
Abstract
Wilson’s disease (WD), which might lead to acute liver failure, is an inherited disorder characterized by accumulation of copper (Cu2+) in the brain, the liver, and other vital organs. In the clinic, decreased serum alkaline phosphatase (ALP) concentration is used for WD diagnosis. But to the best of our knowledge, using a fluorescent probe to simultaneously detect multiple factors in WD (e.g., Cu2+, pyrophosphate (PPi), and ALP) has not been reported. Herein, we rationally designed a fluorescent switch (E)-8-((4-methylbenzylidene)amino)napthalen-1-amine (L) and successfully applied it for sequential and selective detections of Cu2+, PPi, and ALP in vitro, in living cells and synovial fluid samples with “Off,” “On,” and “Off” fluorescence signals, respectively. Considering the obvious correlations among Cu2+, PPi, and ALP in WD, we envision that our fluorescent probe L could be applied to in vitro diagnosing WD in the near future.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Copper is present as trace element in the human body. It is crucial for various physiological activities like functioning of proteins, expressing genes [1], and operating of the human nerve system [2]. Dearth of copper ion (Cu2+) results in myelopathy [3] and exalted level of Cu2+ may lead to kidney and liver damage [4]. For sensitive discernment of Cu2+, different techniques such as surface plasmon resonance (SPR) [5], atomic absorption/emission spectroscopy (AAS/AES) [6], inductively coupled plasma mass spectroscopy (ICPMS) [7], and surface-enhanced Raman scattering (SERS) [8] have been successfully applied hitherto. However, costly equipment and sophisticated and well-trained personnel are required for these approaches.
Small molecule–based (SMB) systems are potentially able to overcome the obstacles. For examples, nucleic acid sensors [9] and new “Off” (or “On”) fluorescent probes [10,11,12,13,14] have been introduced for the detection of Cu2+ with excellent sensitivity and good accuracy. However, these systems also have some snags. Often they require harsh reaction conditions, tedious sample preparation procedures, and chemical and physical interference with coexisting metal ion or inadequate biocompatibility [15].
Beside cations, anions are also essential in biological processes and industrial units and have gained attention in the last decades [16]. One biologically significant anion is pyrophosphate (PPi) which is product of adenosine triphosphate (ATP) metabolism [17]. PPi concentration provides indispensible information about replication of DNA [18]. The eminent level of PPi in the synovial fluid has been suggested as a disease marker in arthritis patients; hence, PPi serves as biomarker for calcium pyrophosphate dihydrate (CPPD) crystal deposition or chondrocalcinosis [19, 20]. Until now, various techniques had been used for PPi detection, such as colorimetric assays [21], electrooptical methods [22], and fluorescence assays [23]. Advantages of fluorescent chemosensors are their low cost, high spatial resolution and sensitivity, and short response time [24]. The strong binding interaction between metal ion and PPi opens different ways to design metal ion complex–based fluorescent switch as useful approach for PPi detection [25].
Alkaline phosphatase (ALP) is an indispensible enzyme which catalyzes the dephosphorylation of proteins, small molecules, and nucleic acids [26]. Deviated ALP level might induce various disorders such as diabetes, bone diseases, hepatobiliary disease, breast cancer, and prostatic cancer [27]. Hence, it is decisive to establish sensitive and simple methods for accurate detection of ALP level. Up to date, many different approaches have been applied to recognize ALP, such as electrochemical assays [28], fluorescence [29], chemiluminescence [30], SERS [31], and capillary electrophoresis [32]. The fluorescent assays of ALP have captivated much consideration due to their reliability, accessibility, and sensitivity [33]. Su et al. developed a carbon quantum dot–based fluorescent switch for ALP sensing [34,35,36]. Xiang and coworkers reported a fluorescent sensor for ALP quantification utilizing bright fluorescent nanosheets of g-C3N4 [28]. Liang et al. utilize sol-gel transition (hydrogelation) with “Turn-Off” fluorescence for quantitative analysis of ALP both in vitro and in LoVo cells [37].
Wilson’s disease (WD) is an inherited disorder characterized by an excess of copper in the liver and brain. Severe WD might lead to acute liver failure which is often fatal. In genomics, WD is induced by the mutation in the Atp7b gene which codes the copper-transporting P-type ATPase involved in cellular copper excretion [38]. Patients suffering from Wilson’s disease exhibit high urinary copper levels (> 100 mg/day, compared with 20–40 mg/day in healthy individuals) and increased serum-free copper levels (> 25 μg/dL, compared with 11–25 μg/dL in healthy individuals) [38]. As a result, decreased serum ALP concentration (the ratio between serum ALP to bilirubin is < 2) and non-immune hemolytic anemia are often seen in WD and the former is used for WD diagnosis in the clinic [39].
Considering the relationships among Cu2+, PPi, and ALP in WD, we designed a fluorescent switch for the sequential and selective detections of these three analytes. Such a combined detection scheme has already developed before making use of different kinds of nanoparticles. More specifically, Mo oxide quantum dots [40], carbon dots [41,42,43], Au nanoclusters [44], carbon dots and Au nanoclusters [45], silver nanocluster [46], upconversion nanoparticle [47], Eu(DPA)3@Lap nanohybrid material [48], or anionic conjugated polymers [49] were utilized. Several of these have superior sensitivity. However, these are comparably large and thus sensing resolution is limited. Also most of them contain heavy metals which can be a biocompatibility concern. The existing literature is compared with this work in Table 1.
Here, a fluorescent probe (E)-8-((4-methylbenzylidene)amino)napthalen-1-amine (L) which could specifically bind with Cu2+ was rationally designed and synthesized (see Scheme 1). Chelation between Cu2+ and L yields the L-Cu2+ complex. In this configuration, the fluorescence is “Off.” Due to the stronger binding affinity of PPi-Cu2+ than L-Cu2+, addition of PPi to above solution dissociates the L-Cu2+ complex and turns the fluorescence “On” again. Interestingly, further added ALP will hydrolyze PPi in the PPi-Cu2+ complex, freeing Cu2+ to re-chelate with L and turn the fluorescence “Off.” With this fluorescence “Off”-“On”-“Off” property, the fluorescence switch L was successfully applied for sequential and selective detection of Cu2+, PPi, and ALP in vitro and in living cells. A slightly more complex molecule with a similar purpose was reported earlier this year by Pandith et al. [50]. They used a doubly armed hydrazone-based probe (FLRHYDDFP) to continuously detect Cu2+, PPi, and ALP with high sensitivity. Compared with Pandith et al., we designed a Schiff base derivative, which has excellent ability for metal ion detection with highly flexible, efficient, selective, and easily applicable structure [51,52,53]. Our probe was synthesized easily and cheaply by one step. Besides in vitro detection and cell imaging, we also applied our probe for detection of PPi in biological synovial fluid samples. What’s more, we carried out the computational calculations to elaborate the mechanism of fluorescence quenching by ligand to metal charge transfer (LMCT).

Schematic presentation of fluorescence “Off,” “On,” and “Off” of L for selective detections of Cu2+, PPi, and ALP respectively in aqueous buffer and in cells
Experimental section
General methods
Materials
Recombinant intestinal ALP was purchased from BaoMan Inc. (Shanghai, China) (one unit is the enzyme activity that cleaves 1 μmol of the standard substrate per minute at 37 °C). The phosphatase inhibitor complex II was purchased from Sangong Biotech Inc. (Shanghai, China) (every 10 μL ALP inhibitor complex II in culture medium containing 1 × 107 cells). All the starting materials were obtained from Sigma or Sangon Biotech. Commercially available reagents were used without further purification, unless noted otherwise.
Methods
1H NMR and 13C NMR spectra were recorded on a Bruker Model AV 300 system. ESI mass spectra were obtained on a Finnigan LCQ Advantage ion trap mass spectrometer (Thermo Fisher Corporation) that was equipped with a standard ESI source, respectively. A F-4600 fluorescence spectrophotometer (Hitachi High-Technologies Corporation, Japan) was used to record fluorescence spectra with excitation wavelengths set to 310 nm. Cell imaging was carried out on a IX71 fluorescence microscope (Olympus, Japan).
Cell experiments
MTT assay
The cervical cancer HeLa cell lines of passage four were kindly provided by Prof Wang, School of Life Science, University of Science and Technology of China. HeLa cells were cultured in Dulbecco’s modified Eagle medium (DMEM), augmented with streptomycin (100 μg/mL) and 10% fetal bovine serum. The cells were cultured in dishes and stored at 37 °C with 5% CO2. 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay was used to calculate the cytotoxicity of the probe. HeLa cells were grown into a 96-well cell culture plate for 12 h with 3 × 103 cells/well concentration in humid atmosphere. The solutions of L in 100 μL DMEM (100 μL/well) at various concentrations (6.25, 12.5, 25, 50, and 100 μM) were added to each well carefully. These cultured plates were incubated in humid atmosphere for another 24, 48, or 72 h. In total, 5 mg/mL MTT (10 μL/well) solution was added to every well of the 96-well plate. After 4-h incubation, DMSO (100 μL) was added to each well to dissolve the formazan. The data were obtained at 570/680 nm using an enzyme-linked immunosorbent assay (ELISA) reader (VARIOSKAN FLASH). The following formula was used to calculate the percentage viability of cells: viability (%) = (mean of absorbance value of treatment group/mean of absorbance value of control) × 100.
Cell imaging
Cultured HeLa cell dishes were carefully washed with PBS (pH 7.4) and then 10 μM L in serum-free DMEM was added to each cell culture dish. These dishes were stored at 37 °C for 1 h prior to imaging. For Cu2+ imaging, cells were again incubated for 30 min with Cu2+ at a concentration of 10, 20, or 30 μM in serum-free DMEM followed by imaging. For PPi imaging, abovementioned cells were further incubated with various concentrations of PPi (20 μM, 40 μM, or 60 μM) for 30 min prior to imaging. Later, these cells were split into two groups (with/without ALP inhibitor) for time course fluorescence imaging of ALP activity at 30, 60, and 120 min.
Fluorescence sensing of PPi in synovial fluids of arthritis patients
The synovial fluids of arthritis patients were collected from the Shanghai Ninth Hospital, Shanghai Jiao Tong University School of Medicine. The patients have given their consent for the procedure and the doctors have the respective permissions to draw synovial fluids. Two samples were from arthritis patients while one sample was obtained from a healthy person as control. Prior to the fluorescence sensing, the synovial fluid samples were centrifuged with an ultrafiltration device (molecular weight cutoff 50 kDa; Millipore Amicon Ultra) at 6000 rpm for 20 min. Then, these samples were diluted with HEPES buffer (pH 7.4, 10 mM) in order to be consistent with the linear range of our method. For the fluorescent sensing of PPi in the synovial fluids, 10 μL diluted synovial fluid was added to the solution containing 5 μM L and 5 μM Cu2+ in HEPES buffer (pH 7.4, 10 mM) and incubated for 30 min.
Density functional theory calculations
To better evaluate the nature of coordination between L and Cu2+, the optimized structures of L and its corresponding Cu2+ complex have been calculated. All calculations were carried out by density functional theory (DFT) formalism implemented in Gaussian 09 quantum chemistry software package. Time-dependent DFT (TDDFT) calculation at the hybrid function density functional theory B3LYP level was carried out to find the excited states; molecular orbitals were built with the GaussView package.
Syntheses and characterizations of L and L-Cu2+
The study was started with the synthesis of L. Briefly, 1,8-diaminonaphthalene in ethanol was added dropwise to an equivalent 4-methyl benzaldehyde and the colorless solution turned to light yellowish rapidly. Then, the mixture was heated to reflux for 2 h (ESM Scheme S1). The pure light yellowish solid product L was obtained and characterized after purification (ESM Figs. S1–S3). After synthesis, we first investigated the spectroscopic properties of L. The excitation spectrum of L in HEPES buffer (10 mM, 4% DMSO) was recorded (ESM Fig. S4). Further on, the absorption spectrum was observed at various pH values ranging from 5 to 9 in HEPES buffer (10 mM, 4% DMSO) at RT. UV-vis spectra of L at various pH values did not show obvious changes, suggesting that the chemical structure of L was stable to pH variations (ESM Fig. S5).
Results and discussion
In vitro detection of Cu2+
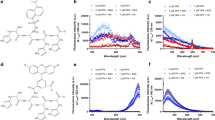
After confirming the fluorescence property of L, we tested its capability for Cu2+ detection. An equivalent amount of Cu2+ was added into the abovementioned L solution. We observed a sharp decrease of the fluorescence (ESM Fig. S6A), probably due to entrapment of Cu2+ in L to form L-Cu2+ complex. Photographs showed that the light yellowish solution of L changed to a blackish green one after Cu2+ addition (ESM Fig. S6B). In detail, with the gradual addition of Cu2+ (from 0 to 40 μM), we observed a continuous decrease of the fluorescence peak of 40 μM L, accompanied by a 23-nm redshift from 415 to 438 nm (Fig. 1a). The FI at 415 nm and Cu2+ concentration showed a linear relation (Y = 3095.32 − 74.35*X, R2 = 0.98) over the range of 0–40 μM and the limit of detection (LOD) of Cu2+ was measured to be 2.60 μM according to the 3σ method (Fig. 1b), which is comparable with those of recently reported fluorescence probes for Cu2+ detection (Tab. S1). When the Cu2+ concentration exceeded 40 μM (L:Cu2+ = 1:1), no further decrease of the FI was observed, suggesting the binding stoichiometry between L and Cu2+ is 1:1. Electrospray ionization mass spectrum (ESI/MS) of L after the addition of 1 equiv. Cu2+ showed the dominant ionic peak in the spectrum has a m/z value of 393.08, which corresponds to [L-Cu2+] (ESM Fig. S7). Job’s plot analysis also indicated that the binding stoichiometry between L and Cu2+ is 1:1 (ESM Fig. S8). According to the reported method [54], the binding constant was calculated to be 1.263 × 105 M−1. To further confirm the formation of the complexes, we directly synthesized CuCl2L complex by heating and stirring the dichloromethane/methanol solution of L with CuCl2 (1:1.2) at 50 °C for 3 h [55]. Transmission electron microscopy (TEM) images revealed the nanorod structure of CuCl2L with an average diameter of 65.3 ± 3.0 nm (ESM Fig. S9A). Energy-dispersive X-ray spectrometer (EDS) spectra of CuCl2L proved the existence of Cu in the complex (ESM Fig. 9B). The X-ray diffraction (XRD) analysis was employed to further identify CuCl2L complex formation. The XRD patterns of ligand, CuCl2, and L-Cu2+ complex were carefully analyzed, We observed that there were no specific peaks for ligand L, suggesting that the ligand is non-crystalline in nature. For CuCl2, an obvious clear peak was observed [56, 57]. However, for L-Cu2+ complex, we observed that some peaks were quite similar to CuCl2 peaks while some new peaks were also observed, suggesting that L-Cu2+ complex is successfully formed (ESM Fig. S10).

a Fluorescence spectra of 40 μM L in the presence of Cu2+ (0, 5, 10, 15, 20, 25, 30, 35, or 40 μM) in HEPES buffer (pH 7.4, 10 mM, 4% DMSO) at RT. λex = 310 nm. b The fitted calibration line in the linear region of 0–40 μM Cu2+. c Fluorescence emissions of Cu2+ at RT were measured with 40 μM L before and after the addition of 3 equiv. of various ions (Mg2+, Cr2+, Cd2+, Ag+, Pb2+, Zn2+, Ni2+, Co2+, Ca2+, Fe2+, and Fe3+ or mix) in HEPES buffer (pH 7.4, 10 mM). λex = 310 nm, λem = 415 nm. d Fluorescence responses of L to various ions under a UV lamp (top row) and photographs of L in presence of different metals (bottom row). These experiments were performed in triplicate; error bar represents standard deviation
To evaluate the activity of newly designed probe, selectivity and interference are two important parameters. Especially for probes having biomedical applications, a selective response toward the target over various competing species is needed. Therefore, the selectivity study of L to Cu2+ over various metal ions, e.g., Co2+, Zn2+, Ni2+, Mg2+, Ag+, Pb2+, Cr2+, and Cd2+, was conducted. Moreover, interference from all above metal ions with the Cu2+ detection by L was investigated. As shown in Fig. 1c, L showed excellent selectivity and interference for Cu2+ detection among abovementioned metal ions. Besides, photographs showed clear color changes in cuvettes which contained Cu2+ alone or mixed with other metal ions; fluorescence of the samples under a UV lamp echoed that only the fluorescence from above two cuvettes was turned “Off” (Fig. 1d). These results confirmed the excellent selectivity and interference of L for Cu2+ detection in vitro.
Computational study
To elaborate the mechanism of fluorescence quenching by the energy and/or charge transfer model, we carried out the computational calculations (Fig. 2). The optimized structures of L and L-Cu2+ were calculated at ground state (Fig. 2a). Time-dependent DFT (TDDFT) calculations at the hybrid DFT B3LYP level were carried out to find out the excited states. From this calculation, we noticed that the fluorescence quenching by Cu2+ could be rationalized in terms of the occupancy of the frontier orbitals. Figure 2 b shows the calculated frontier molecular orbitals (HOMO, LUMO, LUMO+1, LUMO+2) of L and L-Cu2+. These HOMO, LUMO, and LUMO+1 of both L and L-Cu2+ showed similar electron density distribution. The LUMO+2 of L showed equally distributed density over the whole structure while that of L-Cu2+ showed the electron density particularly over methylbenzene ring and Cu2+ ion, suggesting that the electron was transferred from the diaminonaphthalene ring to Cu2+ ion. This ligand to metal charge transfer (LMCT) mechanism caused the fluorescence quenching of L upon addition of Cu2+ [58].

a Optimized structures of L (left side) and L-Cu2+ (right side). b B3LYP/6-31G*-calculated molecular orbitals of L (top row) and L-Cu2+ (bottom row)
In vitro detection of PPi
As mentioned above, the binding affinity between Cu2+ and PPi is larger than that between L and Cu2+ [59]. Thus, addition of PPi to above L-Cu2+ solution will dissociate the L-Cu2+ complex to form new PPi-Cu2+ complex and turns the L fluorescence “On” again for PPi detection. As shown in Fig. 3a, with the increase of PPi concentration, fluorescence emission of the L-Cu2+ complex at 438 nm gradually increased accompanied by a 9-nm blueshift from 438 to 430 nm. Thus, a calibration curve of the FI of the L-Cu2+ complex at 438 nm for the determination of PPi in aqueous buffer was obtained (inset of Fig. 2a). It showed a linear relationship between the FI and PPi concentrations (Y = 421.17 − 8.63*X, R2 = 0.99) over the range of 0–60 μM. The LOD of PPi in this assay was calculated to be 0.31 μM (S/N = 3), which is comparable with those of recently reported fluorescence probes for PPi detection (Tab. S2). Moreover, the selectivity of L-Cu2+ complex to PPi among different common anions (NO3−, SO42−, F−, I−, Br−, Cl−, and AcO− in this work) was carefully studied. As shown in Fig. 3b, among the tested anions, only PPi induced a turned “On” fluorescence, suggesting excellent selectivity of the L-Cu2+ complex for PPi detection.

a Fluorescence spectra of 40 μM L-Cu2+ in the presence of PPi (0, 10, 20, 30, 40, 50, or 60 μM) in HEPES buffer (pH 7.4, 10 mM, 4% DMSO) at RT (λex = 310 nm). Inset: the fitted calibration line in the linear region of 0–60 μM PPi. b FL of 40 μM L-Cu2+ complex at 438 nm with/without the addition of 5 equiv. of various common anions
Detecting pyrophosphate anion in synovial fluid samples
The potential importance of our proposed design is its ability to operate in biological samples. To further demonstrate the potential application of our method in clinical samples, we detected PPi in synovial fluids from arthritis patients and healthy controls. As shown in Fig. 4, when L-Cu2+ complex was added to the synovial fluid of healthy control, we observed a 1.2-fold increase of FI. Compare with the healthy control, the Fl of L-Cu2+ complex with two synovial fluid samples from arthritis patients could further increase ∼ 1.4 and ∼ 1.8-fold, suggesting the elevated level of PPi in synovial fluids from arthritis patients. These results promote our findings that L-Cu2+ complex is quite suitable to apply for PPi detection in arthritis patients due to strong binding interaction between Cu2+ and PPi. Synovial fluid is a very complex medium and the redshift might be attributed to adsorption of medium compounds to the molecule or interaction with medium compounds.

The fluorescence spectra of the reaction system carried out in synovial fluid from L-Cu2+ complex, healthy control, and two arthritis patients
In vitro detection of ALP
Considering that ALP is able to dephosphorylate the PPi in the PPi-Cu2+ complex to free Cu2+ to re-chelate with L, we used the abovementioned mixture solution for quantitative detection of ALP activity with fluorescence “Turn-Off.” Following the formation of Cu2+-PPi, various concentrations of ALP (0.4 to 3.0 U/mL) were added and the fluorescence spectra were recorded after 30-min incubation at RT. As shown in Fig. 5a, with the increase of ALP concentration, Fl of the mixture solution at 406 nm gradually decreased. By correlating the FI the mixture solution at 406 nm with the concentration of ALP, we obtained a calibration curve for the determination of ALP in aqueous buffer (inset in Fig. 5a). A linear relationship between the Fl at 406 nm and ALP concentration (Y = 1006.44 − 208.43*X, R2 = 0.99) was obtained (0.4–3.0 U/mL), with LOD value 0.05 U/mL (S/N = 3) of ALP in this assay, which is comparable with those of recently reported fluorescence probes for ALP detection (Tab. S3). Selectivity study of as-formed Cu2+-PPi complex in the solution to ALP over various enzymes (trypsin, lysozyme, caspase-3, and thrombin in this work) indicated that the mixture solution is selective for ALP detection in vitro with fluorescence “Turn-Off” property (Fig. 5b).

a Fluorescence spectra of 40 μM L-Cu2+ + 60 μM PPi in the presence of ALP at different concentrations (0.4, 0.6, 0.8, 1.2, 1.4, 1.8, 2.2, 2.4, 2.6, or 3.0 U/mL) in HEPES buffer (pH 7.4, 10 mM, 4% DMSO) at RT. λex = 310 nm. Inset: the fitted calibration line in the linear region of 0.4–3.0 U/mL ALP. b FL of 40 μM L-Cu2+ + 60 μM PPi solution at 406 nm in the presence of ALP (3 U/mL), trypsin (50 U/mL), lysozyme (10 mg/ mL), caspase-3 (50 U/mL), and thrombin (50 μM)
Sequential imaging of Cu2+ and PPi in living cells
Before cell imaging studies, cytotoxicity of L was investigated on living HeLa cells. MTT results indicated that, up to 100 μM and 72 h, L did not impose obvious cytotoxicity on the cells, suggesting 10 μM L was safe for live cell imaging (ESM Fig. S11). HeLa cells were incubated with 10 μM L at 37 °C for 1 h in serum-free DMEM and then washed with PBS for three times prior to imaging. Bright blue fluorescence from cell cytoplasm was observed, suggesting our probe L is cell permeable (left column of Fig. 6). Then, the cells were incubated with Cu2+ (0, 10, 20, or 30 μM) at 37 °C for 30 min. Fluorescence cell imaging showed that the Fl gradually decreased with the increase of Cu2+ concentration (ESM Fig. S12) and was effectively quenched at Cu2+ concentration of 30 μM (ESM Fig. S12 and the middle column of Fig. 6). The average FL of HeLa cells in ESM Fig. S12 was measured by using ImageJ and is summarized in ESM Fig. S13. The Fl of L in Hela cells had a 7.4-fold decrease by 30 μM Cu2+. After that, the 30 μM Cu2+-treated cells were incubated with various concentrations of PPi at 0, 20, 40, or 60 μM for another 30 min prior to imaging. Clearly, we found that cell fluorescence gradually turned “On” again (ESM Fig. S14) and reached its plataeu at 60 μM PPi concentration (ESM Fig. S14 and the right column of Fig. 6). The average FL of HeLa cells in ESM Fig. S14 was measured by using ImageJ and is summarized in ESM Fig. S15. The Fl of L-Cu2+ in Hela cells had a 7.6-fold increase by 60 μM PPi. With the “Off” and “On” fluorescence property of L after Cu2+ and PPi additions, we successfully applied L for imaging Cu2+ and PPi in living cells.

Fluorescence images (top row) and merged (DIC + FL) images (bottom row) of HeLa cells incubated with 10 μM L in serum-free DMEM for 1 h at 37 °C, washed with PBS (left column), added with 30 μM Cu2+ for 30 min (middle column) and then 60 μM PPi for another 30 min (right column) in serum-free DMEM at 37 °C prior to imaging. Scale bar: 20 μm
Imaging of ALP in living cells
In order to apply our probe for imaging ALP activity in living HeLa cells, we split above PPi-treated cells into two groups: one group was treated with ALP inhibitor prior to imaging while the other was without any pretreatment. In detail, the 30 μM Cu2+-treated and 60 μM PPi-treated cells were incubated with/without phosphatase inhibitor complex II (10 μL ALP inhibitor complex II in 1 mL culture medium) and fluorescence images were taken at 30, 60, and 120 min at 37 °C after inhibitor addition. The results indicated that the Fl inside the inhibitor-treated cells maintained over the observation time (top row in Fig. 7a and black line in Fig. 7b). However, Fl inside the cells without any pretreatment decreased over time (bottom row in Fig. 7a and red line in Fig. 7b). Above results suggested that the decreased FI in cells without treatment was induced by intracellular ALP which dissociated the PPi-Cu2+ complex to free Cu2+ to re-chelate with L, turning the fluorescence “Off.” With this property, our probe L could be employed for imaging intracellular ALP activity in living cells pretreated with Cu2+ and PPi.

a Fluorescence imaging of the Cu2+- and PPi-pretreated HeLa cells incubated with (top row) or without (bottom row) phosphatase inhibitor complex II and imaged at different time points (0, 30, 60, and 120 min). Scale bar: 50 μm. b FL plots of the cells in A. These experiments were performed in triplicate. Error bars represent standard deviations
Conclusion
In conclusion, we rationally designed a new fluorescent probe (E)-8-((4-methylbenzylidene)amino)napthalen-1-amine (L) for sequential and selective detections of Cu2+, PPi, and ALP in vitro and in living cells with fluorescence “Off,” “On,” and “Off” signals, respectively. The working mechanisms underlying these processes are as follows: (1) chelation between Cu2+ and L yields the L-Cu2+ complex, accompanied by fluorescence “Off” due to the LMCT effect; (2) due to the stronger binding affinity of PPi-Cu2+ than L-Cu2+, addition of PPi to the L-Cu2+ solution dissociates the L-Cu2+ complex and turns the fluorescence “On”; (3) further added ALP hydrolyzes PPi in the PPi-Cu2+ complex, freeing Cu2+ to re-chelate with L and turn the fluorescence “Off.” In physiological solutions, L was successfully applied for selective detections of Cu2+, PPi, and ALP in vitro with LODs of 2.60 μM, 0.31 μM, and 0.05 U/mL, respectively. Moreover, with its property of fluorescence switch, L was successfully applied to image Cu2+, PPi, and ALP in living HeLa cells. Considering the obvious correlations among Cu2+, PPi (product of ATP), and ALP in Wilson’s disease, we envision that our fluorescent probe L could be applied to in vitro diagnose WD in the near future.
References
Kim B, Nevitt T, Thiele DJ. Mechanisms for copper acquisition, distribution and regulation. Nat Chem Biol. 2008;4:176–85.
Ricardo U, Olivares M, Gonzalez M. Essentiality of copper in humans. Am J Clin Nutr. 1998;67:952S–9S.
Schleper B, Stuerenburg HJ. Copper deficiency-associated myelopathy in a 45-year-old woman. J Neurol. 2001;248:705–6.
Zietz BP, Dieter HH, Lakomek M, Schneider H, Keslergaedtke G, Dunkelberg H. Epidemiological investigation on chronic copper toxicity to children exposed via the public drinking water supply. Sci Total Environ. 2003;302:127–44.
Lee S, Choi I, Hong S, Young YI, Lee J, Kang T, et al. Highly selective detection of Cu2+ utilizing specific binding between Cu-demetallated superoxide dismutase 1 and the Cu2+ ion via surface plasmon resonance spectroscopy. Chem Commun. 2009:6171–3.
Tokalioǧlu Ş, Kartal Ş, Elçi L. Determination of heavy metals and their speciation in lake sediments by flame atomic absorption spectrometry after a four-stage sequential extraction procedure. Anal Chim Acta. 2000;413:33–40.
Wu J, Boyle EA. Low blank preconcentration technique for the determination of lead, copper, and cadmium in small-volume seawater samples by isotope dilution ICPMS. Anal Chem. 1997;69:2464–70.
Sarkar S, Pradhan M, Sinha AK, Basu M, Pal T. Selective and sensitive recognition of Cu2+ in an aqueous medium: a surface-enhanced Raman scattering (SERS)-based analysis with a low-cost Raman reporter. Chem Eur J. 2012;18:6335–42.
Liu J, Lu Y. A DNAzyme catalytic beacon sensor for paramagnetic Cu2+ ions in aqueous solution with high sensitivity and selectivity. J Am Chem Soc. 2007;129:9838–9.
Ganguly A, Paul BK, Ghosh S, Kar S, Guchhait G. Selective fluorescence sensing of Cu(II) and Zn(II) using a new Schiff base-derived model compound: naked eye detection and spectral deciphering of the mechanism of sensory action. Analyst. 2013;138:6532–41.
Mei Y, Bentley PA, Wang W. A selective and sensitive chemosensor for Cu2+ based on 8 hydroxyquinoline. Tetrahedron Lett. 2006;47:2447–9.
Wang X, Miao Q, Song T, Yuan Q, Gao J, Liang G. A fluorescent switch for sequentially and selectively sensing copper(II) and L-histidine in vitro and in living cells. Analyst. 2014;139:3360–4.
Jo J, Lee HY, Liu W, Olasz A, Chen CH, Lee D. Reactivity-based detection of copper(II) ion in water: oxidative cyclization of azoaromatics as fluorescence turn-on signaling mechanism. J Am Chem Soc. 2012;134:16000–7.
Grinyte R, Barroso J, Díez-Buitrago B, Saa L, Möller M, Pavlov V. Photoelectrochemical detection of copper ions by modulating the growth of CdS quantum dots. Anal Chim Acta. 2017;986:42–7.
Fu Y, Ding C, Zhu A, Deng Z, Tian Y, Jin M. Two-photon ratiometric fluorescent sensor based on specific biomolecular recognition for selective and sensitive detection of copper ions in live cells. Anal Chem. 2013;85:11936–43.
Gale PA, Gunnlaugsson T. Preface: supramolecular chemistry of anionic species themed issue. Chem Soc Rev. 2010;39:3595–6.
Lee HN, Xu Z, Kim S, Swamy KM, Kim Y, Kim SJ, et al. Pyrophosphate-selective fluorescent chemosensor at physiological pH: formation of a unique excimer upon addition of pyrophosphate. J Am Chem Soc. 2007;129:3828–9.
Hirose M, Abe-Hashimoto J, Ogura K, Tahara H, Ide T, Yoshimura T. A rapid, useful and quantitative method to measure telomerase activity by hybridization protection assay connected with a telomeric repeat amplification protocol. J Cancer Res Clin Oncol. 1997;123:337–44.
Timms AEZ, Russell YRG, Brown MA. Genetic studies of disorders of calcium crystal deposition. Rheumatology. 2002;41:725–9.
Terkeltaub RA. Inorganic pyrophosphate generation and disposition in pathophysiology. Am J Physiol: Cell Physiol. 2001;281:C1–C11.
Quinlan E, Matthews SE, Gunnlaugsson T. Colorimetric recognition of anions using preorganized tetra-amidourea derived calix [4] arene sensors. J Org Chem. 2007;72:7497–503.
Anzenbacher P, Palacios MA, Jursíková K, Marquez M. Simple electrooptical sensors for inorganic anions. Org Lett. 2005;7:5027–30.
Su X, Zhang C, Xiao XJ, Xu AQ, Xu ZD, Zhao MP. A kinetic method for expeditious detection of pyrophosphate anions at nanomolar concentrations based on a nucleic acid fluorescent sensor. Chem Commun. 2013;49:798–800.
Caltagirone C, Bazzicalupi C, Isaia F, Light ME, Lippolis V, Montis R, et al. A new family of bis-ureidic receptors for pyrophosphate optical sensing. Org Biomol Chem. 2013;11:2445–51.
Zhang JF, Kim SJ, Han JH, Lee SJ, Pradhan T, Cao QY, et al. Pyrophosphate- selective fluorescent chemosensor based on 1,8-naphthalimide–DPA–Zn(II) complex and its application for cell imaging. Org Lett. 2011;13:5294–7.
Lallès JP. Intestinal alkaline phosphatase: novel functions and protective effects. Nutr Rev. 2014;72:82–94.
Goggins S, Naz C, Marsh BJ, Frost CG. Ratiometric electrochemical detection of alkaline phosphatase. Chem Commun. 2015;51:561–4.
Zhang L, Hou T, Li H, Li F. A highly sensitive homogeneous electrochemical assay for alkaline phosphatase activity based on single molecular beacon-initiated T7 exonuclease-mediated signal amplification. Analyst. 2015;140:4030–6.
Xiang MH, Liu JW, Li N, Tang H, Yu RQ, Jiang JH. A fluorescent graphitic carbon nitride nanosheet biosensor for highly sensitive, label-free detection of alkaline phosphatase. Nanoscale. 2016;8:4727–32.
Jiang H, Wang X. Alkaline phosphatase-responsive anodic electrochemiluminescence of CdSe nanoparticles. Anal Chem. 2012;84:6986–93.
Ruan C, Wang W, Gu B. Detection of alkaline phosphatase using surface-enhanced Raman spectroscopy. Anal Chem. 2006;78:3379–84.
Craig DB, Wong JY, Dovichi NJ. Detection of attomolar concentrations of alkaline phosphatase by capillary electrophoresis using laser-induced fluorescence detection. Anal Chem. 1996;68:697–700.
Pei H, Zhu S, Yang M, Kong R, Zheng Y, Qu F. Graphene oxide quantum dots@silver core–shell nanocrystals as turn-on fluorescent nanoprobe for ultrasensitive detection of prostate specific antigen. Biosens Bioelectron. 2015;74:909–14.
Liu SY, Pang S, Na W, Su X. Near-infrared fluorescence probe for the determination of alkaline phosphatase. Biosens Bioelectron. 2014;55:249–54.
Liang J, Yin BC, Wu X, Ye BC. Copper-mediated DNA-scaffolded silver nanocluster on–off switch for detection of pyrophosphate and alkaline phosphatase. Anal Chem. 2016;88:9219–25.
Qian ZS, Chai LJ, Huang YY, Tang C, Shen JS, Chen JR, et al. A real-time fluorescent assay for the detection of alkaline phosphatase activity based on carbon quantum dots. Biosens Bioelectron. 2015;68:675–80.
Dong L, Miao Q, Hai Z, Yuan Y, Liang G. Enzymatic hydrogelation-induced fluorescence turn-off for sensing alkaline phosphatase in vitro and in living cells. Anal Chem. 2015;87:6475–8.
Roelofsen H, Wolters H, Van MJ, Luyn A, Miura N, Kuipers F, et al. Copper-induced apical trafficking of ATP7B in polarized hepatoma cells provides a mechanism for biliary copper excretion. Gastroenterology. 2000;119:782–93.
Lee S, Barin G, Ackerman CM, Muchenditsi A, Xu J, Reimer JA, et al. Copper capture in a thioether-functionalized porous polymer applied to the detection of Wilson’s disease. J Am Chem Soc. 2016;138:7603–9.
Zhang L, Hong MF, Chu ZJ, Xu H, Wang SP, Zhao XJ, et al. A new copper mediated on-off assay for alkaline phosphatase detection based on MoOx quantum dots. Microchem J. 2018;141:170–5.
Choi CA, Mazrad ZAI, Ryu JH, In I, Lee KD, Park SY. Membrane and nucleus targeting for highly sensitive cancer cell detection using pyrophosphate and alkaline phosphatase activity-mediated fluorescence switching of functionalized carbon dots.J. Mater. Chem. B.2018; 6: 5992–6001.
Dai Y, Liu Z, Bai Y, Chen Z, Qin J, Feng F. A novel highly fluorescent S, N, O co-doped carbon dots for biosensing and bioimaging of copper ions in live cells. RSC Adv. 2018;8:42246–52.
Hu Y, Geng X, Zhang L, Huang Z, Ge J, Li Z. Nitrogen-doped carbon dots mediated fluorescent on-off assay for rapid and highly sensitive pyrophosphate and alkaline phosphatase detection. Sci Rep. 2017;7:5849.
Chen Y, Li W, Wang Y, Yang X, Chen J, Jiang Y, et al. Cysteine-directed fluorescent gold nanoclusters for the sensing of pyrophosphate and alkaline phosphatase. J Mater Chem C. 2014;2:4080–5.
Liu H, Jia L, Wang Y, Wang M, Gao Z, Ren X. Ratiometric fluorescent sensor for visual determination of copper ions and alkaline phosphatase based on carbon quantum dots and gold nanoclusters. Anal Bioanal Chem. 2019;411:2531–43.
Ma JL, Yin BC, Wu X, Ye BC. Copper-mediated DNA-scaffolded silver nanocluster on–off switch for detection of pyrophosphate and alkaline phosphatase. Anal Chem. 2016;88:9219–25.
Wang F, Zhang C, Xue Q, Li H, Xian Y. Label-free upconversion nanoparticles-based fluorescent probes for sequential sensing of Cu2+, pyrophosphate and alkaline phosphatase activity. Biosens Bioelectron. 2017;95:21–6.
Zhao J, Wang S, Lu S, Sun J, Yang X. A luminescent europium-dipicolinic acid nanohybrid for the rapid and selective sensing of pyrophosphate and alkaline phosphatase activity. Nanoscale. 2018;10:7163–70.
Malik AH, Hussain S, Tanwar AS, Layek S, Trivedi V, Iyer PK. An anionic conjugated polymer as a multi-action sensor for the sensitive detection of Cu2+ and PPi, real-time ALP assaying and cell imaging. Analyst. 2015;140:4388–92.
Panditha A, Bhattarai KR, Siddappaa KG, Chaeb HJ, Seo YJ. Novel fluorescent C2-symmetric sequential on-off-on switch for Cu2+ and pyrophosphate and its application in monitoring of endogenous alkaline phosphatase activity. Sens Actuator B-Chem. 2019;282:730–42.
Zhang C, Gao B, Zhang Q, Zhang G, Shuang S, Dong C. A simple Schiff base fluorescence probe for highly sensitive and selective detection of Hg2+ and Cu2+. Talanta. 2016;154:278–83.
Das K, Patra C, Sen C, Datta A, Massera C, Garribba E. EPR interpretation, magnetism and biological study of a Cu(II) dinuclear complex assisted by a schiff base precursor. J Biol Inorg Chem. 2017;22:481–95.
Naskar B, Modak R, Maiti DK, Drew MB, Bauzá A, Frontera A. A Schiff base platform: structures, sensing of Zn(II) and PPi in aqueous medium and anticancer activity. Dalton Trans. 2017;46:9498–510.
Tang XL, Peng XH, Dou W, Mao J, Zhang JR, Qin W, et al. Design of a semirigid molecule as a selective fluorescent chemosensor for recognition of Cd(II). Org Lett. 2008;10:3653–6.
Willson RA, Clayson KJ, Leon S. Unmeasurable serum alkaline phosphatase activity in Wilson’s disease associated with fulminant hepatic failure and hemolysis. Hepatology. 1987;7:613–5.
Zhang Q, Shi L, Meng X. Deep adsorption desulfurization of liquid petroleum gas by copper-modified bentonite. RSC Adv. 2016;6:9589–97.
Kobayashi Y, Ishida S, Ihara K, Yasuda Y, Morita T, Yamada S. Synthesis of metallic copper nanoparticles coated with polypyrrole. Colloid Polym Sci. 2009;287:877–80.
Jung HS, Kwon PS, Lee JW, Kim JI, Hong CS, Kim JW, et al. Coumarin-derived Cu2+-selective fluorescence sensor: synthesis, mechanisms, and applications in living cells. J Am Chem Soc. 2009;131:2008–12.
Feng X, An Y, Yao Z, Li C, Shi G. A turn-on fluorescent sensor for pyrophosphate based on the disassembly of Cu2+−mediated perylene diimide aggregates. ACS Appl Mater Interfaces. 2012;4:614–8.
Funding
This work was supported by the Ministry of Science and Technology of China (2016YFA0400904), the National Natural Science Foundation of China (Grants 21725505 and 21675145), and the Science and Technology Department of Zhejiang Province (GF18H180002).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The synovial fluid was obtained from the patients recruited at Ninth People’s Hospital, Shanghai Jiao Tong University, School of Medicine, Shanghai. The samples were collected from the patients above 18 years of age who provided written consent and agreed to participate in the study. The protocol of the study was approved by the Ethical Committee of the Ninth People’s Hospital, Shanghai Jiao Tong University, School of Medicine, Shanghai, in compliance with the Helsinki Declaration. The protocol used in this study was also approved by the Ethical Committee in Research of the University of Science and Technology of China (protocol number: USTCACUC1801025) assigned by the President of this committee.
Conflict of interest
The authors declare that they have no conflicts of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(DOCX 2.48 MB)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Kiran, S., Khatik, R. & Schirhagl, R. Smart probe for simultaneous detection of copper ion, pyrophosphate, and alkaline phosphatase in vitro and in clinical samples. Anal Bioanal Chem 411, 6475–6485 (2019). https://doi.org/10.1007/s00216-019-02027-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-019-02027-2