
Abstract
The fluorescence spectrum of bacterially bound acridine orange (AO) was investigated to evaluate its use for the rapid enumeration of bacteria. Escherichia coli ATCC 25922 samples were stained with 2 × 10−2, 2 × 10−3 or 2 × 10−4% w/v AO, followed by 3, 2 or 0 washing cycles, respectively, and fluorescence spectra were recorded using a fibre-based spectroscopic system. Independent component analysis was used to analyse the spectral datasets for each staining method. Bacterial concentration order of magnitude classification models were calculated using independent component weights. The relationship between fluorescence intensity of bound AO and bacterial concentration was not linear. However, the spectral signals collected for AO stain concentration-bacterial concentration pairs were reproducible and unique enough to enable classification of samples. When above 105 CFU ml−1, it was possible to rapidly determine what the order of magnitude of bacterial concentration of a sample was using a combination of two of the sample preparation methods. A relatively inexpensive (around US$10 per test) rapid method (within 25 min of sampling) for enumeration of bacteria by order of magnitude will reduce the time and cost of microbiological tests requiring gross concentration information.

Fluorescence spectra of bacterially bound acridine orange (AO) were used for the rapid enumeration of bacteria. Order of magnitude bacterial concentration classification models were calculated using independent components analysis of these fluorescence spectra. When above 105 CFU ml−1, it was possible to rapidly determine the order of magnitude of bacterial concentration of a sample using a combination of two sample preparation methods
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The enumeration of microorganisms, and especially bacteria, is a core task for many microbiologists. The information obtained may be used to count contaminants, e.g. to evaluate the safety of food products and water [1], to count survivors, e.g. to evaluate the effectiveness of antimicrobial agents and processes [2], and to determine the viability of inocula for applied biological processes, e.g. for bioremediation or fermentations [3].
Agar plate counts are routinely used for bacterial enumeration; a simple technique requiring serial dilution of samples to ensure the growth of isolated colonies from single cells, and incubation for an appropriate amount of time (overnight or longer) for colonies to become visible. Consequently, this method is labour intensive, will only detect cultivable cells and is slow to return results. To overcome these limitations, culture-independent techniques have been developed, but these often increase costs with the need for more skilled staff, expensive equipment and/or consumables, sometimes with a substantial increase in the limit of detection [4, 5].
In pursuit of rapid, cultivation-independent methods to quantify bacteria, fluorescence detection has proven promising. Acridine orange (AO) is one of the most commonly used fluorescence dyes for cell enumeration and has been for the last three decades [6–8]. AO is an inexpensive nucleic acid stain, fluorescing green when it binds to double-stranded DNA and red orange when it binds to single-stranded DNA or RNA. The most popular method known to reliably detect AO-stained bacteria is fluorescence microscopy, as used in the AO direct count (AODC) method (final AO concentration 1 × 10−2% w/v) developed for aquatic samples [9, 10] and the direct epifluorescence filter technique method (final AO concentration 2.5 × 10−2% w/v) developed for milk and food samples [11, 12].
The staining of bacteria with fluorescence dyes is effective for enumeration; however, microscopy is laborious and requires automation for an efficient process. The use of fluorescence dyes, including AO, propidium iodide, ethidium bromide and SYTO BC, with flow cytometry has been successfully automated in the BactoScan system, which allows for the routine assessment of milk for bacterial counts [13, 14]. The detection limits of fluorescence microscopy and flow cytometry have been reported to be in the order of 103–104 CFU ml−1 [11, 15, 16]. However, these techniques require expensive equipment.
In this study, we used a portable and cost-effective fluorimeter to enumerate AO-stained bacteria. We have used independent component analysis (ICA) to analyse and interpret the fluorescent spectra produced. The data acquired has been used to develop a classification model for determining the order of magnitude of bacterial count from the AO-bacteria fluorescence spectra collected to enumerate bacteria in an unknown sample.
Materials and methods
Bacterial strain and growth condition
Escherichia coli American Type Culture Collection (ATCC) 25922 was obtained from the ATCC through Cryosite Ltd. (Granville, NSW, Australia). E. coli was cultured overnight in Difco tryptic soy broth (TSB) (Fort Richard, Auckland, New Zealand) and subsequently sub-cultured in fresh TSB (20× dilution) and grown to reach an optical density of 0.5 at 600 nm (1 cm path length) to give a suspension of the order of 108 CFU ml−1. All broth cultures were grown at 37 °C and aerated with orbital shaking at 200 rpm.
Samples
The calibration data (N = 104 samples and n = 275 spectra, on average three per sample) for each staining protocol was comprised of measurements from at least three samples per order of magnitude of bacterial concentration. The data were collected over a total of 13 days with fresh cultures for each day of experiment and each replicate made on a different day. Each sample was treated with only one staining protocol. Six serial tenfold dilutions of a subculture were prepared in sterile deionised water to yield a series of bacterial suspensions from 102 to 108 CFU ml−1. Calibration samples were prepared using these preparations. Validation samples (N = 13, each stained with the three staining protocols considered in this paper) were measured in 1 day and included a free dye sample and suspensions of bacterial concentrations in the range of 104–108 CFU ml−1.
Standard plate count
Bacterial number in the samples was determined using the standard plate count method. For each dilution series, the sample assumed to have 102 CFU ml−1 was used to inoculate Difco tryptic soy agar (TSA) plates (Fort Richard, Auckland, New Zealand). This assumption was based on the optical density of the most concentrated bacterial solution. For each plate count, a 100-μl aliquot of the sample was spread evenly onto a TSA plate and incubated at 37 °C overnight. For each tested sample, duplicate plate counts were made.
Staining procedures with acridine orange
Stock 2% w/v AO in water was obtained from Sigma-Aldrich Corporation (Sydney, Australia) and diluted 10,100 and 1000 times in distilled water to give working solutions with final concentrations of 0.2, 0.02 and 0.002% w/v of AO. All these staining concentrations were tested with different numbers of washing cycles to determine how free AO can be efficiently removed from the sample. Only three staining/washing combinations yield spectra with significant differences per bacteria concentrations as determined from a principal component analysis of collected spectral data. These three staining protocols were investigated:
-
(1)
Final stain concentration 2 × 10−2% AO followed by three washing cycles,
-
(2)
Final stain concentration 2 × 10−3% AO followed by two washing cycles and
-
(3)
Final staining concentration 2 × 10−4% AO with no washing.
Samples were prepared in amber or aluminium foil-covered microcentrifuge tubes. Each sample consisted of a 900-μL aliquot of bacteria suspension or distilled water for free dye samples, which was incubated with 100 μL of working AO solution. Amber or aluminium foil-covered microcentrifuge tubes were used to limit exposure to external light sources and subsequent photodegradation. Samples were mixed for 3 min by vortexing and then rested in the dark for 15 min. Samples were then subjected to washing, as required, to reduce the concentration of unbound dye in the sample. A single washing cycle consisted of several consecutive steps: centrifuging the sample at 4300×g for 5 min at room temperature, removing 970 μL of supernatant, adding 970 μL of distilled water and finally vortexing the sample for 2 min to resuspend the bacteria.
Fluorescence measurement
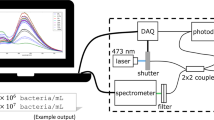
Fluorescence was measured using an all-fibre spectroscopic system (optrode) [17]. The excitation source was a 473-nm solid-state laser placed behind shutter and controlled by a data acquisition (DAQ) card to prevent photobleaching the sample and ensure synchronisation with the spectrometer to allow for exact quantification of the fluorescence signal. To monitor power fluctuations, a 2 × 2 fibre coupler was used to deliver half the excitation light to a photodiode and the signal collected by the DAQ card. The other half was used to illuminate the sample. A single fibre probe was used for excitation and fluorescence collection. All the fibres in the instrument were multimode low OH silica fibre (diameter 200 μm, NA 0.22; Thorlabs Inc., Newton, NJ, USA). A 495-nm long-pass filter before the spectrometer removed the excitation line. Spectra were collected from 400 to 790 nm using an Ocean Optics QE65000 spectrometer. The spectrometer had no slit; the fibre acts as a slit to give an effective slit width of 200 μm. E. coli did not present any native fluorescence using 473 nm excitation.
Each measurement was acquired using a laser power of approximately 10 mW at the sample with 8 ms integration time. The sample was mixed using a vortex before positioning the tip of the sample fibre probe in the middle of the sample and taking a measurement. The fibre probe was washed with 70% ethanol between each measurement. Regularly, a blank measurement was made to ensure the fibre probe was clean.
Data analysis
Dark noise was subtracted from each measurement. Each spectrum was normalised to a laser power of 10 mW and integration time of 8 ms. Subsequently, the background spectrum (saline) was subtracted from sample spectra. Any spectra with saturated signals were noted and were not included in a data matrix containing normalised spectra. All data analyses were executed using MATLAB R2014b version 8.4.0.150421 (The MathWorks, Natick, USA) software.
Independent component analysis
ICA was applied to the fluorescence spectra collected from AO-dyed bacterial solutions. If a sample is composed of multiple fluorophores, then each of these fluorophores will contribute to the fluorescence spectrum. Each fluorophore will have a specific fluorescence signature when using the excitation wavelength of interest. The amount this fluorophore contributes to the fluorescence spectrum from the sample will depend on concentration of fluorophore within the solution. Hence, a sample fluorescence spectrum is the sum of the constituent fluorophores scaled to their respective concentrations. ICA can be used to identify the spectra of the fluorophores within a sample and their proportions in the analysed sample [18].
ICA aims to maximise the statistical independence and non-Gaussianity of the pure underlying signals, which are called the independent components (ICs). There are different methods to define and maximise statistical independence. In this study, we have chosen the joint approximate diagonalization of Eigen matrices (JADE) algorithm [19]. The JADE algorithm was used because it avoids convergence issues encountered with other algorithms (e.g. FastICA) [18]. The Matlab codes of this algorithm were downloaded from http://perso.telecom-paristech.fr/%7Ecardoso/Algo/Jade/jadeR.m. ICA-by-blocks was used to determine how many ICs should be extracted for the data set [20]. In this instance, the calibration spectral dataset was divided into two blocks; data from individual days were collected into the same block. Several ICA models were calculated for each block, with one to five ICs. The models calculated for each block for a given number of ICs were compared. ICs representative of true fluorophore signals should be found in each representative subset of the overall data matrix, and these ICs from the respective blocks will be strongly correlated. If too many ICs are extracted, the ICs will contain noise characteristic of the block, and hence, these ICs from the respective blocks will not be highly correlated. Once the appropriate number of ICs was determined, ICA was applied to the full calibration dataset and the IC weightings for each sample were subsequently used in classification models for bacterial concentration.
The Matlab function mahal was used to calculate the Mahalanobis distances of each data point to clusters of data for each order of magnitude of bacterial concentration. Mahalanobis distance measures the distance between a point and a distribution, i.e. it determines how many standard deviations a sample is away from mean of a group [21]. The Mahalanobis distance calculated, which follows a chi-square distribution, was used to determine significance, p < 0.01 (2 degrees of freedom). The limit of detection was the lowest bacterial concentration that was significantly different from all other orders of magnitude investigated.
Bacterial concentration classification model
The IC weights of each sample were used to calculate classification models for the order of magnitude of the bacterial concentration. For each staining protocol, the corresponding calibration data was clustered by magnitude of bacterial concentration and the mean for each cluster was calculated. Using IC signals, the projection matrix of the validation data set was calculated. Each validation sample measurement was then assigned to an order of magnitude of bacterial concentration based on IC weights. This was done by calculating the squared Euclidean distance from the validation sample to the mean of each cluster. The validation sample was assigned to the closest cluster. Correct classification rates are the percentage of validation samples correctly assigned to magnitude of bacterial concentration.
Results
The AO fluorescence spectra
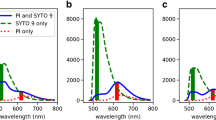
The fluorescence spectra of AO bound to bacteria is dominated by two peaks at approximately 535 and 650 nm. The position and intensity of these peaks shifted with bacterial concentration. Figure 1 represents exemplar spectra for each staining protocol, as indicated in the caption. Solutions containing 106 CFU ml−1 E. coli or less (Fig. 1a) had a single peak at ∼535 nm which shifted to shorter wavelengths at lower concentrations of bacteria. The second peak at ∼650 nm was present for higher bacterial concentrations and shifted to shorter wavelengths to become a shoulder (∼625 nm) of the first peak at lower bacterial concentrations (Fig. 1b, c). Fluorescence signals for all 107 CFU ml−1 E. coli samples (N = 7) stained with 2 × 10−2% AO followed by three washing cycles were saturated and therefore excluded from the ICA.

Exemplar spectra collected for (a) and (b) 2 × 10−2% AO staining followed by 3 washing cycles (c) and (d) 2 × 10−3% AO staining followed by two washing cycles and (e) and (f) 2 × 10−4% AO staining for 0–105 CFU ml−1 E. coli in (a, c and e), and for 106−108 CFU ml−1 E. coli in (b, d and f). Spectra are coloured by concentration: blue 108, green 107, red 106, grey 105, mint green 104, magenta 103, cyan 102, black 0 CFU ml−1
ICA was applied to data from each sample treatment individually. For the treatments 2 × 10−2% AO followed by three washing cycles and 2 × 10−3% AO followed by two washing cycles, two ICs described each spectral dataset (Fig. 2). The two ICs calculated were similar for each dataset and were dominated by features coincident with the two major peaks of AO bound in bacteria. IC1 loading had two peaks of similar intensity at 535 and 650 nm, and IC2 was the difference between these two peaks. For the sample treatment protocol 2 × 10−4% AO with no washing, three ICs were needed (Fig. 3). The third IC was dominated by an intense peak at ∼535 nm.

Independent component (IC) 1 (solid) and IC2 (dashed) signals (a) and weights (b) obtained from the 2 × 10−2% AO stain followed by 3 washing cycles data set (N = 39, n = 113). Note that the 107 CFU ml−1 samples are absent from this independent component analysis since the signal was saturated. IC1 (solid) and IC2 (dashed) signals (c) and weights (d) obtained from the 2 × 10−3% AO stain followed by 2 washing cycles data set (N = 32, n = 76). Samples in (c) and (d) are coloured by concentration following the colour scheme of Fig. 1 and use the following symbols: upward-pointing triangle 108, left-pointing triangle 107, diamond 106, square 105, asterisk 104, circle 103, cross 102, plus sign 0 CFU ml−1.

Independent component (IC) 1 (solid), IC2 (dashed) and IC3 (dotted) signals (a) and weights (b–d) obtained from the 2 × 10−4% AO staining procedure (N = 26, n = 76). Samples in (b), (c) and (d) are coloured by concentration following the colour scheme of Fig. 1 and use the following symbols: upward-pointing triangle 108, left-pointing triangle 107, diamond 106, square 105, asterisk 104, circle 103, cross 102, plus sign 0 CFU ml−1
Across all three staining treatments, fluorescence spectra measured from samples containing 106–108 CFU ml−1 E. coli were significantly different (p < 0.01 based on Mahalanobis distance test) to fluorescence spectra measured from solutions containing free dye and <106 CFU ml−1 E. coli. Furthermore, fluorescence spectra measured from all samples containing 105 CFU ml−1 E. coli and stained with 2 × 10−3% AO followed by two washing cycles were significantly different (p < 0.01) from other bacterial concentrations.
Classification models
Classification models for each staining treatment were calculated and validated with an external sample set (Table 1 and Electronic Supplementary Material (ESM) Tables S1–S4). Using the staining treatment 2 × 10−2% AO with three washing cycles, 100% of the validation samples were correctly classified. The correct order for magnitude was predicted for 77% of validation samples when using the staining conditions 2 × 10−3% AO followed by two washing cycles. The classification performance using this staining procedure was improved to 84% by adding a class for 105 CFU ml−1 (Table 2). The 2 × 10−4% AO staining protocol had the lowest agreement rate at 69%.
Discussion
Several staining protocols were trialled in this study. The outcome of AO staining is influenced by a number of variables including the ratio of AO to nucleic acid [22]. In this study, a large bacterial concentration range was investigated (102–108 CFU ml−1); hence, several staining concentrations of AO were investigated. In addition to the concentration of AO, the number of washing steps was altered to eliminate the influence of unbound AO on the fluorescence spectra measured.
The intensity of AO fluorescence was not linear with bacterial concentration, in agreement with previous observations that the ratio of AO dye to nucleic acid affects the fluorescence signal. However, unique fluorescence profiles were observed for several AO dye-bacterial concentrations pairs, and this information was used to calculate classification models for the order of magnitude of bacterial concentration.
ICA was applied to each sample treatment data set individually. ICs with negative peaks were observed for all data sets. The aim of ICA here was to calculate the spectra of the underlying fluorophores and the respective weighting of each in a sample spectrum. If two components are inter-dependent (i.e. fluorophore 1 increases as fluorophore 2 decreases), these signals are not independent [18]. Consequently, one IC will have contributions from both these components; the IC will not represent a ‘pure’ component but rather a single phenomenon. The peak centre around 650 nm increased in intensity as the peak centred around 535 nm decreased in intensity, and this behaviour is illustrated in calculated ICs for each dataset. As bacterial concentration increases, the width of the 535 nm peak increases and develops a higher wavelength shoulder and, subsequently, a peak at 650 nm when a high-enough concentration of AO is used to stain the sample. When using 2 × 10−3% AO followed by two washing cycles to prepare samples, the peak at 650 nm was not obvious in the collected fluorescence spectra; however, the ICA indicates that these spectral changes were occurring within the dataset as both peaks are present within calculated ICs. Three ICs were appropriate to describe the dataset collected from samples prepared with 2 × 10−4% AO. One of these ICs was dominated by a sharp peak at 535 nm, which probably represents the contribution of unbound AO to the collected fluorescence spectra. The other two ICs were similar to the ICs calculated for the other staining treatments, with the exception of a minimum at 535 nm. This minimum is likely a result of interference from unbound AO and potentially indicates that the 2 × 10−4% AO dataset is not as well described by the independent components analysis as the 2 × 10−2% AO followed by three washing cycles and 2 × 10−3% AO followed by two washing cycles datasets.
The staining protocol that classifies the most samples correctly was 2 × 10−2% AO stain followed by three washing cycles. All validation samples down to 105 CFU ml−1 were correctly classified using this staining treatment. The ∼535 nm peak intensities collected from 106 CFU ml−1 solutions were at least twice as large as those from solutions containing less than 106 CFU ml−1. The saturation of signals from 107 CFU ml−1 solutions (not included in the ICA and plots) provided a clear separation from the single peak spectra of 106 CFU ml−1 solutions and from the spectra with a shoulder or two peaks collected from 108 CFU ml−1 solutions. This saturation was a quick indicator of a bacterial concentration of 107 CFU ml−1 without the need for further analysis.
Other staining treatments were investigated where the concentration of AO was reduced, as were the number of washing cycles. The 2 × 10−3% AO followed by two washing cycles staining treatment did not correctly classify two of the validation samples; however, this treatment did show promise with respect to the classification of 105 CFU ml−1 samples. As such, this staining protocol is a good complement to further classify the low concentration sample identified by the previous protocol as below 106 CFU ml−1. The 2 × 10−4% AO staining protocol had the lowest agreement rate, failing to identify solutions in the order of 107 CFU ml−1. The fluorescence of 106 and 107 CFU ml−1 samples was quenched relative to the fluorescence signal of all other bacteria concentrations, and since the solutions were not washed, the fluorescence of the free dye in the solution concealed the fluorescence from bound AO. The difference in the fluorescence of 106 and 107 CFU ml−1 samples were noticed when samples were washed. The sample containing 6.8 × 107 CFU ml−1 was predicted to be 108 CFU ml−1, similar to the 2 × 10−3% AO and two washing cycles staining protocol.
The major drawback in the method we describe is the strong signal from free AO when the concentration of bacteria is low compared to the concentration of dye. Our method (Fig. 4) therefore requires several washing steps that may lead to precision and/or accuracy issues. To overcome this limitation, we are currently investigating (a) other dyes that have a higher quantum yield when bound to nucleic acid [23] and (b) automating the staining/washing protocol by using microfluidic devices [24, 25]. We believe that both of the above should contribute to the development of a device with a sensitivity relevant for industrial applications. To date, the device described in this study has been developed to offer a competitive alternative to plate counts in terms of time and costs. The actual costs of our technique relate to initial setup costs for the device and ongoing reagent costs. At present, a single prototype device can be constructed for US$20,000, but larger scale production and replacement of expensive elements may substantially reduce this. We anticipate consumable tests will be low cost; at present, these are around US$10 per test, which is competitive with agar plate testing, but substantially faster (20 min compared to days) and requiring less technician time.

Flow chart of the final method to be used to quantify samples from 108 CFU ml−1 down to 105 CFU ml−1, or below 105 CFU ml−1, using our device and technique
Overall, we conclude this study showed that our cost-effective fluorescence spectroscopy method can be used to determine the order of magnitude of bacterial concentrations greater than 105 CFU ml−1 using the procedure, as illustrated in Fig. 4. Staining with 2 × 10−2% AO stain followed by three washing cycles can be used to determine whether a solution contains <106, 106, 107 or 108 CFU ml−1. Samples with less than 106 CFU ml−1 can be tested using the staining protocol 2 × 10−3% AO stain followed by two washing cycles to determine whether the sample has a bacterial concentration in the order of 105 CFU ml−1 or less than 105 CFU ml−1. We expect that increasing the amount of calibration data will improve the classification accuracy and enable the better prediction of bacterial content of a sample. In addition, future studies should incorporate multiple species of bacteria to calculate a general model for the enumeration of bacteria by order of magnitude. We also propose to validate the protocol developed on real samples in the laboratory and then in the field in collaboration with the food industry.
The method developed in this study is fast in comparison with traditional counting methods such as AODC, where it has been reported that it can take a well-trained individual approximately 1 h per sample (using triplicate filters) to perform a count [26]. Other prototype small-scale spectrophotometers are described in the literature; however, these examples have been applied to either measuring bacterial growth by turbidity [27] or identifying the load of specific organisms by quantifying enzyme activities with fluorogenic substrates [28]. In both cases, the limit of detection and time to detection do not compare favourably with the combination of stain and spectrophotometer we describe here. Our method requires minimal operator expertise; measurement of the sample requires simply immersing a probe into the solution. The longest form of sample preparation trialled here required 25 min, and measurements required less than a minute. Fast methods for the detection of bacterial concentration by order of magnitude could be useful in biocide testing or other areas of microbiology where the Miles-Misra plating technique [29] is typically employed.
References
Reich F, Schill F, Atanassova V, Klein G. Quantification of ESBL-Escherichia coli on broiler carcasses after slaughtering in Germany. Food Microbiol. 2016;54:1–5.
Hasan J, Raj S, Yadav L, Chatterjee K. Engineering a nanostructured super surface with superhydrophobic and superkilling properties. RSC Adv. 2015;5(56):44953–9.
Mani-López E, Palou E, López-Malo A. Probiotic viability and storage stability of yogurts and fermented milks prepared with several mixtures of lactic acid bacteria. J Dairy Sci. 2014;97(5):2578–90.
Jimenez L. Rapid methods for the microbiological surveillance of pharmaceuticals. PDA J Pharm Sci Technol. 2001;55(5):278–85.
Wang Y, Salazar JK. Culture-independent rapid detection methods for bacterial pathogens and toxins in food matrices. Compr Rev Food Sci F. 2016;15(1):183–205.
McCarthy LR, Senne JE. Evaluation of acridine orange stain for detection of microorganisms in blood cultures. J Clin Microbiol. 1980;11(3):281–5.
Bhakdi SC, Sratongno P, Chimma P, Rungruang T, Chuncharunee A, Neumann HPH, et al. Re-evaluating acridine orange for rapid flow cytometric enumeration of parasitemia in malaria-infected rodents. Cytom Part A. 2007;71(9):662–7.
Viana KA, Das Graças Carvalho M, Dusse LMS, Fernandes AC, Avelar RS, Avelar DMV, et al. Flow cytometry reticulocyte counting using acridine orange: validation of a new protocol. Jornal Brasileiro de Patologia e Medicina Laboratorial. 2014;50(3):189–99.
Hobbie JE, Daley RJ, Jasper S. Use of nuclepore filters for counting bacteria by fluorescence microscopy. Appl Environ Microbiol. 1977;33(5):1225–8.
Carr SA, Vogel S, Dunbar R, Brandes J, Spear JR, Levy R, et al. Bacterial abundance and composition in marine sediments beneath the Ross Ice Shelf, Antarctica. Geobiology. 2013;11:377–95.
Pettipher GL, Mansell R, McKinnon CH, Cousins CM. Rapid membrane filtration-Epifluorescent microscopy technique for direct enumeration of bacteria in raw milk. Appl Environ Microbiol. 1980;39(2):423429.
Sierra M-L, Sheridan JJ, McGuire L. Microbial quality of lamb carcasses during processing and the acridine orange direct count technique (a modified DEFT) for rapid enumeration of total viable counts. Int J Food Microbiol. 1997;36(1):61–7.
Loss G, Apprich S, Kneifel W, Von Mutius E, Genuneit J, Braun-Fahrländer C. Short communication: appropriate and alternative methods to determine viable bacterial counts in cow milk samples. J Dairy Sci. 2012;95(6):2916–8.
Douglas Cunningham J, Connie LS. Collaborative study of the Bactoscan—an automated method for determinations of total bacteria in raw milk. Can I Food Sc Tech J. 1988;21(5):464–6.
Tortorello ML, Stewart DS, Raybourne RB. Quantitative analysis and isolation of O157: H7 in a food matrix using flow cytometry and cell sorting. FEMS Immunol Med Microbiol. 1997;19(4):267–74.
Flint S, Drocourt J-L, Walker K, Stevenson B, Dwyer M, Clarke I, et al. A rapid, two- hour method for the enumeration of total viable bacteria in samples from commercial milk powder and whey protein concentrate powder manufacturing plants. Int Dairy J. 2006;16(4):379–84.
Hewitt BM, Singhal N, Elliot RG, Chen A, Kuo J, Vanholsbeeck F, et al. Novel fiber optic detection method for in situ analysis of fluorescently labeled biosensor organisms. Environ Sci Technol. 2012;46(10):5414–21.
Rutledge DN, Jouan-Rimbaud Bouveresse D. Independent components analysis with the JADE algorithm. TrAC Trends Anal Chem. 2013;50:22–32.
Cardoso JF, Souloumiac A. Blind beamforming for non-Gaussian signals. IPRSP. 1993;140(6):362–70.
Jouan-Rimbaud Bouveresse D, Moya-González A, Ammari F, Rutledge DN. Two novel methods for the determination of the number of components in independent components analysis models. Chemometrics Intellig Lab Syst. 2012;112:24–32.
Flury B. Discrimination and classification, round 1. A first course in multivariate statistics. New York: Springer New York; 1997. p. 279–374.
McFeters GA, Singh A, Byun S, Callis PR, Williams S. Acridine orange staining reaction as an index of physiological activity in Escherichia coli. J Microbiol Methods. 1991;13(2):87–97.
Tawakoli PN, Al-Ahmad A, Hoth-Hannig W, Hannig M, Hannig C. Comparison of different live/dead stainings for detection and quantification of adherent microorganisms in the initial oral biofilm. Clin Oral Investig. 2013;17(3):841–50.
Bridle H, Miller B, Desmulliez MPY. Application of microfluidics in waterborne pathogen monitoring: a review. Water Res. 2014;55:256–71.
Jokerst JC, Emory JM, Henry CS. Advances in microfluidics for environmental analysis. Analyst. 2012;137(1):24–34.
Cragg BA, Parkes RJ. Bacterial and Archaeal direct counts: a faster method of enumeration, for enrichment cultures and aqueous environmental samples. J Microbiol Methods. 2014;98:35–40.
Maia MR, Marques S, Cabrita AR, Wallace RJ, Thompson G, Fonseca AJ, et al. Simple and versatile Turbidimetric monitoring of bacterial growth in liquid cultures using a customized 3D printed culture tube holder and a miniaturized spectrophotometer: application to facultative and strictly anaerobic bacteria. Front Microbiol. 2016;7:1381.
Tait E, Stanforth SP, Reed S, Perry JD, Dean JR. Analysis of pathogenic bacteria using exogenous volatile organic compound metabolites and optical sensor detection. RSC Adv. 2015;5(20):15494–9.
Miles AA, Misra SS, Irwin JO. The estimation of the bactericidal power of the blood. J Hyg. 1938;38(6):732–49.
Acknowledgements
The authors are grateful to the New Zealand Ministry of Business, Innovation and Employment for funding the Food Safe: real time bacterial count research programme (UOAX1411) which supported this research. Rachel Guo acknowledges the University of Auckland for the provision of a doctoral scholarship. The authors would like to thank Janesha Perera, Fang Ou and Neelam Hari for assistance in the laboratory.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
ESM 1
(PDF 119 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Guo, R., McGoverin, C., Swift, S. et al. A rapid and low-cost estimation of bacteria counts in solution using fluorescence spectroscopy. Anal Bioanal Chem 409, 3959–3967 (2017). https://doi.org/10.1007/s00216-017-0347-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-017-0347-1