
Abstract
Lyme borreliosis (LB) is one of the most common tick-borne diseases in the northern hemisphere. It is a chronic inflammatory disease caused by the spirochaete Borrelia burgdorferi. In its early stages, pathological skin lesions, namely erythema chronicum migrans, appear. The lesions, usually localised at the site of the bite, may become visible from a few weeks up to 3 months after the infection. Predominant clinical symptoms of the disease also involve joint malfunctions and neurological or cardiac disorders. Lyme disease, in all its stages, may be successfully treated with antibiotics. The best results, however, are obtained in its early stages. In order to diagnose the disease, numerous medical or laboratory techniques have been developed. They are applied to confirm the presence of intact spirochaetes or spirochaete components such as DNA or proteins in tick vectors, reservoir hosts or patients. The methods used for the determination of LB biomarkers have also been reviewed. These biomarkers are formed during the lipid peroxidation process. The formation of peroxidation products generated by human organisms is directly associated with oxidative stress. Apart from aldehydes (malondialdehyde and 4-hydroxy-2-nonenal), many other unsaturated components such as isoprostenes and neuroprostane are obtained. The fast determination of these compounds in encephalic fluid, urine or plasma, especially in early stages of the disease, enables its treatment. Various analytical techniques which allow the determination of the aforementioned biomarkers have been reported. These include spectrophotometry as well as liquid and gas chromatography. The analytical procedure also requires the application of a derivatization step by the use of selected reagents.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
In Europe and North America, Ixodes ricinus ticks are common ectoparasites (external parasites). Many species of viruses, bacteria and spirochaetes are found in tick vectors. They belong to a phylum of distinctive Gram-negative bacteria, which have long, helically coiled (spiral-shaped) cells. Therefore, ticks are vectors of a number of diseases, including Lyme disease, Colorado tick fever, tularaemia, tick-borne relapsing fever, babesiosis, ehrlichiosis and tick-borne meningoencephalitis, as well as anaplasmosis in cattle and canine jaundice [1, 2]. The cause of Lyme borreliosis is the activity of a single spirochaete species, Borrelia burgdorferi. Lyme borreliosis or Lyme disease is one of the most often identified diseases on the northern hemisphere. In Europe and North America, approximately 85,000 and 15,000–20,000 cases are identified each year, respectively. Noticeably, numerous cases of Lyme borreliosis are reported in Poland, Sweden, Lithuania, Latvia, Belarus, Austria, the Czech Republic and Slovenia [3].
When a human organism is infected with Lyme borreliosis, the fastest occurring symptoms are inflammation and changes in the immunological system. The disease causes noticeable damage in various organs including the brain, central nervous system (CNS), heart, eyes, skeleton and joints. The effects of the disease depend on the genotype of the spirochaetes, individual immunological system predispositions and other factors [3]. Usually, antibiotics eliminate symptoms of the infection, especially if the illness is treated in its early stage. If the disease is identified long after the infection took place, delayed or inadequate treatment can lead to more serious symptoms, which can even become irreversible (post-Lyme borreliosis syndromes [4]).
In this paper, methods used for diagnosing Lyme borreliosis (LB) are reviewed. The developments in laboratory and analytical methods used for the diagnosis of the disease are taken into consideration and discussed in detail. Procedures applied for the determination of LB biomarkers are also presented. Apart from aldehydes such as malondialdehyde (MDA) and 4-hydroxy-2-nonenal (HNE), many other unsaturated components, e.g. isoprostenes and neuroprostane, are separated and determined in biological samples. The formation of peroxidation products generated by a human organism is directly associated with oxidative stress. An analysis of human encephalic fluid, blood, urine or plasma, provides much important data. The presence of the MDA, HNE and isoprostanes was confirmed in various types of tissues as well as human body fluids. The fast separation of these compounds from encephalic fluid, urine and plasma, especially in early stages of the disease, makes LB treatment possible. Various analytical techniques which allow the determination of the aforementioned biomarkers have been reported. These include spectrophotometry as well as liquid and gas chromatography. The analytical procedure also requires the application of a derivatization step by the use of selected reagents. Moreover, modern analytical techniques including liquid chromatography coupled with tandem mass spectrometry (LC/MSn) or a fluorescence detector (FLD) have been used for the determination of such compounds.
Spectrum of Lyme borreliosis
Lyme borreliosis is a multisystemic disease caused by the spirochaete Borrelia burgdorferi sensu lato (in Poland these are B. burgdorferi, B. garinii, and B. afzelii), transmitted by Ixodes ticks. Lesions are associated primarily with the injury of the skin, joints, nervous system, and heart infections [5–12].
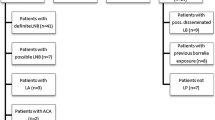
Patients infected with B. burgdorferi sensu lato may experience one or more clinical syndromes of early or late LB. The basis for the LB diagnosis in humans is a series of its clinical forms [13–15]. One of these is erythema migrans (EM, creeping rash). The diagnosis is based solely on the clinical picture. Typically, immunoserological tests towards anti-B. burgdorferi antibodies are unnecessary. Erythema migrans may appear at the site of a tick bite usually after 1–3 weeks (and even after 3 months). A quickly appearing syndrome takes the form of a macula, and quickly expands showing central clearing. Erythema migrans disappears within a few days of the proper antibiotic therapy, but this fact is not tantamount to the elimination of infection. Untreated EM may last several months, and its spontaneous regression does not eliminate the infection either. Multiple secondary EM, which shows the infection is spreading, occurs rarely. The changes are usually smaller than those at the primary stage and uniformly coloured.
Diagnosing Borrelia lymphocytoma (BL) requires the determination of IgM or IgG presence in serum and its histological confirmation. BL symptoms are noted in less than 1% of patients, usually within a few weeks after a tick bite, as a single, bluish red, painless nodule. BL is most frequently localised in earlobes, nipples, and the scrotum. BL changes may be accompanied by lymph node enlargement.
Another kind of LB clinical form is chronic atrophic dermatitis on limbs (acrodermatitis chronica atrophicans, ACA). ACA diagnosis requires the determination of IgM or IgG presence in serum and its histological confirmation. In ACA, bluish red changes, initially with the features characteristic of inflammatory edema, and later skin atrophy, are frequently observed. They appear even many years after infection. ACA is most frequently localised on certain parts of the limbs, especially legs. Less commonly, it can be the trunk. These changes may be accompanied by leg pain which often appears simultaneously with the main symptom of peripheral neuropathy and changes in degenerative arthritis beneath the affected skin. ACA is usually diagnosed in elderly people, mostly women.
Moreover, cases of arthritis (Lyme arthritis, LA) are very often observed. The morbidity requires laboratory confirmation by the determination of IgM antibodies in serum at an early stage, or IgG antibodies at the late stage of the disease. LA can take various clinical forms, e.g. wandering pain in bones, joints, muscles and tendons, or chronic arthritis (asymmetric, occurring even a few years after infection, usually preceded by recurrent pain or arthritis). Flu-like symptoms, including pain in joints, of varied intensity are present in 40% of cases with erythema migrans. They usually recede spontaneously and have no connection with subsequent LA symptoms observed.
In B. burgdorferi spirochaete infection, myocarditis (Lyme carditis) may also occur. A diagnosis requires the confirmation of the presence of IgM antibodies against B. burgdorferi in serum and cardiac dysfunction confirmed by an ECG examination.
One of the most common effects caused by the spirochaete B. burgdorferi is neuroborreliosis. When diagnosing, it is necessary to identify other typical LB symptoms, particularly erythema migrans. It is necessary to confirm the presence of IgM or IgG antibodies against B. burgdorferi in serum, and in the case of the brain and spinal cord inflammation, additionally their intrathecal production. These confirmations are needed to exclude the passive transfer of antibodies through the blood–brain barrier. Some patients with neuroborreliosis in the early stages of LB (the first weeks after getting infected) show no presence of antibodies in serum. In such cases, the test should be repeated 2 weeks after the symptoms of the disease were observed. The seroconversion occurring confirms the diagnosis. In the course of neuroborreliosis, cerebrospinal fluid (CSF) of patients with meningitis is characterized by lymphocytic pleocytosis, a moderate increase in protein concentration, and normal glucose concentration in serum. In its first period, neuroborreliosis may proceed as follows: cranial nerve palsy, nerve roots or individual peripheral nerves paralysis, lymphocytic meninges inflammation (meningitis), brain inflammation (encephalitis) or brain and spinal cord (encephalomyelitis) inflammation. In the late stage of neuroborreliosis, the following symptoms may occur: inflammation of the brain and spinal cord (encephalomyelitis), peripheral neuropathy, chronic encephalopathy (with dominant memory and/or concentration impairment, irritability, sleepiness and personality changes).
Laboratory diagnosis
Many achievements in the field of LB laboratory diagnosis have been discussed and published in numerous reviews [16–20]. In order to identify each of the Lyme disease clinical forms (except EM), laboratory diagnosis requires a two-stage diagnostic protocol. The first step should confirm the presence of specific IgM or IgG (depending on the clinical form) on the basis of the immunoenzymatic method. In the second stage, for patients with positive or dubious results, Western blotting methods should provide unambiguous results. Both methods complement each other as immunoenzymatic tests are usually characterized by high sensitivity and relatively low specificity. Meanwhile, the Western blot method is characterized by high specificity with lower sensitivity. IgM antibodies can be detected as early as in the second week of illness, but in most patients, their presence can be observed a few weeks later. The late stage of LB is usually characterized by the presence of IgG antibodies.
Herpes virus infections (especially Epstein–Barr virus) or other spirochaetes, and autoimmune diseases can cause false positive serological test results. A seropositive result without clinical symptoms typical of LB does not provide the basis for diagnosing the disease and its treatment.
One of the optimal techniques for a potential diagnosis in the early stage of LB is the polymerase chain reaction (PCR) [16, 21]. The true frequency and clinical correlates of PCR-documented, blood-borne infection in the spread of Lyme disease are widely determined. PCR detection of B. burgdorferi is a method at least three times more sensitive than the cultivation of microorganisms for identifying spirochaetaemia in early LB. The PCR detection may be useful for a rapid diagnosis.
PCR-based methods are useful for the laboratory diagnosis of LB [16]. The first PCR assays for specific detection of a chromosomally encoded B. burgdorferi sensu lato gene were reported 20 years ago. Various other PCR assays were subsequently developed for detection of B. burgdorferi sensu lato DNA in clinical specimens. Given that the number of spirochaetes in infected tissues or body fluids of patients is very low, appropriate procedures for sample collection and transport and preparation of DNA from clinical samples are critical for yielding reliable and consistent PCR results. A variety of clinical specimens from patients with suspected Lyme disease have been analysed by PCR assays. Skin biopsy samples taken from patients with EM or ACA have been the most frequently tested specimens. Depending on the clinical manifestations of the patients, appropriate body fluid samples can be collected and analysed by PCR. The sensitivity of PCR assays may be reduced by degradation of the B. burgdorferi sensu lato DNA during sample transport, storage and processing [16]. The sensitivities of the PCR assays varied from 36 to 88% for patients with EM and from 54 to 100% for patients with ACA. The median sensitivities of the reported PCR assays were in the range from 69 to 76%. The sensitivity of PCR assays for detection of B. burgdorferi sensu lato in whole blood (plasma or serum) and CSF specimens is low [16]. By contrast, higher PCR sensitivities were reported with synovial fluid samples from patients with LA. Also PCR is a very sensitive approach when it is employed to detect B. burgdorferi sensu lato DNA in skin biopsy and synovial fluid specimens from patients with LB, whereas the diagnostic value of PCR assays for detection of B. burgdorferi sensu lato DNA in blood (plasma or serum) and CSF specimens is low. Moreover, PCR assays have not been widely accepted for laboratory diagnosis of LB because of low sensitivity in CSF and blood. PCR as a diagnostic tool may be hampered by potential false-positive results due to accidental contamination of samples with a small quantity of target DNA. Although PCR is highly sensitive for detection of DNA of B. burgdorferi sensu lato in skin biopsy samples from patients with EM, such testing is rarely necessary, as a clinical diagnosis can be easily made if the characteristic skin lesion is present [16].
Bloodstream invasion is an important and common mechanism for spreading the Lyme disease spirochaete. As a result of insufficient standardization in diagnosing infections that occur in Poland, PCR is not routinely used. PCR can detect spirochaete DNA without specifying whether it comes from living organisms, so a positive result does not mean an active infection.
Generally, in a laboratory diagnosis of LB, various methods for direct or indirect detection of B. burgdorferi can be applied. Particular data and some examples of methods are listed in Table 1.
The decision about diagnosis and further treatment of patients with Lyme disease should be made only by a physician who interprets the symptoms taking into account the results of additional tests. A therapy at least 21 days long is based on putting a patient on an antibiotic. The choice of the antibiotic depends on the clinical form of the disease and an individual’s tolerance.
Others methods of diagnosis
Laboratory diagnosis needs CSF (liquor cerebrospinalis) or joint fluids from patients suffering from LB. The detection of B. burgdorferi microscopically or by the cultivation of spirochaetes derived from blood or CSF is difficult and burdened with high rates of false results. Currently in Poland, the study of cerebrospinal or joint fluid, in the case of infections caused by B. burgdorferi spirochaetes, is nonspecific and does not belong to routine diagnostic procedures. However, it is known that in the early stages of inflammation, the interaction between B. burgdorferi and multinucleated granulocytes and endothelial cells is observed, which leads to the generation of reactive oxygen species, lipid peroxidation products and other inflammatory mediators. Therefore, it is extremely important to develop new analytical methodologies aimed at the effective determination of lipid peroxidation products and other mediators of inflammation, as biomarkers of LB and other diseases.
The reaction of an organism during the infection or persistent disease is an inflammation, which is responsible for the damage of organs and systems. An inflammation of various organs is linked to the production of free radicals including reactive oxygen species (ROS). Free radicals play an important role in numerous biological processes, some of which are necessary for life, such as the intracellular killing of bacteria. On the other hand, because of their high reactivity, free radicals also can participate in unwanted side reactions resulting in cell damage [22, 23].
When patients suffer from various diseases including LB, free radical activity rapidly increases. This process is also connected to lipid peroxidation, which refers to the oxidative degradation of lipids, and is especially dangerous for brain cells, resulting in their damage. Metabolism processes in brain cells require more oxygen and glucose concentrations than other cells and tissues. Moreover, brain cells contain large amounts of polyunsaturated fatty acids (PUFAs) and iron ions. In addition, these ions are catalysts of certain reactions in which free radicals take part. All these processes are induced by free radicals and their reaction with lipids in cell membranes, which has an effect on cell damage. This reaction involves the oxidation of PUFAs, which are biological membrane components. The peroxidation of cell constituents is highly correlated with the formation of reactive aldehydes. Beside aldehydes, many other unsaturated components are formed during lipid peroxidation [24]. Important biomarkers of lipid peroxidation process are MDA and HNE. Particular MDA and HNE formation steps from PUFAs are presented in Figs. 1 and 2, according to refs. [25, 26].

Formation of MDA during peroxidation of PUFAs [25]

Pathways of PUFA peroxidation and the formation of HNE [26]
MDA and HNE are generated during all physiological processes. However, their concentrations are sometimes three times higher in inflammation, because free radicals induce their synthesis [27]. These compounds display reactive character toward nucleic acids, proteins and others [28]. Some products of lipid peroxidation react with DNA leading to both genotoxic and mutagenic action. In particular, MDA is the most mutagenic, but HNE the most genotoxic. The aforementioned compounds resulting in DNA damage caused by adducts of lipid peroxidation products with DNA can be removed by the repairing action of glycosylases [28].
The increase of membrane phospholipid peroxidation products in the course of LA has been reported [29]. ROS generated by stimulated phagocytes located near the joints during B. burgdorferi infection influence the high concentration of peroxidation products [30]. During inflammation, the lipid peroxidation process is additionally intensified by the decrease in the antioxidants concentration in the lipid phase of the biomembrane [31]. The reaction of ROS with phospholipid PUFAs has an effect on the increased production of superoxides and low molecular weight aldehydes [32]. Results concerning the determination of MDA and HNE in the plasma as well as in the urine of patients with B. burgdorferi infection have been published [29]. During LA the level of free form of MDA and HNE in the plasma, as well as in the urine, is about twofold higher in comparison with the control group [29]. The aforementioned compounds have a longer lifetime than ROS and can diffuse through biomembranes and cause greater cell damage. MDA and HNE are very reactive electrophilic compounds. These compounds react with antioxidants participating in diminishing lipid peroxidation, such as glutathione (GSH) and glutathione peroxidase (GSHPO) [29, 33]. High reactivity of aldehydes generated during lipid peroxidation contributes to the fact that the estimation of their total level in biological samples is difficult. For that reason, other lipid peroxidation products such as prostaglandin F2-like compounds (including more stable isoprostane 8-isoPGF2α) are determined in the plasma and urine samples obtained from patients with LA [29].
Main reactions occurring during peroxidation of PUFAs, which result in the synthesis of isoprostanes, are presented in Fig. 3.

Proposed pathways of lipid peroxidation which lead to the synthesis of isoprostanes [24]
Many classes of F2-isoprostanes can arise from arachidonic acid (AA). On the contrary, peroxidation of eicosapentaenoic acid (EPA) is predicted to lead to the synthesis of six classes of F3-isoprostanes, but α-linolenic and γ-linolenic acids to two classes of E1- and F1-isoprostanes, and docosahexaenoic acid to eight classes of D4-isoprostanes and eight classes of E4-isoprostanes. The formation of F2-isoprostanes involves the formation of positional peroxyl radical isomers of AA, which undergo endocyclization to PGG2-like compounds, which are subsequently reduced to PGF2-like compounds. F2-isoprostanes are formed in situ in phospholipids by free radical-catalysed peroxidation of esterified AA and subsequently are released in free form, presumably by phospholipases. The scheme of the formation of various classes of compounds from AA is presented in Fig. 4.

Formation of various classes of compounds from AA [34]
Methodology use for the determination of biomarkers of Lyme borreliosis and other inflammatory diseases
Malondialdehyde
MDA exists predominantly in the enol form, and it is highly reactive. Therefore, in laboratories MDA is obtained by hydrolysis of 1,1,3,3-tetramethoxypropane (commercially available compound). MDA is easily deprotonated to give the sodium salt of the enolate.
Various methods have been developed for the determination of MDA including spectrophotometry, fluorimetric detection, immunotests, high-performance liquid chromatography (HPLC) and gas chromatography (GC) [25, 35, 36].
However, the preparation of biological samples before analysis is the most critical point. In case of plasma analysis, various types of anticoagulant can be used for MDA assay like sodium heparin [37, 38], sodium citrate [37] and tripotassium EDTA [37, 39, 40]. Nevertheless, EDTA is more preferable than citrate and heparin as an anticoagulant, because it provides the lowest limit of detection (LOD) of MDA. This is probably related to iron chelation by EDTA and its weak activity e.g. as an antioxidant [41]. Moreover, the Fe–EDTA complex can participate in the Fenton reaction, forming OH· radicals and overestimating the lipid peroxidation [42]. Biological samples are stable at low temperature, which is why a suitable storage temperature is −20 °C for a short period of time, or even −80 °C for storage for more than 1 month [25, 37, 43]. Moreover, the antioxidant butylated hydroxytoluene (BHT) can be used to prevent further formation of MDA during the assay [38, 44–46]. The preparation of biological samples requires the application of an acid to precipitate proteins. Various inorganic acids can be used, e.g. sulphuric acid [39], perchloric acid [47] and (ortho)phosphoric acid [37, 38, 40].
The determination of MDA can be performed on various biological samples. The thiobarbituric acid reactive substances (TBARS) assay has been carried out in plasma [37, 39, 40, 44, 45, 48], serum [47], different tissues [44, 49, 50, 51] and urine [52]. Some investigations report the reaction of TBA with MDA and linked chromogens to lipoperoxides in biomaterials [53]. Reaction products can be detected by spectrophotometry (λ = 532–535 nm) or colorimetry as well as by fluorimetry (excitation at λ = 532 nm and emission at λ = 553 nm) [54]. The principal reaction between MDA and thiobarbituric acid (TBA) is presented in Fig. 5. Formation of MDA–TBA2 adduct occurs by a nucleophilic attack involving carbon-5 of TBA and carbon-1 of MDA, followed by dehydration and similar reaction with a second molecule of TBA to produce a red pigment [55]. The intensity of the pink MDA–TBA product formed from this condensation indicates the extent of lipid peroxidation.

Formation of MDA–TBA2 adduct after reaction between MDA and TBA [25]
Nevertheless, the MDA assay by means of TBARS is limited by the fact that MDA is unstable. Additionally, its oxidation yields organic alcohols and acid [56, 57]. The use of colorimetric or fluorimetric techniques may give some mistakes, especially in the initial stages of human diseases in which lipid peroxidation is lower, when compared with the nonspecific background reaction between TBA and products not derived from lipid peroxidation [50]. Concentrations MDA or TBARS in plasma obtained with the methods previously developed varied over a very wide range (0–50 mmol/L) [32, 58].
The determination of MDA by use of HPLC utilizes a derivatization as pretreatment. TBA is also used as a reagent for this derivatization. The separation of the MDA–TBA2 adduct from other compounds by reversed-phase HPLC might be difficult. Several researchers have adapted HPLC methods so that the procedure offers some advantages in terms of specificity, recovery and reproducibility. HPLC assay for MDA is a useful method to measure concentrations of lipid peroxidation products in biological samples [25, 35, 37–39]. Besides, the analytical procedure consisting of TBA addition to the sample followed by extraction with n-butanol has been performed. This step avoids interference formation and extends the lifetime of the column by removing contaminants from the incubation mixture [38, 59]. Moreover, the difference between detection of free and total plasma MDA is discussed [59]. Only low amounts of free MDA are present in biological samples, and an alkaline hydrolysis step by use of sodium hydroxide is necessary to obtain a more complete and uniformed release of MDA from protein–MDA complexes, which results in a higher MDA value, when total MDA is being considered [38, 40].
Useful detectors for the determination of MDA are UV-Vis (λ = 532 or 535 nm) [37, 38, 45, 51] and FLD [38, 46, 59, 60].
Certain methods consist of plasma sample pretreatment by the acidic hydrolysis of amino acid-bound MDA and consequent protein precipitation [47]. The protein-free plasma is directly injected into the HPLC system and MDA is quickly eluted (monitored at λ = 254 nm) thereby allowing rapid analyses. The LOD of some methods is relatively low (0.012 mmol/L). Another advantage is the low amount of plasma needed for the analysis (50 mL).
GC is also a technique that can be used in the determination of MDA. However, a derivatization step is necessary prior to GC analysis. Some of the main derivatization agents are 2,3-propanediol [61], 2-hydrazinobenzothiazole [62] and pentafluorophenylhydrazine [63]. Depending on the method, either free or bound MDA may be determined and aldehydes other than MDA can be identified. In addition, headspace solid-phase microextraction (HS-SPME) used for the analyses of volatile aldehydes in biological matrices has been recently proposed [64]. The HS-SPME has been applied to prepare samples for MDA analysis, improving the sample preparation steps in lipid peroxidation research. A modified MDA assay involves derivatization and HS-SPME. The product of reaction of MDA and N-methylhydrazine is 1-methylpyrazole, which was analysed by GC with a nitrogen–phosphorus detector, and its detection limit was 0.0103 nmol/mL [64].
The most recent and precise methods used to assess MDA in human plasma or serum are briefly reported in Table 2.
4-Hydroxy-2-nonenal
Alkenals, including HNE, are strong electrophiles that react in tissues by alkylating nucleophiles (Michael addition), particularly sulfhydryl groups, amino groups (e.g. lysine), and the imidazole group of histidine [73, 74]. Such reactions can lead to structural alteration and/or cross-linking of proteins, which in turn can cause impairment of protein function. HNE is a relatively stable aldehyde that can diffuse to different subcellular compartments and interact with many different cell proteins [73]. Increased production of HNE has been shown to indicate an increased level of oxidative damage, which may not always be evident using the more common marker MDA [75].
Usually, the conventional analytical method for the quantitative determination of HNE is by derivatization with 2,4-dinitrophenylhydrazine (DNPH) that reacts with HNE to generate 2,4-dinitrophenylhydrazones with characteristic absorbance maxima at λ = 360–390 nm. Afterwards, separation by thin-layer chromatography (TLC) or HPLC is possible [76–78]. This two-step method is efficient and applicable to blood plasma samples. Moreover, the quantitative determination of HNE by derivatization with 3H-labelled sodium borohydride (NaB[3H]H4), which converts HNE Michael adducts into 3H-labelled dihydroxy derivatives, has been used as a measure of protein-bound HNE [79, 80]. Michael adducts generated in peptides and proteins, e.g. involving HNE–cysteine, HNE–histidine and HNE–lysine, can be analysed by HPLC with o-phthaldehyde derivatization [79, 80]. Reduction of the aldehyde group of primary HNE Michael addition products with NaBH4, which converts them to hydroxy derivatives that are stable to strong acid hydrolysis, forms the basis of methods for the identification and quantification of the HNE adducts by HPLC. By means of these techniques, it has been established that at least 80% of the histidine residues that are lost when low density lipoprotein (LDL) is treated with HNE can be accounted for as the Michael addition products, whereas the Michael addition product accounted for 49% of the lysine residues that disappeared upon HNE treatment. This method has also allowed one to quantitate the Michael addition-type HNE–histidine adducts (7–9 mol/mol LDL) and trace amounts of HNE–lysine adducts in Cu2+-oxidized LDL [79].
Currently, LC-ESI-MS/MS methodology is being developed. Chromatography coupled to MS is more sensitive and selective in the quantification of HNE [73, 81]. The determination of the sites of linkage of HNE to protein has been carried out by use of LC/MSn. The aforementioned method has also allowed for the characterization of HNE-adducted apomyoglobin [82]. The procedure involves an initial analysis by ESI-MS of the adducted protein to determine the stoichiometry of HNE incorporation. The adducted protein is then subjected to proteolytic digestion, followed by direct analyses of the unfractionated digest by ESI-MS for the localization of the sites of HNE adduction. Proteolytic digestion using trypsin produces fragments of suitable length for analysis by tandem MS with low-energy collision-induced dissociation. The components containing HNE-adducted histidine residues are identified by scanning for precursors of m/z 266, which correspond to an immonium ion derived from HNE adducted histidine. Last, product ion scanning of each modified tryptic fragment is performed to provide additional structural detail and to confirm the results obtained by precursor ion scanning. Application of this approach to the characterization of HNE-modified apomyoglobin indicated that there were between three and ten HNE adducts per protein molecule and that the adduction occurred solely to histidine residues [82].
An interesting aspect in the detection of HNE adducts is the use of antibodies. Some reports concern antibodies to HNE-treated LDL [75, 79]. Others raised the anti-HNE-LDL antibodies interacting with epitopes on Cu2+-oxidized LDL and other modified proteins [74, 83, 84]. It was observed that the antibody–protein interactions can be partly blocked by the HNE-modified forms of several polyamino acids, including polylysine, polytyrosine, polyarginine and polyhistidine [79].
With regard to the fact that HNE is very reactive with proteins and forms stable Michael addition-type adducts, a novel immunochemical procedure has been developed [85]. It is based on the detection of HNE trapped by a protein that has been coated on an immunoplate. The HNE-derived epitopes generated in the coating protein are then detected by an ELISA using a monoclonal antibody (mAbHNEJ2) specific to the haptenic groups of the HNE–protein conjugates [85]. Using this method, it was observed that a considerable amount of HNE was released from human plasma low density lipoproteins treated with copper ions or endothelial cells. Furthermore, a simple and rapid ELISA method for quantitation of HNE-modified proteins has been reported. During this method microtiter plate wells are precoated and blocked simultaneously with epitope-bound bovine caseins as matrix proteins, and aldehyde-modified proteins are quantitated by competition assay with a monoclonal antibody (mAbHNEJ2) specific for the HNE-histidine Michael adduct [86].
The stability of HNE in whole venous blood stored for different time intervals was studied [87]. The HNE content in venous blood stored in ice was determined (Fig. 6). The obtained results show a rapid decrease of HNE concentration, especially within the first hour after drawing the blood.

Content of HNE in venous blood stored in ice vs. time of storage [87]. Methodology: after 3 h of derivatization with DNPH the concentration of HNE was determined by HPLC
Physiological levels of HNE in human plasma are successfully determined by use of HPLC. The measurement of HNE in human plasma should be done within 30 min after collection. It was established that HNE is a normal constituent of human blood plasma, but other investigations on its occurrence under various conditions may help to clarify the relevance of lipid peroxidation in physiological and pathological processes [76]. The description of some methods applied for the determination of HNE is presented in Table 3.
Iso- and neuroprostanes
Isoprostanes (Iso-Ps) and neuroprostanes (Neuro-Ps) are formed by radical mechanisms and accordingly exist in several forms which in biological matrices undergo additional transformation to other metabolites. This fact creates a necessity to find appropriate methods to enable their analysis. The most often mentioned techniques among researchers involved in trying to develop new methodologies allowing the quantification and qualification of Iso-Ps and Neuro-Ps are GC/MS and LC/MS/MS. However, sample preparation seems to be the most critical point. The most preferable methods are SPE. Also, TLC and derivatization in case of GC analysis are widely used. Choosing LC imposes the use of only the SPE with conventional or specific immunoaffinity sorbent (immunoaffinity separation, IFS). Such a method simplification and the accompanying reduction of cost and time needed for sample preparation have created wide interest in this method. Among useful analytical methods, ELISA [89, 90], which may be also called enzyme immunoassay (EIA) in the literature, should be first discussed because of its utility as a clinical screening test.
ELISA
ELISA is a biochemical technique which is often used in immunology to quantify an antibody or an antigen in samples. A more specific application of this method is in the detection of isoprostanes in clinical analysis [90–95]. Because of its low cost and easy performance, ELISA is used widely in medicine and plant pathology [96–98]. Nevertheless, when we consider the application of ELISA to isoprostanes we see that the estimated level of these compounds is only semiquantitative. The main reason for this fact may be, in compliance with Yan et al. [99], cross-reactivity. Such an interaction may also be observed in future investigations of other prostaglandins. Application of GC allows one to see that ELISA overestimates the isoprostanes level by twofold [99].
GC analysis of prostanoids
GC/MS should be sensitive enough to quantify all desirable analytes after appropriate sample preparation. Because of their properties, prostanoids need a derivatization step prior to analysis [91, 100, 101]. SPE has to be performed twice in order to obtain high purity of selected compounds or greater enrichment.
Iso- and neuroprostanes from human plasma and urine are firstly extracted with use of SPE. This step of sample preparation may be performed using a C-18 Sep-Pak cartridge [100, 102, 103]. Other methodologies use two extraction steps. In the first step, reversed-phase C-18 is applied to remove protein and lipids. Subsequently, a Sep-Pak containing NH2 groups is used [98]. After the prostanoids are eluted from the appropriate cartridge, they are evaporated and then dissolved in a small amount of methanol [100, 104]. Dissolved compounds are applied to a silica TLC plate and developed to the top in a chosen solvent system. According to Morales et al. [100], the separation of isoprostane (15-F2t-IsoP-M) is performed with a mixture of ethyl acetate/acetic acid/methanol (80:0.1:20 v/v/v). The visualization of PGF2α may be done with a 10% solution of phosphomolybdic acid in ethanol followed by heating of the plates. Compounds, which migrated close to the acid, are scraped and extracted from the silica with the use of ethyl acetate/ethanol (50:50 v/v). The obtained samples of 15-F2t-IsoP-M after drying under vacuum or nitrogen are converted to the pentafluorobenzyl ester (PFB) [101]. A second purification with the use of TLC is performed usually in a solvent mixture containing ethyl acetate/methanol (98:2 v/v). Visualization, similarly to the first step, is performed by spraying with phosphomolybdic acid followed by heating. All compounds which migrate near the PGF2α methyl ester are then scraped and eluted with a defined volume of ethyl acetate [100] or other solvent. The eluate is dried (under nitrogen or vacuum) and converted to the corresponding trimethylsilyl (TMS) ether derivative. Such prepared samples are able to be analysed by GC/MS [101, 102, 104, 105].
A shorter and easier method for sample preparation prior to GC/MS analysis was developed and described by Lee et al. in 2004 [106]. Their approach is based on mixed anion exchange solid-phase extraction (MAX-SPE), which may be followed directly by GC/MS analysis by use of various ionization techniques.
Various ionization techniques are frequently used in analysis of both iso- and neuroprostanes by MS. Electron capture negative ionization (GC/ECNI-MS) is preferable due to its sensitivity and because it allows lower LODs [101–103, 107–110].
In the case of very complex matrices a single-ion monitoring (SIM) mode is often used. Additionally, multiple reaction monitoring (MRM) enhances sensitivity and selectivity of tandem instruments.
HPLC analysis of prostanoids
HPLC seems to be a more appropriate method for the analysis of prostanoids. After sample treatment, the obtained extracts may be directly analysed by LC without any derivatization which was necessary in GC. However, the purification of samples is an important step. The sample containing prostanoids needs extraction using e.g. silica-based SPE sorbents [29, 91, 111, 112] or immunoaffinity sorbents [91, 111].
The plasma or other biological samples initially have to be prepared according to an appropriate given protocol. Biological samples with BHT and triphenylphosphine have to be homogenized thoroughly. Then, hydrolysis of esterified prostanoids is performed (heating after adding of KOH solution). Afterwards, samples are diluted with water to prevent precipitation. In the case of urine, only alkaline solution followed by acetic acid has to be added. Such prepared samples may be loaded on the C-18 Sep-Pak columns [112]. Because of the low recovery obtained with conventional C-18 columns (near 20%), it was better to use Oasis HLB cartridges for sample extraction as, depending on the kind of compounds, recoveries were about 80% [111]. The eluate after SPE is evaporated to dryness under vacuum or nitrogen and after reconstitution (usually in mobile phase) may be analysed by HPLC.
Extraction by immunoaffinity bed
In the case of prostanoids, deuterated internal standards and KOH have to be added to the freshly thawed plasma. After short incubation at increased temperature the alkali is neutralized by addition of potassium dihydrophosphate [111]. But urine samples need only dilution in potassium dihydrophosphate and addition of internal deuterated standards. Samples may be analysed by LC/MS after evaporating to dryness and dissolving in mobile phase.
The literature contains examples of applications using C-18 [29, 91, 111–114] and polar end-capped C-18 [89] columns. Similarly to GC where the most useful tool in prostanoids determination was MS, in LC the same situations are occurring. In LC/MS the most common ionization technique is electrospray (ESI). Also atmospheric pressure chemical ionization (APCI) in prostanoids analysis is recommended [112]. The analysers applied to this aim may be different but in almost all cases a triple quadrupole and quadrupole ion trap (QTRAP) are preferable [89, 91, 111–113]. Beside MS, applications of other detectors are also presented. An interesting example is the application of electrochemical and spectrophotometric detectors [29]. Such detection methods may be applied as an alternative to MS only in limited cases.
The recent methods used to assess isoprostanes and neuroprostanes are presented in Table 4.
Conclusions
Several Central European countries are characterised by the highest incidence of tick-borne diseases in the northern hemisphere. Molecular methods including PCR give possibilities for the detection of B. burgdorferi sensu lato and these are the most valuable detection methods. For patients with LB involving systems other than skin, PCR sensitivity is in general low, with the exception of patients with LA. Beside this, PCR is not routinely used in Poland. Therefore, an interesting alternative could be methods for the determination of lipid peroxidation products. The lipid peroxidation process is enhanced during LB and other human body infections. The determination of these products is essential in pathologies including LB and in toxicology associated with oxidative stress. MDA, HNE, isoprostanes and neuroprostanes are suitable biomarkers of various diseases, which refer to viruses and bacteria attack on an organism and to the increased formation of free radicals and oxidative stress as a consequence. If LB is detected in its early stage, it may be successfully treated. One of the possibilities is the development of methods used for the determination of biomarkers mentioned above. These analytical methods are characterised by high sensitivity, precision, accuracy and are not time consuming.
The concentration of 8-isoPGF2α in plasma and urine of LA patients could be over threefold and eightfold higher, respectively, than that in healthy people. The ratio of free to esterified form of 8-isoPGF2α is significantly smaller if compared with the levels in healthy people. This indicates that the ratio of free to esterified form of 8-isoPGF2α may be useful as an indicator of LA. Moreover, the complementary determination of three lipid peroxidation products (MDA, HNE and 8-isoPGF2α) may be very helpful in the diagnosis of LB.
From an analytical point of view, chromatography techniques are the preferred methods to determine the true amount of biomarkers in biological materials, once they are reliable, specific and, moreover, without the presence of artefacts or methodological mistakes. Comparing CG and HPLC methodologies, both are reliable and specific, but CG requires small sample volumes. HPLC UV-VIS is widely used in clinical laboratories and there are methodologies utilizing small sample volume or mass with total reliability and specificity.
Abbreviations
- AA:
-
arachidonic acid
- ACA:
-
acrodermatitis chronica atrophicans
- APCI:
-
atmospheric pressure chemical ionization
- BHT:
-
butylated hydroxytoluene
- BL:
-
Borrelia lymphocytoma
- CNS:
-
central nervous system
- DAN:
-
diaminonaphthalene
- DNPH:
-
2,4-dinitrophenylhydrazine
- ECD:
-
electron capture detector
- EIA:
-
enzyme immunoassay
- ELFA:
-
enzyme-linked fluorescent assay
- ELISA:
-
enzyme-linked immunosorbent assay
- EM:
-
erythema migrans
- EPA:
-
eicosapentaenoic acid
- ESI:
-
electrospray ionization
- FLD:
-
fluorescence detector
- GC:
-
gas chromatography
- GSH:
-
glutathione
- GSHPO:
-
glutathione peroxidase
- HNE:
-
4-hydroxy-2-nonenal
- HPCE:
-
high-performance capillary electrophoresis
- HS-SPME:
-
headspace solid-phase microextraction
- IFA:
-
immunofluorescent antibody assay
- IFS:
-
immunoaffinity separation
- Iso-Ps:
-
isoprostanes
- LA:
-
Lyme arthritis
- LB:
-
Lyme borreliosis
- LC:
-
liquid chromatography
- MDA:
-
malondialdehyde
- MRM:
-
multiple reaction monitoring mode
- MS:
-
mass spectrometry
- Neuro-Ps:
-
neuroprostanes
- PCR:
-
polymerase chain reaction
- PFB:
-
pentafluorobenzyl ester
- PH:
-
phenylhydrazine
- PUFAs:
-
polyunsaturated fatty acids
- ROS:
-
reactive oxygen species
- SIM:
-
single-ion monitoring mode
- SPE:
-
solid-phase extraction
- TBARS:
-
thiobarbituric acid reactive substances
- TCPH:
-
2,4,6-trichlorophenylhydrazine
- TLC:
-
thin-layer chromatography
References
Burgdorfer W (1984) Yale J Biol Med 57:515
Rahn DW, Malawista SE (1991) West J Med 154:706
Pancewicz SA, Hermanowska-Szpakowicz T, Makarewicz-Płońska M, Witek A, Farbiszewski R, Zajkowska J, Michalska B (2002) Neurol Neurochir Pol 36:767
Cairns V, Godwin J (2005) Int J Epidemiol 34:1340
Nadelman RB, Wormser GP (1998) Lancet 352:557
Stanek G, Strle F (2003) Lancet 362:1639
Steere AC (2001) N Engl J Med 345:115
Steere AC, Bartenhagen NH, Craft JE (1983) Ann Intern Med 99:76
Steere AC, Batsford WP, Weinberg M, Alexander J, Berger H, Wolfson SXMSE (1980) Ann Intern Med 93:8
Wormser GP, Nadelman RB, Nowakowski J, Schwartz I (2001) Med Hypotheses 57:435
Weber K, Pfister HW (1993) In: Weber K, aBurgdorfer W (eds) Aspects of Lyme borreliosis. Springer-Verlag, Berlin, pp 1–20
Wilske B (2003) Vector Borne Zoonotic Dis 3:215
Pfister HW, Wilske B, Weber K (1994) Lancet 343:1013
Stanek G, O’Connell S, Cimmino M, Aberer E, Kristoferitsch W, Granstrom M, Guy E, Gray J (1996) Wien Klin Wochenschr 108:741
Steere AC (1989) N Engl J Med 263:201
Aguero-Rosenfeld ME, Wang G, Schwartz I, Wormser GP (2005) Clin Microbiol Rev 18:3484
Barbour AG (1988) Clin Microbiol Rev 1:399
Bunikis J, Barbour AG (2002) Med Clin North Am 86:311
Schmidt BL (1997) Clin Microbiol Rev 10:185
Wilske B, Preac-Mursic V (1993) In: Weber K, Burgdorfer W (eds) Aspects of Lyme borreliosis. Springer-Verlag, Berlin, Germany, pp 267–300
Goodman JL, Bradley JF, Ross AE, Goellner P, Lagus A, Vitale B, Berger BW, Luger S, Johnson RC (1995) Am J Med 99:6
Pacher P, Beckman JS, Liaudet L (2007) Physiol Rev 87:315
Karthikeyan R, Manivasagam T, Anantharaman P, Balasubramanian T, Somasundaram ST (2011) J Appl Phycol 23:257
Marnett LJ (1999) Mutat Res Fundam Mol Mech Mutagen 424:83
Grotto D, Santa Maria L, Valentini J, Paniz C, Schmitt G, Garcia SC, Pomblum VJ, Rocha JBT, Farina M (2009) Quím Nova 32:169
Awasthi S, Singhal SS, Awasthi YC, Martin B, Woo JH, Cunningham CC, Frankel AE (2008) Clin Cancer Res 14:4372
Griffiths HR, Moller L, Bartosz G, Bast A, Bertoni-Freddari C, Collins A, Cooke M, Coolen S, Haenen G, Hoberg AM, Loft S, Lunec J, Olinski R, Parry J, Pompella A, Poulsen H, Verhagen H, Astley SB (2002) Mol Aspects Med 23:101
Łuczaj W, Skrzydlewska E (2003) Cell Mol Biol Lett 8:391
Łuczaj W, Moniuszko A, Rusak M, Pancewicz S, Zajkowska J, Skrzydlewska E (2011) Eur J Clin Microbiol Infect Dis 30:415
Pancewicz SA, Skrzydlewska E, Hermanowska-Szpakowicz T, Zajkowska JM, Kondrusik M (2001) Med Sci Monit 7:1230
Bradford A, Atkinson J, Fuller N, Rand RP (2003) J Lipid Res 44:1940
Comporti M (1998) Free Radic Res 28:623
Kinter M, Roberts RJ (1996) Free Radic Biol Med 21:457–462
de Zwart LL, Meerman JH, Commandeur JN, Vermeuln NH (1999) Free Radical Biol Med 26:202
Bird RP, Hung SSO, Hadley M, Draper HH (1983) Anal Biochem 128:240
Ichinose T, Miller MG, Shibamoto T (1989) Lipids 24:895
Nielsen F, Mikkelsen BB, Nielsen JB, Andersen HR, Grandjea P (1997) Clin Chem 43:1209
Hong Y, Yeh S, Chang C, Hu M (2000) Clin Biochem 33:619
Del Rio D, Pellegrini N, Colombi B, Bianchi M, Serafíni M, Torta F, Tegoni M, Musci M, Brighenti F (2003) Clin Chem 49:690
Grotto D, Santa Maria LD, Boeira S, Valentini J, Charão MF, Moro AM, Pomblum VJ, Nascimento PC, Garcia SC (2007) J Pharm Biomed Anal 43:619
Wong SHY, Knight JA, Hopfer SM, Zaharia O, Leach CN, Sunderman FW (1987) Clin Chem 33:214
Tamura H, Kitta K, Shibamoto T (1991) J Agric Food Chem 39:439
Gonçalves TL, Erthal F, Corte CLD, Muller LG, Piovezan CM, Nogueira CW, Rocha JBT (2005) Clin Biochem 38:1071
Lepage G, Munoz G, Champagne J, Roy CC (1991) Anal Biochem 197:277
Templar J, Kon SP, Milligan TP, Newman DJ, Raftery MJ (1999) Transplantation 14:946
Lykkesfeldt J (2001) Clin Chem 47:1725
Carbonneau MA, Peuchant E, Sess D, Canioni P, Clerc M (1991) Clinical Chem 37:1423
Scott B, Deman A, Peeters P, Van den Branden C, Stolear JC, Van Camp G, Verbrrlen D (2003) Nephrol Dial Transplant 18:737
Liu J, Yeo HC, Doniger SJ, Ames BN (1997) Anal Biochem 245:161
Folmer V, Santos FW, Savegnago L, Brito VB, Nogueira CV, Rocha JB (2004) Toxicol Lett 153:333
Cirak B, Inci S, Palaoglu S, Bertan V (2003) Clin Chim Acta 327:103
Draper HH, Csallany AS, Hadley M (2000) Free Radical Biol Med 29:1071
Yagi K, Nishigaki I, Ohama H (1968) Vitamins 37:105
Knight JA, Pieper RK, Mc Clellan L (1988) Clin Chem 34:2433
Janero DR (1990) Free Radical Biol Med 9:515
Almandos ME, Giannini DH, Ciarlo AS, Boery RL (1986) J Sci Food Agric 37:54
Seo CW (1976) J Food Sci 41:594
Del Rio D, Stewart AJ, Pellegrini N (2005) Nutr Metab Cardiovasc Dis 15:316
Fukunaga K, Takama K, Suzuki T (1995) Anal Biochem 230:20
Londero D, Lo Greco P (1996) J Chromatogr A 729:207
Lakshminarayana G, Cornwell DG (1986) Lipids 21:175
Beljean-Leymarie M, Bruna E (1988) Anal Biochem 173:174
Tomita M, Okuyama T, Hatta Y, Kawai SJ (1990) Chromatogr 526:174
Fujioka K, Shibamoto T (2005) J Agric Food Chem 53:4708
Agarwal R, Chase SD (2002) J Chromatogr B Analyt Technol Biomed Life Sci 775:121
Del Rio D, Pellegrini N, Colombi B, Bianchi M, Serafini M, Torta F (2003) Clin Chem 49:690
Wilson DW, Metz HN, Graver LM, Rao PS (1997) Clin Chem 43:1982
Sim AS, Salonikas C, Naidoo D, Wilcken DE (2003) J Chromatogr B Analyt Technol Biomed Life Sci 785:337
Steghens JP, van Kappel AL, Denis I, Collombel C (2001) Free Radic Biol Med 31:242
Cighetti G, Debiasi S, Paroni R, Allevi P (1999) Anal Biochem 266:222
Stalikas CD, Konidari CN (2001) Anal Biochem 290:108
Cighetti G, Allevi P, Anastasia L, Bortone L, Paroni R (2002) Clin Chem 48:2266
Williams TI, Lynn BC, Markesbery WR, Lovell MA (2006) Neurobiol Aging 27:1094
Subramaniam R, Roediger F, Jordan B, Mattson MP, Keller JN, Waeg G (1997) J Neurochem 69:1161
McGrath LT, McGleenon BM, Brennan S, McColl D, Mc IS, Passmore AP (2001) Oxford J Med QJM 94:485
Poli G, Schaur RJ, Siems WG, Leonarduzzi G (2008) Med Res Rev 28:569
Esterbauer H, Cheeseman K, Dianzani MU (1982) Biochem J 208:129
Winkler P, Lindner W, Esterbauer H, Schaunstein E, Schaur RJ (1984) Biochim Biophys Acta 796:232
Uchida K (2003) Progr Lipid Res 26:318
Uchida K, Stadtman ER (1993) J Biol Chem 268:6388
Williams IT, Lovell MA, Lynn BC (2005) Anal Chem 77:3383
Bolgar MS, Gaskell SJ (1996) Anal Chem 68:2325
Palinski W, Rosenfeld ME, Yla-Herttuala S, Gurtner GC, Socher SS, Butler SW (1989) Proc Natl Acad Sci U S A 86:1372
Chen Q, Esterbauer H, Jürgens G (1992) Biochem J 288:249
Uchida K, Osawa T, Hiai H, Toyokuni S (1995) Biochem Biophys Res Commun 212:1068
Satoh K, Yamada S, Koike Y, Toyokuni S, Kumano T, Takahata T (1999) Anal Biochem 270:323
Strohmaier H, Hinghofer-Szalkay H, Schaur RJ (1995) J Lipid Mediat Cell Signal 11:51
Selley ML (1997) J Chromatogr B 691:263
Monneret D, Pepin JL, Godin-Ribuot D, Ducros V, Baguet JP, Levy P, Faure P (2010) Free Radical Bio Med 48:619
Faure P, Tamisier R, Baguet JP, Favier A, Halimi S, Levy P, Pepin JL (2008) Eur Respir J 31:1046
Sircar D, Subbaiah PV (2007) Clin Chem 53:251
Liu HM, Liu KX, Cheng MH, Liu Y, Lei S, Irwin MG, Xia Z (2011) J Surg Res 168:18
Lucidi V, Ciabattoni G, Bella S, Barnes PJ, Montuschi P (2008) Free Radical Bio Med 45:913
Samitas K, Chorianopoulos D, Vittorakis S, Zervas E, Economidou E, Papatheodorou G, Loukides S, Gaga M (2009) Resp Med 103:750
Nishibe A, Kijima Y, Fukunaga M, Nishiwaki N, Sakai T, Nakagawa Y, Hata T (2008) Prostag Leukotr Ess 78:257
Gong Y, Yi M, Fediuk J, Lizotte PP, Dakshinamurti S (2010) Free Radical Bio Med 48:882
Dalaveris E, Kerenidi T, Katsabeki-Katsafli A, Kiropoulos T, Tanou K, Gourgoulianis KI, Kostikas K (2009) Lung Cancer 64:219
Kitano S, Hisatomi H, Hibi N, Kawano K, Harada S (2006) World J Gastroenterol 12:5846
Yan W, Byrd GD, Ogden MW (2007) J Lipid Res 48:1607
Morales CR, Terry ES, Zackert WE, Montine TJ, Morrow JD (2001) Clin Chem Acta 314:93
Morrow JD, Zackert WE, Yang JP, Kurhts EH, Callewaert D, Dworski R, Kanai K, Taber D, Moore K, Oates JA, Roberts LJ (1999) Anal Biochem 269:326
Roberts LJ, Montine TJ, Markesbery WR, Tapper AR, Hardyi P, Chemtobi S, Dettbarn WD, Morrow JD (1998) J Biol Chem 273:13605
Reich EE, Zackert WE, Brame CJ, Chen Y, Roberts LJ, Hachey DL, Montine TJ, Morrow JD (2000) Biochem 39:2376
Obata T, Sakurai Y, Kase Y, Tanifuji Y, Horiguchi T (2003) J Chromatogr B 792:131
Thomas MJ, Chen Q, Sorci-Thomas MG, Rudel LL (2001) Free Radical Bio Med 30:1337
Lee CY, Jenner AM, Halliwell B (2004) Biochem Biophys Res Commun 320:696
Mori TA, Croft KD, Puddey IB, Beilin LJ (1999) Anal Biochem 268:117
Lawson JA, Rokach J, FitzGerald GA (1999) J Biol Chem 274:24441
Nourooz-Zadeh J, Gopaul NK, Barrow S, Mallet AI, Anggard EE (1995) J Chromatogr B 667:199
Montine TJ, Montine KS, Reich EE, Terry ES, Porter NA, Morrow JD (2003) Biochem Pharmacol 65:611
Liu X, Whitefield PD, Ma Y (2010) Talanta 81:1599
Yin H, Porter NA, Morrow JD (2005) J Chromatogr B 827:157
Waugh RJ, Morrow JD, Roberts LJ, Murphy RC (1997) Free Radical Bio Med 23:943
Łuczaj W, Welerowicz T, Skrzydlewska E, Buszewski B (2008) Toxicol Mech Method 18:483
Durand T, Bultel-Poncé V, Guy A, El Fangour S, Rossi JC, Galano JM (2011) Biochimie 93:52
Musiek ES, Cha JK, Yin H, Zackert WE, Terry ES, Porter NA, Montine TJ, Morrow JD (2004) J Chromatogr B 799:95
Acknowledgments
This work was supported financially by the Polish Ministry of Science and Higher Education (No N N 401 315739).
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
Published in the special issue Analytical and Bioanalytical Science in Poland with guest editor Marek Biziuk.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Ligor, M., Olszowy, P. & Buszewski, B. Application of medical and analytical methods in Lyme borreliosis monitoring. Anal Bioanal Chem 402, 2233–2248 (2012). https://doi.org/10.1007/s00216-011-5451-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-011-5451-z