to first limitations. Since 2009, perfluorooctanesulfonic acid (PFOS) and, since 2019, perfluorooctanoic acid (PFOA) are listed in annexes of the Stockholm Convention on Persistent Organic Pollutants [19]. For these reasons, the production and use of PFOS and PFOA must be reduced respectively avoided [19]. Since then, PFOS and PFOA are substituted with shorter- (≤ 6 perfluorinated carbons [20]) and longer-chained (≥ 7 perfluorinated carbons [20]) PFASs, which aren’t potentially less persistent or risky [21]. The huge amount of different PFASs (> 4700 [22]) and this replacement lead to new analytical challenges. Because of their extreme persistence and vast anthropogenic emission, PFASs are emerging pollutants.
In this context, the environmental compartment water is particularly interesting. Especially through the ineffective removal of PFASs by conventional treatments of waste water treatment plants (WWTP) [23], PFASs accumulate in the aquatic environment and lead to contamination of ground and drinking water, thus entering the food chain (e.g., plants [24], animals [25, 26], and humans [27]).
PFASs target analytic in aqueous samples by liquid chromatography coupled with mass spectrometry (LC-MS) and gas chromatography coupled with mass spectrometry (GC-MS) methods cover only a small proportion (7–53 PFASs for LC-MS [28,29,30], and 10–13 PFASs for GC-MS [31, 32]) of the over 4700 different PFASs and vastly underestimate the quality and quantity of total organically bound fluorine (OF) [33]. This results in a huge gap in the PFAS mass balance with an unknown amount of potentially toxic and persistent PFASs. Consequently, a sum parameter method for organically bound fluorine is inevitable to cover the unknown proportion of PFASs.
PFASs in aqueous samples can be extracted using a sorbent (adsorbable organically bound fluorine, AOF) or alternatively using an organic solvent (extractable organically bound fluorine, EOF) [34]. Usually activated carbon (AC) is used as sorbent for AOF determination. Hence, AOF represents all PFASs present in water samples which are adsorbable on AC. Which PFASs are summed up in the EOF depends on the solid-phase material used during the extraction. Often, a weak anion exchanger (WAX) is used. Using WAX-solid-phase extraction (SPE), the EOF covers only neutral and anionic PFASs. In the literature, there is also the use of hydrophilic-lipophilic balance (HLB) materials described, resulting in a wider range of extracted PFASs [9].
The sample preparation for the determination of AOF is carried out according to the standardized adsorbable organically bound halogen (AOX) method ISO 9562 (adsorption on AC and argentometric determination) [35]. Based on ISO 9562, Wagner et al. developed a combustion ion chromatography (CIC) method, applicable to determine the AOF as well. Upon adsorption, AC is combusted and fluorine is converted into hydrogen fluoride (HF), which is then adsorbed in a trapping solution. Subsequently, the analysis of fluoride was carried out using ion chromatography (IC) [36]. A complementary target analysis by Willach et al. connoted that the AOF of some highly contaminated aqueous samples can only be explained by < 5% with LC-MS approaches, which underlines the importance of an organically bound fluorine sum parameter [33].
The determination of EOF in aqueous samples was firstly described by Miyake et al. in 2007. For sample preparation, they used an SPE method comprising a WAX phase. The eluent was measured via CIC in accordance with the aforementioned AOF CIC approach. With a different analytical approach utilizing high resolution-continuum source-graphite furnace molecular absorption spectrometry (HR-CS-GFMAS), Metzger et al. developed a method for the determination of EOF using in situ formation of GaF in the graphite furnace for detection. GaF is the most sensitive diatomic molecule for fluorine analysis using HR-CS-GFMAS in surface water analysis [37]. In contrast to the previously developed method by Miyake et al. using WAX as SPE material, Metzger et al. used an HLB material for SPE.
The overall aim of this study is the comparison of fluorine analysis using either CIC or HR-CS-GFMAS. Additionally, a mass balance and sum parameter analysis of OF is applied, which is schematically described in Fig. 1. This approach involves the determination of TF, followed by the complementary adsorption respectively extraction of organic fluorine. By comparing the concentration and composition of EOF/AOF, it can be estimated, which sum parameter reflects OF better and which sum parameter is therefore advantageous in risk evaluation and understanding of the environmental prevalence of the emerging pollutant PFASs. Coherently, the accurate and sensitive determination of these sum parameters using either CIC or HR-CS-GFMAS plays an equally important role for risk evaluation. Revealing the most advantageous sum parameter for organically bound fluorine with the most sensitive analytical method is therefore the aim of this study.

Scheme of a fluorine mass balance approach applying organically bound fluorine sum parameters
Materials and methods
Chemicals
Deionized water was produced using a Milli-Q® Advantage A10 System (Merck KGaA, Darmstadt, Germany) for HR-CS-GFMAS experiments (at Federal Institute for Materials Research and Testing (BAM), HR-CS-GFMASB), and for CIC experiments at the Federal Institute of Hydrology (BfG; instrumental setup in the following abbreviated as CICKO); a LaboStar DI 2 system (Siemens Evoqua Water Technologies GmbH, Günzburg, Germany) generating ultrapure water for CIC experiments at the Federal Institute for Materials Research and Testing (BAM; instrumental setup in the following abbreviated as CICB). Nitric acid (65%, p.a.) was purchased from ChemSolute® (Th. Geyer GmbH & Co. KG, Renningen, Germany) and subboiled prior use via a two-stage custom made subboiling system (PicoTrace Subboiling Kuppel-Destille SCD, PicoTrace GmbH, Göttingen, Germany). Zirconium, calcium, magnesium, and palladium solutions (all Certipur® grade purchased from Merck KGaA, Darmstadt, Germany) were used as HR-CS-GFMASB modifiers and PIN platform (Analytik Jena AG, Jena, Germany) coating reagents respectively. Aqueous sodium acetate solution 10 g/L as HR-CS-GFMASB modifier was prepared from sodium acetate trihydrate (BioChemica grade, AppliChem GmbH, Darmstadt, Germany). Gallium(III) nitrate hydrate (trace metals basis, 99.999%) was purchased from Sigma-Aldrich. Fluoride standard solution and ortho-phosphate standard solution were obtained from Merck (all 1000 mg/L; Certipur®, Merck KGaA, Darmstadt, Germany). Methanol was purchased from Merck (hypergrade for LC-MS; LiChrosolv®, Merck KGaA, Darmstadt, Germany). Sodium carbonate and sodium hydrogen carbonate stock solutions were obtained from Thermo Fisher Scientific (0.5 M, Thermo Fisher Scientific GmbH, Dreieich, Germany). Activated carbon was purchased from Analytik Jena (50–150 μm, from Analytik Jena AG, Jena, Germany). 4-Fluorobenzoic acid (99%; purified by sublimation) was purchased from Merck (Merck KGaA, Darmstadt, Germany). Sodium nitrate (99.5%) was purchased from ChemSolute® (Th. Geyer GmbH & Co. KG, Renningen, Germany). Ammonium fluoride (p.a.) was obtained from Merck (Supelco®, Merck KGaA, Darmstadt, Germany). Ammonium solution (25%) was purchased from Merck (Suprapur®, Merck KGaA, Darmstadt, Germany). For determination of accuracy during HR-CS-GFMASB measurement, the following fluoride-containing-certified reference material (CRM) was used: ION-96.4 environmental matrix reference material (c(F) = 0.123 ± 0.034 mg/L, Environment and Climate Change Canada, Canada).
Sampling
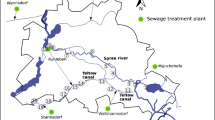
Water samples from the river Spree were taken on 4th of June 2020 on ten spots along its way through Berlin, Germany. Coordinates of the sampling locations were tracked using a GPSMAP® 64SX (Garmin Ltd., Olathe, USA) and are listed in Table 1. A map of the sampling locations is shown in Fig. 2. Each sample was collected at 20–30 cm depth under the water surface and 1.5–2.0 m distance to the riverbank using a leached sample bottle (LDPE high performance bottles, VWR, Darmstadt, Germany) mounted on a telescope pole. Sample bottles were conditioned with river water before filling. On each spot, 6 samples of 500 mL were collected. Water temperature, conductivity, pH value, and O2 concentration were measured in a separate vessel using a Multi 3430 Set G (Wissenschaftlich-Technische Werkstätten, Weilheim, Germany). Measured environmental parameters for each location are summarized in Table 1. Water samples and two blanks (deionized water) were filtered on the day of sample collection using nitro cellulose membrane filters with a pore size of 0.45 μm (LABSOLUTE®, Th. Geyer GmbH & Co. KG, Renningen, Germany) and stored in a refrigerator at 4 °C in the dark to reduce the potential growth of microorganisms.

Sampling locations along the river Spree in Berlin
Total fluorine analysis
To determine the amount of total fluorine (TF), 1 mL of each filtrated sample was directly analyzed in triplicate by means of contrAA 800 HR-CS-GFMAS system (Analytik Jena AG, Jena, Germany) and the software ASpect CS 2.2.2.0 (Analytik Jena AG, Jena, Germany). HR-CS-GFMASB measurements were performed following a protocol of Metzger et al. [9]. Zirconium-coated graphite furnaces with PIN platforms were prepared and conditioned as described previously [9]. Absorption of GaF formed in situ in the graphite furnace was measured at a wavelength of 211.248 nm. Injection of the sample as well as modifiers was conducted as follows: 2 μL deionized water, 16 μL sample, 9 μL 1 g/L gallium solution, 3 μL 10 g/L sodium acetate solution, 3 μL modifier mix (consisting of 0.1% (v/v) of palladium, 0.05% (v/v) of magnesium matrix modifier, and 20 mg/L zirconium standard) and 2 μL deionized water. For quantification, an external calibration of aqueous fluoride standard with concentrations of 0, 40, 80, 120, 160, 180, 200, 220, and 250 μg/L was used. To prevent enrichment of the analytes through evaporation of solvents, each sample vessel was covered with Parafilm® M purchased from Th. Geyer (Th. Geyer GmbH & Co. KG, Renningen, Germany). Samples were measured in instrumental triplicates.
Additionally, TF analysis was carried out using CICB consisting of a combustion system (AQF-2100H, Mitsubishi Chemical, Tokyo, Japan) and an IC (ICS Integrion, Thermo Fisher Scientific GmbH, Dreieich, Germany) controlled by the software Chromeleon 7.2.10 (Thermo Fisher Scientific GmbH, Dreieich, Germany). The combustion unit consisted of an autosampler (ASC-210) connected to the induction furnace unit (AQF-2100H) maintained at constant 1050 °C operated by the NSX 2100 software (instrumental setup is summarized in Table 2 and Table 3). Before use, all ceramic boats were thermolytically cleaned for at least 15 min at 1050 °C to avoid organic contamination. A liquid sample (0.5 mL) was loaded on a ceramic boat with a pipet (Transferpette, Brand GmbH + CO KG, Wertheim, Germany) and investigated via CICB. Hydropyrolysis during combustion was enabled by a constant flow of dry O2 (300 mL/min) and water supplied argon (150 mL/min). Combustion gases were absorbed in 5 mL of a freshly prepared 0.1 mM NH3 absorption solution within the gas absorption unit (GA-210). Ion chromatography was performed using Dionex IonPac AG22 guard column (2 × 50 mm) as guard column and Dionex IonPac AS22 (2 × 250 mm) as analytic column (column temperature 30 °C), operated with an eluent consisting of 4.5 mM Na2CO3 and 1.4 mM NaHCO3 and flow rate of 0.3 mL/min. Fluoride ions were detected by a conductivity detector using 250 mM H3PO4 as suppressor regenerant. For calculation of detected peak areas and fluoride concentrations, the chromatography data system Chromeleon 7.2.10 was used. A seventeen-point standard calibration curve was prepared at 0.002, 0.004, 0.006, 0.008, 0.01, 0.02, 0.04, 0.06, 0.08, 0.1, 0.2, 0.4, 0.6, 0.8, 1.0, 2.0, and 5.0 mg/L fluoride using stock NH4F solutions (c(F) = 1000 mg/L).
Solid-phase extraction of extractable organically bound fluorine
SPE was carried out following the optimized protocol of Metzger et al. [9] and was done in triplicate for each sample and in duplicate for deionized water as blanks. Therefore, HLB-SPE cartridges (OASIS®, Waters, Eschborn, Germany) and a vacuum chamber (HyperSep™, Thermo Fisher Scientific GmbH, Schwerte, Germany) were used. The SPE cartridges were rinsed with 3.0 mL methanol and twice with 3.0 mL of an acidified aqueous solution (deionized water acidified with HNO3 to pH 2). The valves were closed, and the solid phases were covered with 2.5 mL of acidified aqueous solution (pH 2). Before the extraction step, the pH value of each filtrated sample was adjusted to pH 2 using HNO3. 250 mL of each sample was vacuumed through the cartridges; the solid phase was rinsed two times with 3.0 mL of the acidified aqueous solution (pH 2) and vacuum dried for 30 min. The extracted compounds were eluted by means of 1 mL methanol. Eluates were then evaporated to dryness in a vacuum spin evaporator system (RVC 2-25 CDplus, Christ Martin Gefriertrocknungsanlagen GmbH, Osterode am Harz, Germany) and stored until further analysis in a refrigerator at 4 °C in the dark. Before the measurement using HR-CS-GFMASB as described above (see Total fluorine analysis), samples were re-dissolved in 1 mL of methanol/water (1:1; v/v). For EOF calibration, a mixture of methanol/water (1:1; v/v) was used as solvent, resulting in concentrations at 0, 5, 10, 20, 40, 60, 80, 100, 120, 160, 200, and 250 μg/L fluoride.
Combustion ion chromatography analysis was conducted at the Federal Institute for Materials Research and Testing (CICB). Therefore, 0.2 mL aliquots of the re-dissolved EOF samples were loaded on quartz wool (0.2 g)-packed ceramic boats, carefully evaporated prior to combustion and combustion gases absorbed in 4 mL of a freshly prepared 0.1 mM NH3 absorption solution within the gas absorption unit (GA-210). The same calibration curve was used as for TF determination.
Adsorption and combustion of organic bound fluorine
Determination of the AOF in river water samples was divided in three steps, following a modified protocol of ISO 9562:2004-09: (i) adsorption of the organic fluorine on AC columns, (ii) combustion of the AC and absorption of released hydrogen fluoride in a trapping solution, and (iii) quantitative measurement of fluoride in the trapping solution using both IC and HR-CS-GFMAS.
-
(i)
For the enrichment step, the pH value of each filtrated river water sample and each methodic blank, consisting of deionized water, was adjusted to pH 2 using HNO3. Samples were prepared as triplicates. Aliquots of 100 mL were automatically vacuum pumped through triplex quartz containers (Analytik Jena AG, Jena, Germany) packed with two times 55–60 mg AC (50–150 μm, from Analytik Jena AG, Jena, Germany) and once with a cotton pellet (4.0 mm, Orbis Dental Handelsgesellschaft mbH, Münster, Germany). The adsorption columns were washed with 25 mL of an aqueous sodium nitrate solution (c(NaNO3) = 0.01 mol/L) to remove ionic fluoride.
-
(ii)
For combustion, the AC was transferred quantitatively into ceramic sample boats and hydropyrolyzed in a combustion device (AQF-2100H, A1 Enviroscience GmbH, Düsseldorf, Germany) at 1000 °C (CICKO). During the combustion process, a carrier gas flow of 200 mL/min (Ar) and oxygen gas flow of 400 mL/min were applied. Additionally, an argon stream-supported water supply (100 mL/min) was used according to Wagner et al. [36]. CICKO combustion parameters are summarized in Table 4. During combustion, the adsorbed organically bound fluorine compounds were converted into HF, which was trapped in 10 mL of an aqueous phosphate solution (5 mg/L).
-
(iii)
The trapping solution was split for analysis via IC and HR-CS-GFMASB.
(i) and (ii) steps of AOF analysis were conducted at the BfG in Koblenz, Germany. One set of trapping solutions out of the sample triplicates was shipped to Berlin by overnight express in cooled boxes and stored immediately in a refrigerator at 4 °C in the dark to be measured by means of HR-CS-GFMASB as described above (see Total fluorine analysis). HR-CS-GFMASB calibration solutions for AOF measurements contained additionally 5 mg/L phosphate, prepared by dilution of an ortho-phosphate standard solution to match the matrix of the trapping solution, resulting in concentrations at 0, 1, 2, 5, 10, 15, 20, 30, 40, 60, 80, and 100 μg/L fluoride.
For IC analysis of the AOF trapping solutions at the BfG, an 881 compact IC pro system (Metrohm GmbH & Co. KG, Filderstadt, Germany) equipped with a conductivity detector was used. IC was performed using Metrosep A Supp 5 Guard/4.0 as guard column and Metrosep A Supp 5 250/4.0 as analytical column (column temperature 45 °C), operated with an eluent consisting of 3.2 mM Na2CO3 and 1.0 mM NaHCO3. The flow rate was 0.7 mL/min. Parameters of the method are summarized in Table 5. For external calibration and quantification of fluoride, 4-fluorobenzoic acid purchased from Merck (Merck KGaA, Darmstadt, Germany) was dissolved in methanol. Calibration solutions were prepared by dilution of the 4-fluorobenzoic acid solution with deionized water to end up with the following calibration curve: 0, 4, 6, 8, 10, 20, 40, 60, 80, 100, 200, 400, 600, 800, and 1000 μg/L fluoride.
Limit of detection/limit of quantification
Instrumental limit of detection (LOD) and limit of quantification (LOQ) were determined for fluorine analysis using HR-CS-GFMASB and CIC. Calculation was conducted according to DIN 32645 [38]. Therefore, 10 blank measurements (deionized water) and a calibration of the same day were taken. Subsequently, blank standard deviation (SD) was calculated, divided by the slope of the calibration curve, and multiplied by 3, resulting in the instrumental LOD value. Factor 10 was used for the determination of the instrumental LOQ.
Data analysis
All data plots were created using Origin®2020 software (OriginLab Corporation, Northampton (MA), USA). Linear regressions and confidence intervals for the scatter plot (see Comparison of AOF determined via CIC and EOF determined via HR-CS-GFMAS, Results and discussion section) were calculated also using Origin®2020. (Relative-)Standard deviations and mean values were calculated using Microsoft Excel (Office 365 ProPlus, Redmond (WA), USA).
Figure 3 displays the sample pretreatment and analysis scheme deployed within this study.

Sample pretreatment and analysis scheme
Results and discussion
Determination of LOD and LOQ
Results for LOD and LOQ are shown in Table 6. HR-CS-GFMASB LOQ was the lowest with 2.7 μg/L, while CIC LOQs were around 10 μg/L. TF, EOF, and AOF concentrations mostly exceeded the LOQ of all instruments. Only one EOF sample (sample location 8) out of a triplicate was below CICB LOQ and ten AOF samples (sample locations 1, 2, 3, 4, 6, and 10) out of the triplicates were below the CICKO LOQ. Thus, according to the obtained LOD and LOQ, all instrumental methods are suitable for TF and EOF determination. For the analysis of AOF via CIC, a higher concentration factor should be chosen for quantitative measurements.
Total fluorine analysis
TF was determined using HR-CS-GFMASB and CICB and results are shown in Fig. 4. Samples were measured in technical and methodical triplicates. For HR-CS-GFMASB, mean concentrations varied around ~ 190 μg/L with maximum concentrations of 213.5 μg/L at sampling location 8 and minimum concentration of 169.8 μg/L at location 3. Relative SD of three independent samples was between 4.2 and 10.6%. For CICB, mean concentrations varied around ~ 210 μg/L with maximum concentrations of 269.8 μg/L at sampling location 3 and minimum concentration of 147.6 μg/L at location 9. Relative SD was between 0.9 and 8.3%. Similar concentration ranges were published for fluoride by Berliner Wasserbetriebe along the river Spree in Berlin [39]. On average, CICB concentrations for TF were ~ 20 μg/L higher than using HR-CS-GFMASB. Additionally, relative SD was lower during CICB TF determination compared with HR-CS-GFMASB. As mentioned above, TF concentration mostly depends on inorganic fluoride concentration [40]. Recently published data for the rivers Rhine and Moselle indicated that maximum concentrations were around ~ 130 μg/L respectively ~ 180 μg/L fluoride [41]. Hence, concentrations at Spree with ~ 200 μg/L were of the same order of magnitude.

Total fluorine concentrations in Spree river water samples determined using HR-CS-GFMASB (square) and CICB (triangles). Error bars are based on n = 3 ± SD (methodical triplicate). The mean CRM fluorine concentration was 146.31 μg/L (acceptable according to the manufacturer). For sample locations, refer to Fig. 2 and Table 1
Concentrations of sample locations 5, 6, 8, and 10 were in good agreement between the instrumental methods, while for sample locations 1, 2, 3, and 4, CICB tended to provide higher TF concentrations in comparison to HR-CS-GFMASB. For samples 7 and 9, CICB tended to provide lower TF concentrations in comparison to HR-CS-GFMASB. Overall, TF concentrations between the two instrumental methods were in the same order of magnitude. Higher TF concentrations provided by CICB at sample locations 1, 2, 3, and 4 could be due to carry-over effects or cross contamination during the measurements. A higher uncertainty arises due to the instrumental methodology of CICB while trapping HF, which results from the dilution of the samples by a factor of 20 during the trapping process. Variations of the trapping solution volume could also lead to variations, especially for sample locations 7 and 9, in which lower concentrations were detected for CICB in comparison to HR-CS-GFMASB. While a comparison of IC and HR-CS-GFMAS for fluoride was published before and results were in better agreement compared to this study [41], combustion-coupled IC might result in higher uncertainty compared to fluoride determination using IC solely. The intended purpose of using CIC rather than IC in this study was to provide a better comparison of total fluorine, because IC analysis provides only the detection of inorganic fluorine species, while CIC provides results for both inorganic and organic fluorine species summarized as TF.
AOF analysis
During the first two steps of AOF determination (see Adsorption and combustion of organic bound fluorine, Materials and methods section), analytes were adsorbed onto AC and, during combustion, converted and absorbed as HF in a trapping solution. For ICKO measurements, trapping solutions were directly analyzed. The trapping solution of one of the methodic triplicates was collected and analyzed using HR-CS-GFMASB (see Fig. 5). Furthermore, all AOF values are corrected for the methodic blank value according to von Abercron et al. [34]. Methodic blanks were relatively high compared to the instrumental blank values of both systems, resulting in low corrected analyte values (some were even negative). This means that the determined concentrations are lower than the methodological blank concentrations; hence, concentrations are below the methodological detection limit. Samples were measured in technical triplicates using HR-CS-GFMASB and results are shown in Fig. 5. The dashed line (Fig. 5) indicates the trend of the methodical triplicate for CICKO AOF concentrations. Due to the higher standard deviation of the methodical mean values, the overall trend that HR-CS-GFMASB and CICKO AOF results were similar along the river Spree in Berlin could not be shown (see inlay Fig. 5). To better compare the results generated using CICKO and HR-CS-GFMASB, only one sample out of the triplicate (values above LOQ) was analyzed by means of both methods (see Fig. 5).

Blank-corrected AOF concentrations in Spree river water samples determined using HR-CS-GFMASB (square) and CICKO (circle). The dashed line shows mean values of methodical triplicates of AOF sample analyzed using CICKO. The inlay figure shows AOF concentrations analyzed using CICKO in methodical triplicate. Error bars are based on n = 2 and n = 3 ± SD for HR-CS-GFMASB as well as CICKO (for the inlay figure in the top right corner); n = 1 for CICKO (without error bars). The mean CRM (diluted 1:1 in a 10 ppm aqueous phosphate solution) fluorine concentration was 63.34 μg/L (acceptable according to the manufacturer). For sample locations, refer to Fig. 2 and Table 1. + = n = 2; * = n = 1; # = value was set to “0”—due to negative values upon blank correction
AOF values determined by HR-CS-GFMASB varied up to about 0.9 μg/L with the lowest concentration at sampling location 7 and the highest concentration at location 2. Those determined by CICKO varied between about 0.3 and 1.5 μg/L also with the lowest concentration at sampling location 7 and the highest concentration at location 2. HR-CS-GFMASB and CICKO AOF results showed a similar trend along the river Spree in Berlin. The higher AOF concentrations for all samples determined using CICKO compared to HR-CS-GFMASB could be due to potential systematic blank value problems. Additionally, all AOF values were nearby the instrumental LOQ of CICKO, which enhanced the uncertainty. Overall, the trends for samples 1–8 were in good agreement between HR-CS-GFMASB and CICKO, while AOF concentrations determined using CICKO shifted upwards because of potential systematic blank value problems.
CICKO AOF concentrations with about 0.3–1.5 μg/L were in good agreement with previously determined AOF concentrations by Wagner et al. with 0.45–2.5 μg/L for WWTPs, surface waters, and ground waters, with concentrations near the LOQ of 0.3 μg/L [36]. Furthermore, Willach et al. determined similar AOF concentrations in the range of 0.88–1.98 μg/L for WWTP effluents and surface waters [33].
EOF analysis
Organic fluorine was extracted using HLB-SPE and methanol as eluent in methodic triplicate. The resulting extracts were aliquoted and analyzed by means of HR-CS-GFMASB and CICB. Results are shown in Fig. 6. In order to assure the same quality level, EOF values were corrected for methodic blank values according to von Abercron et al. [34]. EOF concentrations determined using HR-CS-GFMASB varied around 0.05–0.55 μg/L, while CICB EOF concentrations were lower, ranging up to 0.22 μg/L. EOF concentrations between CICB and HR-CS-GFMASB were in best agreement at sample locations 1, 3, and 4 with mean differences < 0.05 μg/L. The highest differences were observed at sample locations 8, 9, and 10 with mean differences > 0.3 μg/L. SDs of CICB EOF triplicates were relatively high compared to HR-CS-GFMASB as shown in Fig. 6. While CICB EOF values were relatively consistent, HR-CS-GFMASB analysis revealed potential EOF hot spots at sample locations 2, 8, 9, and 10.

Blank-corrected EOF concentrations in Spree river water samples determined using HR-CS-GFMASB (square) and CICB (triangle). Error bars according to n = 3 ± SD (methodical triplicate). The mean CRM (diluted 1:1 with methanol) fluorine concentration was 56.4 μg/L (acceptable according to the manufacturer). For sample locations, refer to Fig. 2 and Table 1. *Two samples out of the triplicate were negative after blank correction and hence, values were omitted
Although Miyake et al. used WAX-SPE and CIC, the herein published EOF concentrations determined using HLB-SPE and HR-CS-GFMASB with about 0.05–0.55 μg/L were in good agreement with their EOF values (0.093 μg/L in unpolluted water and 0.562 μg/L at a contaminated site) [40].
EOF values published by Metzger et al. determined using the same SPE method and HR-CS-GFMAS were also in a similar range (0.05–0.30 μg/L for river water samples) [9].
While EOF concentrations determined using HR-CS-GFMASB and CICB showed the same trend for sample locations 1 and 3–7, EOF concentrations determined using HR-CS-GFMASB provided higher concentration values at all sampling locations. The highest differences observed between EOF concentrations determined using HR-CS-GFMASB and CICB at sampling locations 2, 8, 9, and 10 could possibly be related to potential loss of volatile HF during CICB measurements, possibly because of a leaky device or incomplete combustion of the samples. Furthermore, the variation of the trapping solution could be an important factor to consider, which might lead to higher variations of EOF concentrations determined using CICB. The trend of the EOF concentrations determined using HR-CS-GFMASB cannot be accurately recovered using CICB probably because the concentrations were near the LOQ of CICB. This could possibly lead to a higher uncertainty for concentrations near the LOQ. Therefore, HR-CS-GFMASB provided a lower LOQ and was more sensitive especially while conducting EOF analysis. Overall, EOF concentrations were in the same order of magnitude while determined using either HR-CS-GFMASB or CICB. EOF concentrations presented in this study compared to EOF concentrations described in the literature were also in the same order of magnitude.
Comparison of AOF determined via CIC and EOF determined via HR-CS-GFMAS
As shown above, different analytical methods (CIC ↔ HR-CS-GFMAS) are providing comparable results that were in the same order of magnitude for each sum parameter (TF, AOF, as well as EOF). Methodical triplicates of EOF and AOF are compared for the following discussion as well as in Fig. 7.

Comparison of EOF determined via HR-CS-GFMASB (square) and AOF determined via CIC (triangle). Means of methodical triplicates are shown; error bars refer to n = 2 and n = 3 ± SD (methodical triplicates). The mean CRM (diluted 1:1 with methanol) fluorine concentration was 56.35 μg/L (acceptable according to the manufacturer). For sample locations, refer to Fig. 2 and Table 1. + One sample out of the triplicate was negative after blank correction. * Two samples out of the triplicate were negative after blank correction and hence not taken into account
On average, the quotient between AOF determined via CICKO and EOF determined via HR-CS-GFMASB on each sampling location was a factor of about 4, resulting in a slope of about 0.25 in the scatter plot (see Fig. 8). SDs of AOF values were notably higher compared to the SDs of EOF values (average SD values were for AOF 0.54 μg/L and for EOF 0.02 μg/L). A similar trend between AOF and EOF along the sampling locations was observed. The mean values are in best agreement at sampling locations 3 and 7. Highest differences of the mean sum parameter values were observed at sampling locations 2 and 10, which could be due to different selectivity of AOF and EOF, resulting in varying compositions of the measured samples for AOF and EOF. HR-CS-GFMASB provided noticeably less variation while EOF concentration ranges were similar compared to CICKO AOF concentrations. Inferring, EOF analysis using HR-CS-GFMAS is less time consuming, more sensitive, and more precise, and for the future prospective, possibly more relevant than AOF analysis conducted by CIC.

Scatter plot for the comparison of AOF (determined using CICKO) and EOF (determined using HR-CS-GFMASB) with linear regression (black line) and 95% confidence interval (gray area). The equation for the linear regression is y = 0.284 (± 0.143)x + 0.039 (± 0.111)
By plotting the EOF against the AOF, the scatter plot in Fig. 8 was obtained. The slope of 0.284 (± 0.143) expresses that AOF values are on average systematically higher than EOF values. All values but two (sample locations 2 and 9) were inside the 95% confidence interval. Overall, results were in good agreement and similar trends between determined sum parameters and instrumental approaches could be observed (see Fig. 7).
Mass balance
Proportionally, AOF determined by CICKO made up 0.11–0.51% of mean TF (determined using CICB) along the river Spree in Berlin. EOF determined by HR-CS-GFMASB made up 0.04–0.28% of mean TF (determined using HR-CS-GFMASB) along the river Spree in Berlin. In this study, it could be shown that TF is mainly depended on the inorganic fluoride content. In this context, the results are consistent with the previously published data by Miyake et al. [40]. Despite its small proportion, the OF is crucial due to the extreme environmental persistence and bioaccumulation of PFASs as well as potential severe negative health effects.
Conclusion
HR-CS-GFMAS vs. CIC
HR-CS-GFMAS and CIC are both powerful devices in (organically bound) fluorine trace analysis. HR-CS-GFMAS analysis is faster, more sensitive, and more precise compared to CIC respectively IC for fluorine analysis in the low microgram per liter range. When using combustion-coupled IC, the injection of a sample aliquot (~ 200–500 μL) in the sample boat and trapping of HF after combustion in a volume of ~ 10 mL results in high dilution factors (~ 1:20–1:50), which is disadvantageous for detection of low concentrations. Furthermore, the volume of the trapping solution varies, which results in different dilution factors during triplicate measurements of a sample. Additionally, potential loss of volatile HF, possibly because of a leaky device, or incomplete combustion, leads to an underestimation of the fluorine concentration, which can be reduced by means of a basic trapping solution. Consequently, the direct analysis of samples via HR-CS-GFMAS is preferable for EOF determination. Since only < 1% of TF depends on EOF or AOF, the sensitivity and precision in the lower microgram per liter concentration range of the analytical setup is more relevant for risk evaluation. Therefore, the outcome of our comparison study is that HR-CS-GFMAS is beneficial compared to CIC to determine OF.
AOF vs. EOF
Because of the higher AOF values compared to the EOF values, the AOF seems to represent a higher proportion of the OF. It could be concluded that even lower concentrations of OF are thus better recorded. On the other hand, determined EOF values scattered less and blank value correction had a negligible effect, making the EOF the more precise parameter. The overall higher concentrations of AOF samples could be due to contamination during adsorption of analytes on AC, washing off of inorganic fluoride and the subsequent combustion of AC. Since the determined EOF values are systematically lower than determined AOF values, the OF extraction could be incomplete using HLB phase SPE. According to the systematically lower EOF values, HLB-SPE is indeed more effective than WAX-SPE but further optimization for more accurate determination of OF is needed. With further optimization, EOF might be the superior sum parameter than AOF, but currently, EOF and AOF are equally important in risk evaluation.
The herein presented study is the first comparative study on HR-CS-GFMAS⇔CIC as well as AOF⇔EOF.
Change history
27 September 2021
A Correction to this paper has been published: https://doi.org/10.1007/s00216-021-03675-z
References
Berger R, Resnati G, Metrangolo P, Weber E, Hulliger J. Organic fluorine compounds: a great opportunity for enhanced materials properties. Chem Soc Rev. 2011;40(7):3496–508. https://doi.org/10.1039/c0cs00221f.
Steele M, Griffith C, Duran C. Monthly variations in perfluorinated compound concentrations in groundwater. Toxics. 2018;6(3):56. https://doi.org/10.3390/toxics6030056.
Buck RC, Franklin J, Berger U, Conder JM, Cousins IT, de Voogt P, et al. Perfluoroalkyl and polyfluoroalkyl substances in the environment: terminology, classification, and origins. Integr Environ Assess Manag. 2011;7(4):513–41. https://doi.org/10.1002/ieam.258.
Wu Y, Romanak K, Bruton T, Blum A, Venier M. Per- and polyfluoroalkyl substances in paired dust and carpets from childcare centers. Chemosphere. 2020;251:9. https://doi.org/10.1016/j.chemosphere.2020.126771.
Mumtaz M, Bao YX, Li WC, Kong LX, Huang J, Yu G. Screening of textile finishing agents available on the Chinese market: an important source of per- and polyfluoroalkyl substances to the environment. Front Env Sci Eng. 2019;13(5):10. https://doi.org/10.1007/s11783-019-1145-0.
Trier X, Granby K, Christensen JH. Polyfluorinated surfactants (PFS) in paper and board coatings for food packaging. Environ Sci Pollut Res. 2011;18(7):1108–20. https://doi.org/10.1007/s11356-010-0439-3.
Laitinen JA, Koponen J, Koikkalainen J, Kiviranta H. Firefighters’ exposure to perfluoroalkyl acids and 2-butoxyethanol present in firefighting foams. Toxicol Lett. 2014;231(2):227–32. https://doi.org/10.1016/j.toxlet.2014.09.007.
Sharma BM, Bharat GK, Tayal S, Larssen T, Becanova J, Karaskova P, et al. Perfluoroalkyl substances (PFAS) in river and ground/drinking water of the Ganges River basin: emissions and implications for human exposure. Environ Pollut. 2016;208:704–13. https://doi.org/10.1016/j.envpol.2015.10.050.
Metzger M, Ley P, Sturm M, Meermann B. Screening method for extractable organically bound fluorine (EOF) in river water samples by means of high-resolution-continuum source graphite furnace molecular absorption spectrometry (HR-CS GF MAS). Anal Bioanal Chem. 2019;411(19):4647–60. https://doi.org/10.1007/s00216-019-01698-1.
Shigei M, Ahren L, Hazaymeh A, Dalahmeh SS. Per- and polyfluoroalkyl substances in water and soil in wastewater-irrigated farmland in Jordan. Sci Total Environ. 2020;716:8. https://doi.org/10.1016/j.scitotenv.2020.137057.
Huber S, Haug LS, Schlabach M. Per- and polyfluorinated compounds in house dust and indoor air from northern Norway - a pilot study. Chemosphere. 2011;84(11):1686–93. https://doi.org/10.1016/j.chemosphere.2011.04.075.
Fauconier G, Groffen T, Wepener V, Bervoets L. Perfluorinated compounds in the aquatic food chains of two subtropical estuaries. Sci Total Environ. 2020;719:11. https://doi.org/10.1016/j.scitotenv.2019.135047.
Gronnestad R, Vazquez BP, Arukwe A, Jaspers VLB, Jenssen BM, Karimi M, et al. Levels, patterns, and biomagnification potential of perfluoroalkyl substances in a terrestrial food chain in a Nordic skiing area. Environ Sci Technol. 2019;53(22):13390–7. https://doi.org/10.1021/acs.est.9b02533.
Mottaleb MA, Petriello MC, Morris AJ. High-throughput UHPLC-MS/MS measurement of per- and poly-fluorinated alkyl substances in human serum. J Anal Toxicol. 2020;44(4):339–47. https://doi.org/10.1093/jat/bkz097.
Duffek A, Conrad A, Kolossa-Gehring M, Lange R, Rucic E, Schulte C, et al. Per- and polyfluoroalkyl substances in blood plasma – results of the German environmental survey for children and adolescents 2014–2017 (GerES V). Int J Hyg Environ Health. 2020;228:113549. https://doi.org/10.1016/j.ijheh.2020.113549.
Abdallah MAE, Wemken N, Drage DS, Tlustos C, Cellarius C, Cleere K, et al. Concentrations of perfluoroalkyl substances in human milk from Ireland: implications for adult and nursing infant exposure. Chemosphere. 2020;246:6. https://doi.org/10.1016/j.chemosphere.2019.125724.
Muir D, Bossi R, Carlsson P, Evans M, De Silva A, Halsall C, et al. Levels and trends of poly- and perfluoroalkyl substances in the Arctic environment – an update. Emerg Contam. 2019;5:240–71. https://doi.org/10.1016/j.emcon.2019.06.002.
Sunderland EM, Hu XDC, Dassuncao C, Tokranov AK, Wagner CC, Allen JG. A review of the pathways of human exposure to poly- and perfluoroalkyl substances (PFASs) and present understanding of health effects. J Expo Sci Environ Epidemiol. 2019;29(2):131–47. https://doi.org/10.1038/s41370-018-0094-1.
UNEP - United Nations Environment Programme. The new POPs under the Stockholm Convention. Stockholm Convention. http://www.pops.int/TheConvention/ThePOPs/TheNewPOPs/tabid/2511/Default.aspx. 2019. Accessed July 1st, 2020.
Ritscher A, Wang ZY, Scheringer M, Boucher JM, Ahrens L, Berger U, et al. Zurich statement on future actions on per - and polyfluoroalkyl substances (PFASs). Environ Health Perspect. 2018;126(8):5. https://doi.org/10.1289/ehp4158.
OECD. Working Towards A Global Emission Inventory of PFASs: Focus on PFCAs – status quo and the way forward. In: OECD Environment, Health and Safety Publications Series on Risk Management, No. 30, Paris, 2015.
OECD. Toward a new comprehensive global database of per- and polyfluoroalkyl substances (PFASs). In: OECD Environment, Health and Safety Publications Series on Risk Management, No. 39, Paris, 2018.
Appleman TD, Higgins CP, Quinones O, Vanderford BJ, Kolstad C, Zeigler-Holady JC, et al. Treatment of poly- and perfluoroalkyl substances in US full-scale water treatment systems. Water Res. 2014;51:246–55. https://doi.org/10.1016/j.watres.2013.10.067.
Ghisi R, Vamerali T, Manzetti S. Accumulation of perfluorinated alkyl substances (PFAS) in agricultural plants: a review. Environ Res. 2019;169:326–41. https://doi.org/10.1016/j.envres.2018.10.023.
Zafeiraki E, Vassiliadou I, Costopoulou D, Leondiadis L, Schafft HA, Hoogenboom R, et al. Perfluoroalkylated substances in edible livers of farm animals, including depuration behaviour in young sheep fed with contaminated grass. Chemosphere. 2016;156:280–5. https://doi.org/10.1016/j.chemosphere.2016.05.003.
Zafeiraki E, Gebbink WA, Hoogenboom R, Kotterman M, Kwadijk C, Dassenakis E, et al. Occurrence of perfluoroalkyl substances (PFASs) in a large number of wild and farmed aquatic animals collected in the Netherlands. Chemosphere. 2019;232:415–23. https://doi.org/10.1016/j.chemosphere.2019.05.200.
Ingelido AM, Abballe A, Gemma S, Dellatte E, Iacovella N, De Angelis G, et al. Serum concentrations of perfluorinated alkyl substances in farmers living in areas affected by water contamination in the Veneto Region (Northern Italy). Environ Int. 2020;136:10. https://doi.org/10.1016/j.envint.2019.105435.
Janda J, Nodler K, Brauch HJ, Zwiener C, Lange FT. Robust trace analysis of polar (C-2-C-8) perfluorinated carboxylic acids by liquid chromatography-tandem mass spectrometry: method development and application to surface water, groundwater and drinking water. Environ Sci Pollut Res. 2019;26(8):7326–36. https://doi.org/10.1007/s11356-018-1731-x.
Coggan TL, Anumol T, Pyke J, Shimeta J, Clarke BO. A single analytical method for the determination of 53 legacy and emerging per- and polyfluoroalkyl substances (PFAS) in aqueous matrices. Anal Bioanal Chem. 2019;411(16):3507–20. https://doi.org/10.1007/s00216-019-01829-8.
Lee YM, Lee JY, Kim MK, Yang H, Lee JE, Son Y, et al. Concentration and distribution of per- and polyfluoroalkyl substances (PFAS) in the Asan Lake area of South Korea. J Hazard Mater. 2020;381:13. https://doi.org/10.1016/j.jhazmat.2019.120909.
Scott BF, Moody CA, Spencer C, Small JM, Muir DCG, Mabury SA. Analysis for perfluorocarboxylic acids/anions in surface waters and precipitation using GC−MS and analysis of PFOA from large-volume samples. Environ Sci Technol. 2006;40(20):6405–10. https://doi.org/10.1021/es061131o.
Dimzon IK, Westerveld J, Gremmel C, Fromel T, Knepper TP, de Voogt P. Sampling and simultaneous determination of volatile per- and polyfluoroalkyl substances in wastewater treatment plant air and water. Anal Bioanal Chem. 2017;409(5):1395–404. https://doi.org/10.1007/s00216-016-0072-1.
Willach S, Brauch H-J, Lange FT. Contribution of selected perfluoroalkyl and polyfluoroalkyl substances to the adsorbable organically bound fluorine in German rivers and in a highly contaminated groundwater. Chemosphere. 2016;145:342–50. https://doi.org/10.1016/j.chemosphere.2015.11.113.
von Abercron E, Falk S, Stahl T, Georgii S, Hamscher G, Brunn H, et al. Determination of adsorbable organically bound fluorine (AOF) and adsorbable organically bound halogens as sum parameters in aqueous environmental samples using combustion ion chromatography (CIC). Sci Total Environ. 2019;673:384–91. https://doi.org/10.1016/j.scitotenv.2019.04.068.
German Institute for Standardization. DIN EN ISO 9562:2005-02. Water quality - determination of adsorbable organically bound halogens (AOX). Berlin: Beuth; 2005.
Wagner A, Raue B, Brauch HJ, Worch E, Lange FT. Determination of adsorbable organic fluorine from aqueous environmental samples by adsorption to polystyrene-divinylbenzene based activated carbon and combustion ion chromatography. J Chromatogr A. 2013;1295:82–9. https://doi.org/10.1016/j.chroma.2013.04.051.
Gleisner H, Welz B, Einax JW. Optimization of fluorine determination via the molecular absorption of gallium mono-fluoride in a graphite furnace using a high-resolution continuum source spectrometer. Spectroc Acta Pt B-Atom Spectr. 2010;65(9–10):864–9. https://doi.org/10.1016/j.sab.2010.08.003.
German Institute for Standardization. DIN 32645:1994–05. Decision limit, detection limit and determination limit. Berlin: Beuth; 1994.
Berliner Wasserbetriebe (BWB). Analysendaten der Wasserwerke. In: Jahresmedianwerte 2019 | Annual average values. https://www.bwb.de/de/analysedaten-nach-postleitzahlen.php. 2020. Accessed July 9th, 2020.
Miyake Y, Yamashita N, Rostkowski P, So MK, Taniyasu S, Lam PKS, et al. Determination of trace levels of total fluorine in water using combustion ion chromatography for fluorine: a mass balance approach to determine individual perfluorinated chemicals in water. J Chromatogr A. 2007;1143(1–2):98–104. https://doi.org/10.1016/j.chroma.2006.12.071.
Ley P, Sturm M, Ternes TA, Meermann B. High-resolution continuum source graphite furnace molecular absorption spectrometry compared with ion chromatography for quantitative determination of dissolved fluoride in river water samples. Anal Bioanal Chem. 2017;409(30):6949–58. https://doi.org/10.1007/s00216-017-0647-5.
Funding
Funding was from the German Research Foundation (DFG - Deutsche Forschungsgemeinschaft; reference number: ME 3685/4-1, project number 358057020), the German Environment Agency (Umweltbundesamt—UBA; project number: FKZ: 3719343070 and FKZ: 3718263200), and the German Federal Ministry of Economic Affairs and Energy (BMWi; ZIM program 16KN076702 “PerFluSan-PFTSan” and 16KN076724 “MIDRAPA”).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original online version of this article was revised: Retrospective open access order.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gehrenkemper, L., Simon, F., Roesch, P. et al. Determination of organically bound fluorine sum parameters in river water samples—comparison of combustion ion chromatography (CIC) and high resolution-continuum source-graphite furnace molecular absorption spectrometry (HR-CS-GFMAS). Anal Bioanal Chem 413, 103–115 (2021). https://doi.org/10.1007/s00216-020-03010-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-020-03010-y