
Abstract
A liquid chromatography–tandem mass spectrometry method for the analysis of seven antiviral drugs, zanamivir, ribavirin, oseltamivir, oseltamivir carboxylate, amantadine, rimantadine and arbidol, in poultry muscle is reported. The antiviral drugs were extracted from the homogenized poultry muscle sample using methanol. The extract was purified using tandem solid-phase extraction combining a cation exchange cartridge and a phenylboronic acid cartridge. To prevent excessive matrix effects, the analytes were separated from the matrix constituents using a column-switch liquid chromatography system combining a reversed-phase and a Hypercarb analytical column. Detection was carried out using tandem mass spectrometry. The method was fully validated according to 2002/657/EC [1] and proved to be adequate for quantification and confirmation of zanamivir and ribavirin at 10 μg kg−1, oseltamivir, oseltamivir carboxylate, amantadine and rimantadine at levels below 1.0 μg kg−1 and for qualitative confirmatory analysis of arbidol at levels below 1 μg kg−1.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Influenza infections continue to pose a serious problem. Recent outbreaks of avian influenza (H5N1) have had a huge economic effect in the poultry breeding sector [2, 3] but besides the economic effect, human health is at stake because avian influenza can also infect mammals including humans [4, 5]. The increasing incidence of resistance to antiviral drugs is a major concern to public health. Because strains tend to become more aggressive, adequate therapeutic strategies are needed [6].
Progress has been made by identifying several compounds that aid in the treatment of viral infections and besides the treatment of humans, these are used by farmers to protect their flocks [7]. However, concerns arose about the increasing incidence of resistance to antiviral drugs [7–12]. Several causes have been suggested including the inappropriate use of antiviral drugs in the treatment of humans during the outbreaks of avian influenza and severe acute respiratory syndrome (SARS) [13, 14]. Furthermore, it has been shown that oseltamivir carboxylate (Fig. 1) is not removed or degraded in normal sewage water treatment and thus enters the environment [15]. Waterfowl are then in a position to ingest the active oseltamivir carboxylate. This could drive the selection of resistant avian influenza A strains in the gut. Especially in countries (e.g. Southeast Asia) where waterfowl and humans come in close contact, this could lead to the spread of other strains incorporating the resistance genes and change of host [16, 17]. Finally, amantadine, oseltamivir and arbidol (Fig. 1) are allegedly being administered to poultry and other livestock in China, Southeast Asia and Russia [7, 18, 19]. To prevent excessive use of antiviral drugs the Food and Drug Administration (FDA) prohibited the non-consistent use of two types of antiviral drugs in chickens, turkeys and ducks: the amantadines, including amantadine and rimantadine, and the neuramidase inhibitors, including zanamivir and oseltamivir (Fig. 1) [20].

Chemical structures of a zanamivir, b ribavirin, c oseltamivir, d oseltamivir carboxylate, e rimantadine, f amantadine and g arbidol
To be able to detect extra-label use of antiviral drugs, analytical methods are needed that not only detect the presence of antiviral drugs in poultry muscle, but are also able to quantify the concentration present and to confirm the identity of the compound present at levels as low as reasonably possible. Single-residue methods for the quantitative analysis of antiviral drugs in biological matrices using mass spectrometry have been published for zanamivir [21, 22], ribavirin [23–26], oseltamivir [17, 27–30] (some of them including oseltamivir carboxylate [27, 30]), oseltamivir carboxylate only [31], amandatine [32–34], some of them including rimantadine [33, 35, 36], and arbidol [37, 38]. Recently, Chan et al. [39] reported a multi-compound method for the analysis of antiviral drugs in poultry muscle using zwitterionic hydrophilic interaction liquid chromatography (zic-HILIC) in combination with mass spectrometric detection. However, this method is suitable for screening purposes only and it is limited to the analysis of basic and amphoteric antiviral drugs.
For efficient control of antiviral drug usage, a quantitative confirmatory multi-method is needed that is adequate for analysis of the antiviral drugs that are applied in poultry breeding including zanamivir, ribavirin, oseltamivir including its active metabolite oseltamivir carboxylate, amantadine, rimantadine and arbidol. The main difficulty in the development of a multi-method for the analysis of antiviral drugs is their broad range of physical and chemical properties. The antiviral drugs include the extremely polar zanamivir and ribavirin, whereas arbidol is reasonably non-polar. Furthermore, the selection includes basic, amphoteric and neutral drugs.
This paper presents a tandem solid-phase extraction clean-up in combination with a column-switch liquid chromatography (LC) system for the quantitative confirmatory analysis of a broad range of antiviral drugs in poultry muscle. The necessity of using tandem SPE and two analytical columns to adequately remove matrix effects and thus to enable quantitative analysis of the antiviral drugs is demonstrated. The method was fully validated for zanamivir, ribavirin, oseltamivir, oseltamivir carboxylate, amantadine, rimantadine and arbidol. This is the first time a quantitative confirmatory method for the analysis of a broad range of antiviral compounds is presented.
Experimental
Reagents and equipment
HPLC grade methanol (MeOH) and acetonitrile (ACN) were obtained from Biosolve (Valkenswaard, the Netherlands). Formic acid (FA) and ammonia (25% solution in water) were obtained from Merck (Darmstadt, Germany). Milli-Q water was prepared using a Milli-Q system at a resistivity of at least 18.2 MΩ cm−1 (Millipore, Billerica, MA, USA). Rimantadine HCl (99% purity) and amantadine HCl (100% purity) were obtained from Sigma-Aldrich (St. Louis, MO, USA) and arbidol (99.3% purity) from Amplachem (Carmel, IN, USA). Zanamivir (100% purity), ribavirin (99.5% purity), oseltamivir phosphate (98% purity) and oseltamivir carboxylate (98% purity), and the internal standards zanamivir-13C15N2, ribavirin-13C5, oseltamivir-d 3 phosphate, oseltamivir-d 3 carboxylate, rimantadine-d 4 HCl and amantadine-d 15 HCl were obtained from Toronto Research Chemicals (North York, ON, Canada). Stock solutions of oseltamivir, amantadine, rimantadine, their corresponding isotopically labelled internal standards and arbidol were prepared at 100 mg L−1 in MeOH. Stock solutions of zanamivir, ribavirin, oseltamivir carboxylate and their isotopically labelled internal standards were prepared at 100 mg L−1 in water. All dilutions of the stock solutions were prepared fresh daily in Milli-Q water.
Sample preparation
One gram of a poultry tissue was transferred into a polypropylene (PP) centrifuge tube and 4 mL of MeOH was added while shaking using a vortex mixer. Subsequently, the tube was shaken for 15 min on a rotary tumbler. After centrifugation (3,600 g, 15 min), the upper layer was isolated by decanting it into a PP centrifuge tube and acidified by adding 20 μL of FA prior to the tandem solid-phase extraction (SPE) procedure (Fig. 2a).

Schematic representation of (a) the dual SPE procedure and (b) the LC procedure. Z zanamivir, RB ribavirin, O oseltamivir, OC oseltamivir carboxylate, AM amantadine, RM rimantadine, AB arbidol
A Phenomenex (Torrance, CA, USA) Strata-X C 200 mg/6 mL strong cation exchange SPE cartridge was conditioned with 4 mL 0.5 v/v % FA in MeOH, pH 2. A clean PP centrifuge tube was placed under the Strata-X C cartridge before the acidified sample extract, followed by 1 mL of MeOH, pH 2, was applied onto the cartridge. The total breakthrough was collected in the PP centrifuge tube (fraction A). After addition of 200 μL of 25% ammonia, fraction A was centrifuged (3,600 g, 15 min) and set aside. The Strata-X C cartridge was washed with 4 mL of MeOH, pH 2. A clean PP centrifuge tube was placed under the SPE cartridge and the antiviral compounds were eluted from the cartridge using 4 mL 0.75 v/v % ammonia in MeOH, pH 12, which was collected in the PP centrifuge tube (fraction B). An Agilent Technologies (Santa Clara, CA, USA) Bond elute phenylboronic acid (PBA) 100 mg/3 mL SPE cartridge was conditioned with 2 mL 0.5 v/v % FA in H2O, pH 2, followed by 4 mL MeOH, pH 12. The basified fraction A was applied onto the PBA cartridge, which was subsequently washed with 4 mL MeOH, pH 12. The PP centrifuge tube containing fraction B was placed under the PBA cartridge and ribavirin was eluted from the cartridge using 4 mL MeOH, pH 2, which was combined with fraction B. The solvent of the combined fractions was evaporated under a gentle nitrogen stream at 50 °C until dry. The residue was redissolved in 250 μL water and transferred into a liquid chromatography–tandem mass spectrometry (LC–MS/MS) autosampler vial.
LC–MS/MS analysis
A schematic presentation of the column-switch system is presented in the Electronic Supplementary Material Fig. S1. ‘LC 1’ consisted of an Acquity vacuum degasser, autosampler and binary solvent pump (Waters, Milford, MA, USA). ‘LC 2’ consisted of a Separations (Hendrik-Ido-Ambacht, the Netherlands) vacuum degasser, mixing chamber and two type K-1001 HPLC pumps. ‘V1’ and ‘V2’ are 6-way valves (Separations), which are operated by ‘LC 1’. ‘MS’ is a mass spectrometer Quattro Premier XE (Waters, Milford, MA, USA). ‘Column1’ is a Waters Symmetry C18 (3.0 × 150 mm, 5 μm) analytical column and ‘column 2’ is a Thermo Fisher Scientific (Waltham MA, USA) Hypercarb (3.0 × 100 mm, 5 μm) analytical column.
From the initial time ‘LC 1’ runs a gradient (solvent A, 50 mM FA in water; solvent B, 50 mM FA in ACN): 0–6.0 min, linear increase from 0% to 5% B; 6.0–14.0 min, linear increase to 100% B; 14.0–14.5 min, linear decrease to 0% B; 14.5–20.0 min, equilibration at 0% B, operating at a flow of 0.4 mL min−1. ‘LC 2’ runs a gradient using the same solvents: 0–6.0 min, 5% B; 6.0–10.0 min, linear increase to 100% B; 10.0–11.0 min, 100% B; 11.0 – 12.0 min, linear decrease to 0% B; 12.0–20.0 min, equillibration at 0% B, operating at a flow of 0.4 mL min−1. The injection volume (injected at ‘LC 1’) is 50 μL.
During phase 1, running from the initial time until 6 min, both columns are placed in series. At the start of phase 2 (6.0 min) ‘V2’ is switched and the mobile phase of ‘LC 1’ runs solely through the Symmetry C18 column and is diverted to waste, whereas the mobile phase of ‘LC 2’ runs through the Hypercarb column and enters the MS. At the start of phase 3 (10.0 min) ‘V1’ is switched and the mobile phase of ‘LC 1’ runs solely through the Symmetry C18 column and enters the MS, whereas the mobile phase running through the Hypercarb column is diverted to waste.
Detection was carried out using the mass spectrometer in positive electrospray ionization (ESI) mode. The operating parameters were capillary voltage, 1.0 kV; source offset, 50 V; cone voltage, Table 1; source temperature, 150 °C; desolvation temperature, 450 °C; cone gas flow, 100 L h−1; and desolvation gas, 600 L h−1. The antiviral compounds were fragmented using collision-induced dissociation and the selected reaction monitoring (SRM) transitions are given in Table 1. Data were acquired and processed using MassLynx 4.1 software (Waters). Zanamivir-13C15N2, ribavirin-13C5, oseltamivir-d 3, oseltamivir carboxylate-d 3, amantadine-d 15 and rimantadine-d 4 were used as the internal standards. No internal standard for arbidol was available.
Method optimisation
During method development recovery and matrix effects were monitored. Recovery was calculated by dividing the signal of a blank poultry sample with addition of the compounds before sample clean-up (matrix-matched standard, MMS) by the signal of a blank poultry muscle sample with addition of an equivalent amount of the compounds, based upon 100% recovery, after sample clean-up (matrix-matched recovery standard, MMRS). The matrix effect was calculated by dividing the signal of the MMRS by the signal of a standard solution in water at equivalent concentration. No matrix effects are present if the result of this calculation is 100%. If the result is below 100%, suppression occurs and if the result is above 100%, signal enhancement occurs.
As an alternative to the column-switch set-up, different single analytical columns were tested. zic-HILIC (Sequant, Umeå, Sweden) was tested in combination with gradient elution using ammonium formate in water and ACN or MeOH as the mobile phase, at a pH ranging from 3 to 10. Reversed-phase (RP) C18 was tested in combination with gradient elution using water and ACN or MeOH as the mobile phase, at a pH ranging from 3 to 11. Hypercarb was tested in combination with gradient elution using water and ACN, MeOH or tetrahydrofuran as the mobile phase. For each chromatographic condition, the matrix effects were evaluated on the basis of spiked poultry muscle samples using only a methanolic extraction in combination with evaporation of the extraction solvent and redissolution in a suitable solvent. We aimed for a chromatographic system that results in sufficient retention for all compounds and adequate separation of the antiviral drugs from the matrix constituents to minimize matrix effects.
SPE using strong cation exchange and PBA were tested for sample clean-up. A 50 μg kg−1 spiked methanolic sample extract was acidified and applied onto a Strata-X C cartridge. The breakthrough during application and washing were collected separately. These and the eluent were analysed using the LC–MS/MS system. The same procedure was followed for the PBA cartridges, but then the spiked methanolic extract was basified prior to application.
Method validation
A full validation was carried out according to 2002/657/EC [1]. The following parameters were determined: linearity, trueness, repeatability, within-laboratory reproducibility, decision limit (CCα), detection capability (CCβ), selectivity and robustness. The validation was carried out on three different occasions, by two different technicians, using different batches of SPE cartridges and including 21 different poultry muscle samples.
Linearity
On three different days a matrix-matched calibration line was prepared including the following calibration levels: 0, 2.5, 5, 10, 15 and 20 μg kg−1 (six data points including zero) by adding solutions of the antiviral drugs to blank poultry muscle samples. Calibration lines were constructed by plotting the peak areas normalized by the peak areas of the internal standards, versus the added concentration and carrying out least-squares linear regression. The linearity was considered acceptable if the coefficient of correlation was at least 0.990.
Trueness, repeatability and within-lab reproducibility
On each of the 3 days seven different blank poultry muscle samples were selected and analysed as such and spiked at 5, 10 and 15 μg kg−1. For each sample the level of the individual antiviral drugs was calculated using the calibration line constructed on the same day. The trueness was calculated for each level by dividing the overall average calculated concentration by the nominal concentration. According to 2002/657/EC [1] the trueness at the selected validation levels should be between 80 and 110%.
The repeatability and within-lab reproducibility were calculated using single factor analysis of variance (ANOVA). According to 2002/657/EC [1] the relative within-laboratory reproducibility (RSDRL) and relative repeatability (RSDr) are considered acceptable if they are below the value calculated from the Horwitz equation [40] and two thirds of that value, respectively (i.e. below 31.7% and 21.1%, respectively, at a validation level of 10 μg kg−1). Thompson [41], however, demonstrated that the Horwitz equation is not applicable to the lower concentration range (<120 μg kg−1) and suggested a complementary model. On the basis of this model the RSDRL and RSDr are acceptable if they are below 22% and 14.7%, respectively. The latter, more stringent criteria were adopted for this validation study. It is stated that because different poultry muscle samples were used within 1 day, the repeatability is not the pure repeatability as stated in 2002/657/EC [1]. However, using different poultry muscle samples within 1 day provides a better understanding of the between-sample variation and the validation better reflects a routine analysis situation.
Decision limit and detection capability
Within the EU no regulations for the use of antiviral drugs in food production are established. Within the USA, the FDA prohibited extra-label use of antiviral drugs [20]. Therefore, confirmation of the identity of antiviral drugs should be carried out at levels as low as reasonably possible. Therefore, the decision limit (CCα) was calculated on the basis of zero tolerance in accordance with 2002/657/EC [1] to assign samples that contain antiviral drugs and thus deviate from the blank population (α-error is 1%). CCα was calculated on the basis of the bandwidth of the signal in blank samples (n = 21) in the time window in which the analyte is expected. CCβ is the detection capability and expresses the concentration of the antiviral drugs in a poultry muscle at which the sample will be found non-compliant (β-error is 5%). CCβ was determined on the basis of the bandwidth of the signal in blank samples (n = 21) and the bandwidth of the signal in the samples spiked at 10 μg kg−1 (zanamivir and ribavirin, n = 21) or 5 μg kg−1 (other antiviral drugs, n = 21) in the time window in which the analyte is expected. The individual blank and spiked samples were analysed on three different occasions and calculations were carried out as suggested by Antignac et al. [42], but without a normalisation based on the signal of the internal standards.
Selectivity
The selectivity of the method was studied on the basis of a practical and a theoretical study. Firstly, the 21 analysed blank samples were checked for interferences at the retention times corresponding to the antiviral drugs. Secondly, databases were searched for compounds having a molecular mass equal to the antiviral drugs that could possibly be present in poultry muscle samples. Standard solutions of 100 μg L−1 of androsta-1,4-diene-3,17-dione (ADD), 5-hydroxyflunixin and tetrahydrogestrinone (THG), indicated as possibly interfering compounds, were analysed using the developed method. The resulting chromatograms were checked for interferences at the retention times corresponding to the antiviral drugs.
Robustness
The robustness of the method became clear during the optimisation of the sample preparation, chromatographic conditions and MS parameters. Additionally, some minor changes to the method were tested in duplicate: (1) extraction for 30 min instead of 15, (2) dry the PBA cartridges by applying vacuum for 1 min after applying the extract and after washing the cartridge and (3) evaporation of the solvent at 55 °C instead of 50 °C. The trueness and repeatability of the results were compared to the characteristics of the regular method.
Application to real samples
The suitability of the developed method was tested by applying it to affected sample materials. Amantadine-affected poultry and turkey muscle were a kind gift from the Food and Environmental Research Agency (FERA), York, UK. The determined levels using the developed method were compared to the indicated amantadine concentration in the samples, being approximately 4 μg kg−1 for the poultry muscle and 8–25 μg kg−1 for the turkey muscle.
Results and discussion
Chromatography
Stationary phases using three different interaction mechanisms were tested to obtain sufficient separation power and separate the antiviral drugs from matrix constituents. Therefore, a retention factor of at least 2 should be obtained. The use of zic-HILIC was reported by Chan et al. [39] for the screening analysis of some antiviral drugs. However, using zic-HILIC no retention was obtained for arbidol, the most non-polar compound we studied and which was not included in the screening method reported by Chan et al. [39]. Only slight improvement in the retention time (RT) was made by protonating arbidol (pK a 6) using a mobile phase of pH ≤ 4 and lowering the ammonium formate concentration to 5 mM. Nevertheless, insufficient retention was obtained for arbidol and severe matrix effects, even for late-eluting polar compounds like zanamivir and ribavirin, were observed (Fig. 3).

Matrix effects (signal MMRS/signal standard) of the antiviral drugs when using zic-HILIC (light grey), C18 (medium grey), Hypercarb (dark grey) and the column-switch set-up (black). 100% corresponds to no matrix effects
Using RP chromatography applying C18 columns, zanamivir eluted at the dead time of the system and only slight retention was obtained for ribavirin (retention factor of 0.4). Although the matrix effects significantly decreased for ribavirin, zanamivir was completely suppressed by matrix constituents (Fig. 3) and was therefore not detectable, even at a concentration of 50 μg kg−1 in poultry muscle.
When using Hypercarb as the stationary phase, all antiviral drugs, including the most polar compounds zanamivir and ribavirin obtained retention (retention factor of 3.0 and 4.6 respectively). The matrix suppression for zanamivir decreased from 100% using zic-HILIC or C18 to 97% (Fig. 3) resulting in the detection of zanamivir in poultry muscle at a concentration of 50 μg kg−1. For ribavirin, instead of signal suppression, signal enhancement was observed (signal MMRS/signal standard 255%). The main disadvantage of the use of a Hypercarb column is that arbidol, the most non-polar compound studied, does not elute from the column. It contains a free phenyl group, which strongly interacts with the column material, preventing its elution even when using tetrahydrofuran as the mobile phase.
To be able to analyse the antiviral drugs within one run, a column-switch system combining a C18 column with a Hypercarb column was developed. A schematic of the LC procedure is given in Fig. 2b. Zanamivir and ribavirin are not or hardly retained by the C18 column, elute from it within 5 min and are subsequently retained on the Hypercarb column. After that, on the Hypercarb column, zanamivir and ribavirin are separated from the matrix constituents using a gradient and are directly detected by MS. In the meantime the other antiviral compounds are retained on the C18 column. After elution of zanamivir and ribavirin from the Hypercarb column, the other antiviral drugs are eluted from the C18 column by increasing the organic content of the mobile phase, passing by the Hypercarb column and entering the MS directly. The total run time, including equilibration of both columns, is 20 min. As presented in Fig. 3, the matrix effects using the column-switch LC system are tremendously improved for zanamivir.
Sample clean-up
To further reduce the matrix effects and therefore to facilitate detection and confirmation at a concentration of at least 10 μg kg−1 in poultry muscle, additional sample clean-up proved to be necessary. Two different SPE systems were tested: Strata-X C and PBA. The relative amounts of the antiviral drugs in the breakthrough during application of the sample extract on the cartridge, the breakthrough during washing and the eluent are presented in Fig. 4a, b.

Relative amount of antiviral drugs in the breakthrough during application of a 50 μg kg−1 spiked poultry muscle sample (light grey), the breakthrough during washing (dark grey) and the elution (black) of (a) the Strata-X C and (b) the PBA SPE cartridge
Because ribavirin remains neutral at pH 2, it does not interact with the Strata-X C cartridge and elutes during application of the extract and the washing procedure. The other antiviral compounds are positively charged at pH 2 and therefore are retained. Only zanamivir, having the lowest pK a of all the antiviral compounds (pK a 3.8), shows 10% breakthrough during the washing procedure which is considered acceptable. The Strata-X C cartridge is considered suitable for the clean-up of all antiviral drugs included except for ribavirin.
The PBA material has multiple interaction mechanisms. The strongest is the covalent binding at high pH with cis-diol groups, as present in ribavirin. Other mechanisms include van der Waals interaction, π–π bonding and ionic interaction. Therefore, the results using this type of material are hard to predict. Ribavirin, oseltamivir, amantadine, rimantadine and arbidol are retained by the PBA cartridge (Fig. 4b). Zanamivir and oseltamivir carboxylate do not interact with the PBA material and elute during application of the extract and the washing procedure. It stands out that both compounds containing a carboxylic acid moiety are left unretained by the PBA material. No explanation was found for this observation.
From these results it is concluded that a combination of SPE materials is needed to obtain a satisfactory clean-up for all antiviral drugs included in this study. A tandem SPE procedure was developed, a schematic of which is given in Fig. 2a. All compounds except ribavirin were separated from matrix constituents by retaining them on a Strata-X C cartridge. The breakthrough of this cartridge, containing ribavirin, was basified, centrifuged and applied onto a PBA cartridge to retain ribavirin in order to separate this drug from matrix constituents as well. Eventually, the eluents of both cartridges were combined and the solvent was evaporated. Finally, the residue was redissolved in 250 μL of water. The matrix effects of the optimised clean-up procedure were determined using the C18 and the column-switch system (Fig. 5a) and the recovery of the sample preparation procedure (Fig. 5b) was determined.

(a) Matrix effect (signal MMRS/signal standard) obtained for the antiviral drugs after the clean-up procedure when using RP C18 (light grey) and the column-switch set-up (dark grey) as the chromatographic system (n = 5) and (b) the recovery of the sample clean-up procedure (n = 5)
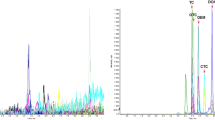
From the comparison of Fig. 5a with Fig. 3, it is concluded that the developed sample clean-up procedure strongly contributes to the decrease of the matrix effect for all compounds. The most striking is the decrease in the suppression for zanamivir from 85% to 29%. After the sample clean-up procedure, for arbidol, signal enhancement (approximately 400%) is observed instead of signal suppression. From Fig. 5a it is concluded that applying the developed sample clean-up procedure in combination with RP chromatography only is adequate for the analysis of ribavirin, oseltamivir, oseltamivir carboxylate, rimantadine, amantadine and arbidol, but the matrix effect for zanamivir is still very pronounced and as a result 50 μg kg−1 of zanamivir in poultry muscle is not detectable using solely RP chromatography. Only when using the combination of the tandem SPE sample clean-up procedure and the column-switch LC–MS/MS system, the matrix effects are sufficiently decreased, enabling detection and identification of all antiviral drugs included at relevant levels. SRM chromatograms of a blank poultry muscle sample spiked at 10 μg kg−1 with zanamivir and ribavirin are presented in Fig. 6 and those of a blank poultry muscle spiked at 2.5 μg kg−1 with the other antiviral drugs are presented in Fig. 7.

SRM chromatograms of (a) blank poultry muscle sample spiked with zanamivir and ribavirin at 10 μg kg−1 including the internal standards and (b) blank poultry muscle sample spiked with the internal standards only

SRM chromatograms of (a) blank poultry muscle sample spiked with oseltamivir, oseltamivir carboxylate, rimantadine, amantadine and arbidol at 2.5 μg kg−1 including the internal standards and (b) blank poultry muscle sample spiked with the internal standards only
Column-switch set-up versus two individual injections
As an alternative to the use of the column-switch set-up, the use of two individual injections on the RP and the Hypercarb column was tested. This solely affects the results for zanamivir and ribavirin, because the RP system remains as it is in the column-switch system. The matrix effects of zanamivir and ribavirin were evaluated after injection of a cleaned extract on the Hypercarb column and were both found to be 39%. It is observed that the matrix effects for zanamivir using injection on solely the Hypercarb column are increased compared to the column-switch set-up. It is concluded that additional matrix interferences are removed in the first phase of the column switch set-up, which is advantageous for the analysis of zanamivir. For ribavirin no statistically significant differences were observed and thus it is concluded that the column-switch system is advantageous in terms of the removal of matrix effects and the analysis time (20 min instead of 40 min). Therefore, the use of the column-switching system is preferred.
Method validation
An overview of the validation results is presented in Table 2.
For zanamivir, ribavirin, oseltamivir, oseltamivir carboxylate, amantadine and rimantadine the linearity in the range of 0–20 μg kg−1 is above the criterion of 0.990; the trueness is between 95 and 108% for all antiviral drugs which is well within the established criterion of 80–110%; the determined maximum RSDr and RSDRL are 14.3% and 15.2%, respectively, and thus below the criteria of 14.7% and 22%, respectively; therefore, the validation results comply with the established criteria. It is concluded that the presented method is adequate for quantitative trace analysis of these six antiviral drugs. For arbidol, inadequate linearity, trueness, repeatability and within-laboratory reproducibility are observed. On two validation days, the calibration lines are adequate, but on 1 day the linearity is insufficient due to two deviating calibration points. The poor trueness, repeatability and within-laboratory reproducibility of arbidol are caused by 10 out of 63 spiked samples showing a quantitative result that is 1.5–3.5 times higher than expected. This is likely to be caused by a difference in matrix interference, which plays a major role for arbidol (Fig. 5a). This effect might be overcome if a suitable internal standard for arbidol becomes available. However, the presented method is suitable for the qualitative analysis of arbidol.
For oseltamivir, oseltamivir carboxylate, amantadine, rimantadine and arbidol the calculated decision limits (CCα) vary from 0.1 to 0.6 μg kg−1 indicating that for these antiviral drugs very low levels are still detectable. The calculated detection capabilities (CCβ) of these compounds are between 0.5 and 2.5 μg kg−1 and samples spiked at 5 μg kg−1 were found to be non-compliant based upon the ion ratio criteria [1] in 100% of the cases (n = 21), indicating that the method is suitable for confirmatory analysis of these antiviral drugs at relevant levels. For zanamivir and ribavirin the calculated CCα values are 8 and 3 μg kg−1, respectively, indicating that for these drugs relevant levels are still detectable. The calculated detection capabilities (CCβ) of zanamivir and ribavirin are 15 and 9 μg kg−1, respectively. Even though the calculated CCβ for zanamivir exceeds 10 μg kg−1, samples spiked at 10 μg kg−1 with zanamivir and ribavirin were found to be non-compliant based upon the ion ratio criteria [1] in 100% of the cases (n = 21) indicating that the calculated CCβ for at least zanamivir is slightly overestimated and that the method is suitable for confirmatory analysis of zanamivir and ribavirin at relevant levels as well.
From the theoretical study hydroxyflunixin, ADD and THG were found to have the same molecular mass as one of the antiviral drugs and might theoretically interfere in the described method. Standard solutions of 100 μg L−1 of these compounds were prepared and injected into the column-switch LC–MS/MS system. No interfering signals were observed in these chromatograms and, furthermore, no significant interferences were detected in the blank samples (n = 21); therefore, the procedure is considered sufficiently selective for the antiviral drugs.
During the method development some critical steps were identified. Firstly the pH values of the solvents used during the SPE clean-up procedure are critical. The acidic methanol should be at pH ≤ 2 to retain zanamivir on the Strata-X C material and the alkaline MeOH should be at least pH 12 for the elution of amantadine and rimantadine. Incorrect pH results in a severe decrease of the recovery. Secondly, it was found critical to centrifuge the basified breakthrough of the Strata-X C cartridge before applying it onto the PBA cartridge to prevent clogging. For ruggedness testing, three slight deviations to the procedure that might occur in practice were specifically tested: (1) extraction for 30 min instead of 15, (2) dry the PBA cartridges by applying vacuum for 1 min after applying the extract and after washing the cartridge and (3) evaporation of the solvent at 55 °C instead of 50 °C. The duplicates analysed by incorporating these deviations in the method showed good trueness and acceptable duplicates, indicating that the tested processes are robust.
Application to real samples
Two amantadine-affected muscle samples were analysed using the developed method. The results were 4.9 μg kg−1 for the poultry muscle and 15.3 μg kg−1 for the turkey muscle sample. These levels are in good agreement with the indicated amantadine concentration, being approximately 4 μg kg−1 for the poultry muscle and 8–25 μg kg−1 for the turkey muscle. From this it is concluded that the developed method is applicable to real muscle samples.
Conclusions
A method using a tandem SPE sample preparation clean-up combined with column-switch LC–MS/MS was developed for the analysis of seven antiviral drugs in poultry muscle, ranging from very polar to reasonably non-polar drugs, and including basic, amphoteric and neutral compounds. The removal of matrix interferences was critical for the detection and quantification of zanamivir and ribavirin in particular. Because neither compound was sufficiently retained by using reversed-phase chromatography, a column-switch LC system combining a reversed-phase and a Hypercarb column was developed. This system showed improved separation power and is relatively easy to set-up, which is of major importance in a routine situation. Furthermore, a tandem SPE procedure proved to be mandatory for additional removal of matrix constituents. The method was fully validated according to 2002/657/EC [1] at concentrations of 5, 10 and 15 μg kg−1. Good linearity, trueness, repeatability and within-lab reproducibility were obtained for zanamivir, ribavirin, oseltamivir, oseltamivir carboxylate, amantadine and rimantadine. For arbidol these paramaters did not comply with the established criteria because the method resulted in a severe overestimation of the concentration for 16% of the spiked samples. The selectivity and robustness proved to be sufficient to apply the presented method in a routine situation enabling parallel analysis of at least 48 samples within 1 day. It is concluded that the presented method is suitable for the quantitative confirmatory analysis of zanamivir and ribavirin at levels as low as 10 μg kg−1 and for oseltamivir, oseltamivir carboxylate, amantadine and rimantadine at a concentration of 1 μg kg−1. Furthermore, the presented method is suitable for the qualitative confirmatory analysis of arbidol at a concentration of 1 μg kg−1.
References
EC (2002) Commission Decision 2002/657/EC of 12 August 2002 implementing Council Directive 96/23/EC concerning the performance of analytical methods and the interpretation of results. Off J Europ Commun L221:8–36
The Writing Committee of the World Health Organization (WHO) Consultation on Human Influenza A/H5 (2005) Avian influenza A (H5N1) infection in humans. N Engl J Med 353:1374–1385
WHO (2011) Avian influenza, fact sheet. WHO, Geneva
Malik Peiris JS, De Jong MD, Guan Y (2007) Avian influenza virus (H5N1): a threat to human health. Clin Microbiol Rev 20:243–267
Mumford E, Bishop J, Hendrickx S, Embarek P, Perdue M (2007) Avian influenza H5N1: risks at the human-animal interface. Food Nutr Bull 28:S357–S363
Von Itzstein M (2007) The war against influenza: discovery and development of sialidase inhibitors. Nat Rev Drug Discov 6:967–974
He G, Qiao J, Dong C, He C, Zhao L, Tian Y (2008) Amantadine-resistance among H5N1 avian influenza viruses isolated in Northern China. Antivir Res 77:72–76
Souza TML, Mesquita M, Resende P, Machado V, Gregianini TS, Fernandes SB, Oliveira ICD, Rosa MdCD, Marinelli R, de Azeredo-Lima CH, Motta FC, Aguiar-Oliveira MdL, Siqueira MM (2011) Antiviral resistance surveillance for influenza A virus in Brazil: investigation on 2009 pandemic influenza A (H1N1) resistance to oseltamivir. Diag Microbiol Infect Dis 71:98–99
Zaraket H, Kondo H, Tabet C, Hanna-Wakim R, Suzuki Y, Dbaibo GS, Saito R, Suzuki H (2011) Genetic diversity and antiviral drug resistance of pandemic H1N1 2009 in Lebanon. J Clin Virol 51:170–174
Pawlotsky J-M (2011) Treatment failure and resistance with direct-acting antiviral drugs against hepatitis C virus. Hepatology 53:1742–1751
Hayden FG, De Jong MD (2011) Emerging influenza antiviral resistance threats. J Infect Dis 203:6–10
Thorlund K, Awad T, Boivin G, Thabane L (2011) Systematic review of influenza resistance to the neuraminidase inhibitors. BMC Infect Dis 11
Lyushina NA, Govorkova EA, Webster RG (2005) Detection of amantadine-resistant variants among avian influenza viruses isolated in North America and Asia. Virology 341:102–106
Weinstock DM, Zuccotti G (2006) Adamantane resistance in influenza A. J Am Med Assoc 295:934–936
Ghosh GC, Nakada N, Yamashita N, Tanaka H (2010) Oseltamivir carboxylate, the active metabolite of oseltamivir phosphate (Tamiflu), detected in sewage discharge and river water in Japan. Environ Health Persp 118:103–107
Singer AC, Nunn MA, Gould EA, Johnson AC (2007) Potential risks associated with the proposed widespread use of Tamiflu. Environ Health Perspect 115:102–106
Fick J, Lindberg RH, Tysklind M, Haemig PD, Waldenström J, Wallensten A, Olsen B (2007) Antiviral oseltamivir is not removed or degraded in normal sewage water treatment: implications for development of resistance by influenza A virus. PLoS One 2:e986
Cyranoski D (2005) China's chicken farmers under fire for antiviral abuse. Nature 435:1009
Delogu I, Pastorino B, Baronti C, Nougairède A, Bonnet E, de Lamballerie X (2011) In vitro antiviral activity of arbidol against Chikungunya virus and characteristics of a selected resistant mutant. Antivir Res 90:99–107
FDA (2006) FDA prohibits use of antiviral drugs in poultry to help keep drugs effective for humans. http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/2006/ucm108620.htm. Accessed 18 Jun 2009
Allen GD, Brookes ST, Barrow A, Dunn JA, Grosse CM (1999) Liquid chromatographic-tandem mass spectrometric method for the determination of the neuraminidase inhibitor zanamivir (GG167) in human serum. J Chromatogr B 732:383–393
Baughman TM, Wright WL, Hutton KA (2007) Determination of zanamivir in rat and monkey plasma by positive ion hydrophilic interaction chromatography (HILIC)/tandem mass spectrometry. J Chromatogr B 852:505–511
Yeh LT, Nguyen M, Lourenco D, Lin CC (2005) A sensitive and specific method for the determination of total ribavirin in monkey liver by high-performance liquid chromatography with tandem mass spectrometry. J Pharm Biomed Anal 38:34–40
Yeh LT, Nguyen M, Dadgostari S, Bu W, Lin CC (2007) LC-MS/MS method for simultaneous determination of viramidine and ribavirin levels in monkey red blood cells. J Pharm Biomed Anal 43:1057–1064
Liu Y, Xu C, Yan R, Lim C, Yeh LT, Lin CC (2006) Sensitive and specific LC-MS/MS method for the simultaneous measurements of viramidine and ribavirin in human plasma. J Chromatography B 832:17–23
Svensson JO, Bruchfeld A, Schvarcz R, Ståhle L (2000) Determination of ribavirin in serum using highly selective solid-phase extraction and high-performance liquid chromatography. Ther Drug Monit 22:215–218
Lindegårdh N, Hanpithakpong W, Wattanagoon Y, Singhasivanon P, White NJ, Day NPJ (2007) Development and validation of a liquid chromatographic-tandem mass spectrometric method for determination of oseltamivir and its metabolite oseltamivir carboxylate in plasma, saliva and urine. J Chromatogr B Anal Technol Biomed Life Sci 859:74–83
Wiltshire B, Citron A, Clarke T, Serpe C, Gray D, Herron W (2000) Development of a high-performance liquid chromatographic-mass spectrometric assay for the specific and sensitive quantification of Ro 64–0802, an anti-influenza drug, and its pro-drug, oseltamivir, in human and animal plasma and urine. J Chromatogr B 745:373–388
Yamanaka T, Yamada M, Tsujimura K, Kondo T, Nagata S, Hobo S, Kurosawa M, Matsumura T (2007) Clinical pharmacokinetics of oseltamivir and its active metabolite oseltamivir carboxylate after oral administration in horses. J Vet Med Sci 69:293–296
Heinig K, Bucheli F (2008) Sensitive determination of oseltamivir and oseltamivir carboxylate in plasma, urine, cerebrospinal fluid and brain by liquid chromatography-tandem mass spectrometry. J Chromatogr B 876:129–136
Bahrami G, Mohammadi B, Kiani A (2008) Determination of oseltamivir carboxylic acid in human serum by solid phase extraction and high performance liquid chromatography with UV detection. J Chromatogr B 864:38–42
Ping W, Yi-Zeng L, Ben-Mei C, Neng Z, Lun-Zhao Y, Yan Y, Zhi-Biao Y (2007) Quantitative determination of amantadine in human plasma by liquid chromatography-mass spectrometry and the application in a bioequivalence study. J Pharm Biomed Anal 43:1519–1525
Xu M, Ju W, Xia X, Tan H, Chen M, Zhang J, Xiong N, Jiang M, Chen L, Gong L (2008) Determination of rimantadine in rat plasma by liquid chromatography/electrospray mass spectrometry and its application in a pharmacokinetic study. J Chromatogr B 864:123–128
Feng S, Tian Y, Zhang Z, Zhang J, Huang M, Chen Y (2009) Rapid simultaneous determination of paracetamol, amantadine hydrochloride, caffeine and chlorpheniramine maleate in human plasma by liquid chromatography/ tandem mass spectrometry. Arzneim Forsch 59:86–95
Higashi Y, Uemori I, Fujii Y (2005) Simultaneous determination of amantadine and rimantadine by HPLC in rat plasma with pre-column derivatization and fluorescence detection for pharmacokinetic studies. Biomed Chromatogr 19:655–662
Shuangjin C, Fang F, Han L, Ming M (2007) New method for high-performance liquid chromatographic determination of amantadine and its analogues in rat plasma. J Pharm Biomed Anal 44:1100–1105
Liu X, Huang YW, Li J, Li XB, Bi KS, Chen XH (2007) Determination of arbidol in human plasma by LC-ESI-MS. J Pharm Biomed Anal 43:371–375
Wang Y, Chen X, Li Q, Zhong D (2008) Metabolite identification of arbidol in human urine by the study of CID fragmentation pathways using HPLC coupled with ion trap mass spectrometry. J Mass Spectrom 43:1099–1109
Chan D, Tarbin J, Sharman M, Carson M, Smith M, Smith S (2011) Screening method for the analysis of antiviral drugs in poultry tissues using zwitterionic hydrophilic interaction liquid chromatography/tandem mass spectrometry. Anal Chim Acta 700:194–200
Horwitz W, Kamps LR, Boyer KW (1980) Quality assurance in the analysis of foods and trace constituents. J Assoc Off Anal Chem 63:1344–1354
Thompson M, Ellison SL, Wood R (2006) The international harmonized protocol for the proficiency testing of analytical chemistry laboratories. Pure Appl Chem 78:145–196
Antignac J-P, Le Bizec BL, Monteau F, Andre F (2003) Validation of analytical methods based on mass spectrometric detection according to the “2002/657/EC” European decision: guideline and application. Anal Chim Acta 483:325–334
Acknowledgements
This project was financially supported by the Dutch Ministry of Economic Affairs, Agriculture and Innovation (project 1217261301). We thank Frédérique van Holthoon for her contribution to the method development and FERA for kindly supplying amantadine-affected muscle samples.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 221 kb)
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Berendsen, B.J.A., Wegh, R.S., Essers, M.L. et al. Quantitative trace analysis of a broad range of antiviral drugs in poultry muscle using column-switch liquid chromatography coupled to tandem mass spectrometry. Anal Bioanal Chem 402, 1611–1623 (2012). https://doi.org/10.1007/s00216-011-5581-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-011-5581-3