
Abstract
With more stringent legislation on brominated flame retardants, it is expected that increasing amounts of substitutes would replace polybrominated diphenylethers (PBDEs). Therefore, the development and optimization of analytical methodologies that allow their identification and quantification are of paramount relevance. This work describes the optimization of an analytical procedure to determine pentabromochlorocyclohexane, tetrabromo-o-chlorotoluene, 2,3,5,6-tetrabromo-p-xylene, tetrabromophthalic anhydride, 2,3,4,5,6-pentabromotoluene, tris(2,3-dibromopropyl)phosphate, decabromodiphenylethane and 1,2-bis(2,4,6-tribromophenoxy)ethane together with PBDEs in sediments and in suspended particulate matter. This method comprises a pressurized liquid extraction followed by three cleanup steps (gel permeation chromatography and solid phase extraction on Oasis™ HLB and on silica cartridges). Gas chromatography–mass spectrometry, using electron capture negative chemical ionization, is used for the final analysis. The proposed method provides recoveries >85%. The method was applied to sediment and suspended particulate matter samples from different locations in the Western Scheldt estuary (the Netherlands). To the best of our knowledge, this is the first time that the occurrence of the additive flame retardants 2,3,5,6-tetrabromo-p-xylene, 3,4,5,6-tetrabromo-o-chlorotoluene and 2,3,4,5,6-pentabromochlorocyclohexane is reported in the literature. The concentrations of these new flame retardants ranged from 0.05 to 0.30 μg/kg dry weight.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Brominated flame retardants (BFRs) are synthetic additives added to diverse materials such as electrical and electronic equipment (E&E), textiles and building materials to slow down or even suppress combustion. The most frequently used BFRs are polybrominated diphenylethers (PBDEs), tetrabromobisphenol A and hexabromocyclododecane [1]. The market for BFRs has been growing rapidly due to stringent fire safety regulations and the increasing use of E&E and polymers. However, restrictive legislation was set in Europe (EC 2002, 2003, 2006, 2008) due to their potential toxicity [2–4], their frequent environmental occurrence [5] and their accumulation in human tissues [6–8].
Collectively, some of US BFR manufacturers voluntarily stopped the production of penta- and octa-PBDEs in 2004. Despite the extensive use of the third commercial mixture (deca-BDE), whose production was reported as high as 56,000 tonnes/year [9, 10], much controversy has arisen over it. The EU has banned the use of deca-BDE in E&E since 1 July 2008. Industries may still apply for temporary exemptions for certain applications under the procedure laid out in article 5 of the RoSH Directive (EC 2002) [11]. On 17 December 2009, as the result of negotiations with the US Environmental Protection Agency (EPA), three companies (Albemarle Corporation, Chemtura Corporation and the largest U.S. importer, ICL Industrial Products Inc.) agreed to phase out the production and sale of deca-BDE for most applications within 3 years in the USA (EPA 2009). Flame retardants taken off the market are likely to be replaced by others. Although in Europe the introduction and utilization of new chemicals is regulated by REACH (Registration, Evaluation, Authorization and restriction of Chemical substances), it is still prudent to monitor the environment for the presence of new BFRs.
Decabromodiphenyl ethane (DBDPE), which was first found in the environment by Kierkegaard et al. [12], is being marketed as an alternative to deca-BDE. It does not produce dioxins or furans under natural sunlight [13] and only traces of 2,3,7,8-tetrabromodibenzofuran under pyrolysis conditions [14]. Another alternative flame retardant is 1,2-bis(2,4,6-tribromophenoxy)ethane (BTBPE), which was announced to replace octa-BDE [15] and was already found in different environmental samples [16] and in E&E waste [17]. Other alternatives such as pentabromochlorocyclohexane (PBCCH), tetrabromo-o-chlorotoluene (TBoCT), 2,3,5,6-tetrabromo-p-xylene (pTBX), tetrabromophthalic anhydride (TBPhA), 2,3,4,5,6-pentabromotoluene (PBT) and tris(2,3-dibromopropyl)phosphate (TDBPP) have been scarcely reported in environmental samples.
The aim of this work was the development and optimization of an analytical method that allows the simultaneous determination of these new alternative BFRs and PBDEs. This method was validated and applied to sediment and suspended particulate matter samples from the Western Scheldt estuary (the Netherlands) to screen their environmental occurrence.
Experimental section
Chemicals and materials
PBCCH (congeners A, B, C and D) and tetrabromo-o-chlorotoluene (PBoCT, 98%) were supplied by Accustandard (New Haven, USA) at 98% purity. pTBX (98%), TBPhA (98%), PBT (99%) and TDBPP were supplied by Sigma-Aldrich Chemie B.V. (Zwijndrecht, the Netherlands). Individual stock solutions of each standard were prepared in toluene at 1,000 μg/mL and diluted to 1 μg/mL. DBDPE (98%) and BTBPE (98%) were purchased respectively in toluene at 25 μg/mL and in nonane/toluene (5%, v/v) at 50 μg/mL from Wellington Laboratories (Guelph, Ontario, Canada). A standard mixture of the target analytes was prepared in toluene at 0.5 μg/mL each. Isotopically labelled (13C) 2,2′,3,3′,4,4′,5,5′,6,6′-decabromodiphenyl ether (13C-BDE209, 99%) in toluene at 25 μg/mL, decabromodiphenylethane (13C-DBDPE, 99%) in toluene at 25 μg/mL and 1,2-bis(2,4,6-tribromophenoxy)ethane (13C-BTBPE) in nonane/toluene (5%, v/v) at 50 μg/mL were supplied by Wellington Laboratories. The PDBE congeners were also purchased as a mixture (BDE-MXE) in a nonane/toluene solution (48.6:51.4, v/v) from Wellington Laboratories at concentrations that ranged from 1.0 to 5.0 μg/mL. All solutions were stored at 4 °C and darkness. Table 1 shows the molecular structure of the target analytes.
n-Hexane, acetone, methanol, toluene, iso-octane and HPLC water were purchased from J.T. Baker (Deventer, the Netherlands), dichloromethane (DCM) for residue analysis from Promochem (Wesel, Germany) and diethyl ether (DEE) from Merck (Darmstadt, Germany). Sulphuric acid (98%) and anhydrous sodium sulphate were obtained from Merck. All solvents and reagents were at least of analytical grade.
Copper powder was purchased from Sigma Aldrich (>99.9%, Sigma Aldrich, Zwijndrecht, the Netherlands).
The following solid phase extraction (SPE) cartridges were tested: 200 mg/3 mL Lichrolut® EN (Merck), 500 mg/3 mL octadecyl (C18) Bakerbond (J.T. Baker), 500 mg/3 mL amino (NH2) Bakerbond (J.T. Baker), 500 mg/6 mL Oasis™ HLB (Waters, Mildford, MA, USA), 500 mg/6 mL Supelclean ENVI-Carb Supelco (Sigma-Aldrich) and 200 mg/5 mL Oasis™ WCX (Waters).
Silica gel (high purity, 70–230 mesh) was obtained from Merck. It was activated at 250 °C overnight and then deactivated either with 3% (w/w) HPLC water or 33% (w/w) sulfuric acid (98%). Silica gel and acidified silica gel columns were manually prepared by filling an amber glass column (i.d. 0.6 cm) with 1.8 g of silica or 5.0 g of acidified silica with a plug of glass wool at the bottom.
Samples
Sediment samples (upper 2–4 cm) were taken from five different locations at the Western Scheldt estuary in the Netherlands (Fig. 1) in 2005 by a Van Veen grab sampler from a square of approx. 100 m2/location. The samples were dried, ground, sieved to <90 μm and stored in amber glass jars at −18 °C until analysis. The suspended particulate matter (SPM) sample was obtained from the surface water of location 3 (Terneuzen, the Netherlands; see Fig. 1). Centrifugation was carried out at 20,000 rpm until 200 g of material was collected. They were stored in amber glass jars at −18 °C.

Sampling locations in the Western Scheldt estuary (the Netherlands). Sediments were collected from 1, 2, 3 and 4 and SPM from 3
Analytical methodology
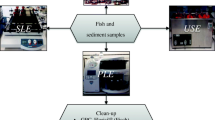
Figure 2 shows a scheme of the optimized method. Twenty grams of freeze-dried sediment samples or 5 g of SPM, which was previously dried with 15 g anhydrous sodium sulphate for 12 h, was spiked with the internal standards (or surrogates) and extracted by pressurized liquid extraction (PLE) with n-hexane/acetone (3:1, v/v) using a Dionex ASE200 Accelerated Solvent Extractor (Dionex Benelux, Amsterdam, the Netherlands). The operational settings were as follows: extraction temperature 70 °C, extraction pressure 2,000 psi, static time 5 min, flush volume 60%, purge time 120 s, and two static cycles. The extracts were concentrated to 0.5 mL.

Scheme of the analytical procedure
The extracts were further cleaned up by gel permeation chromatography (GPC) and two successive SPE procedures: initially on commercial Oasis™ HLB cartridges and then on manually prepared silica gel columns. The final extracts were analysed by gas chromatography with electron capture negative ion mass spectrometry (GC-(ECNI)MS).
The GPC consisted of a Polymer Laboratories (Haarlem, the Netherlands) pre-column PL-Gel (pore size 10 μm) 25 × 25 mm (length × width) coupled online with two columns PL-Gel (pore size 10 μm) 300 × 25 mm (length × width) cross-linked polystyrene divinylbenzene (50 Å porous size). The mobile phase (DCM) was pumped at 10 mL/min by a LC-10 AT Shimadzu pump (Shimadzu Scientific Instruments, Riverwood Drive, Columbia, USA). The collected fraction (15–27 min) was concentrated to 0.5 mL, redissolved in toluene and further purified.
The GPC extract was loaded on an Oasis™ HLB cartridge, which was previously cleaned with 5 mL of DCM and conditioned with 5 mL of methanol. After loading the sample, the cartridge was washed with 0.5 mL methanol and dried under vacuum and/or a nitrogen stream for 15 min. The analytes were finally eluted with 5 mL of a mixture DCM/iso-octane (3:1). The eluate was concentrated to 0.5 mL.
Subsequently, another SPE procedure on silica gel columns was conducted to remove the remaining interferences. The extract was loaded and eluted with 12 mL of iso-octane, 24 mL of 15% DEE in iso-octane and 16 mL of DEE. The eluate was concentrated to 0.5 mL and transferred to a GC injection vial.
Traces of sulphur, common in sediment samples, were removed by a combination of GPC and a reaction with acid-washed copper powder. The copper was activated as follows: A spatula of powder was washed with 1 mL of water and hydrochloric acid (3:1) and centrifuged for 5 min at 3,000 rpm. The upper liquid layer was discarded and the solid was washed and centrifuged as before with 2 mL of methanol (twice) and 2 mL of acetone. The activated powder was dried under a nitrogen atmosphere and placed in the SPE syringe on top of the Oasis™ HLB resin.
Instrumental analysis
The identification and quantification of the BFRs was performed on an Agilent GC 6890N (Agilent Technologies Netherlands BV, Amstelveen, the Netherlands) coupled in-line with a 5975XL MS with a chemical ionization ion source and equipped with a pulsed splitless injector and an Agilent 7683 autosampler. The separation of the analytes was carried out with two different columns. Octa-BDEs, nona-BDEs, deca-BDE and DBDPE were quantified with a J&W DB-5 (15 m × 0.25 mm, 0.25-μm film) column. One microliter was injected at 270 °C in the pulsed pressure mode (pulsed pressure 150 kPa kept for 1.5 min). The oven temperature was programmed from 80 °C, for 1.5 min, then raised with 30 °C/min to 90 °C, then raised with 3 °C/min to 225 °C and then with 7 °C/min to 270 °C, and finally raised with 10 °C/min to 320 °C, which was kept for 10 min. A constant flow of 1 mL/min was set. The following fragments were monitored: m/z = 79/81 (bromine trace), m/z = 486.6/488.7 and 495/497, corresponding to the ion [C6Br5O]− obtained by fragmentation of the BDE209 and 13C-BDE209, respectively [18]. The other PDBEs and the new BFRs were analysed on a CP-Sil8 (Varian, 60 m × 0.25 mm, 0.25-μm film) column. One microliter was injected at 250 °C in the pulsed pressure mode (pulsed pressure 460 kPa kept for 1 min). The oven temperature was programmed at 90 °C for 3 min, then raised with 30 °C/min to 210 °C, kept for 20 min, and finally raised with 5 °C/min to 320 °C, which was kept for 15 min. The flow was constant at 3 mL/min. The fragments m/z = 79/81 and m/z 250.8/252.8 and 256.9/257.9 (which correspond to the ion [C6H2Br3O-Br]− obtained by fragmentation of the BTBPE and 13C-BTBPE, respectively) were monitored. The temperatures of the interface, quadrupole and ion source were 300, 150 and 250 °C, respectively. Prior to any analysis, the optimum mode of operation, either electron impact (EI) or ECNI (using methane as ionization gas at 3.25 mL/min) was selected. Full-scan mass spectra (m/z 50–1,000) of the individual standard solutions were recorded using both EI and ECNI modes. The most abundant ions of each compound were recorded using SIM mode.
The quantification was conducted by internal standard calibration. 13C-BDE209 was used as internal standard for octa-, nona-, deca-BDE and DBDPE, whilst BDE58 and 13C-BTBPE were used to quantify the rest of the analytes.
Validation performance and quality control
The validation of the method was performed according to EURACHEM guidelines [19].
The detection limit (LOD) was estimated as the concentration of the analytes that had a signal-to-noise ratio based on the height of the chromatographic peak (S/N) of 3.0, whilst the quantification limit (LOQ) was estimated as the concentration that provided a S/N ratio based on the peak height of 10.0.
The linearity was established in the 0.2- to 140-ng/mL range, depending on the analyte. Statistical analysis (analysis of variance, ANOVA) was performed to check the goodness-of-fit and linearity.
Recovery (or recovery factor) is defined by IUPAC as the yield of an analytical process for an analyte divided by the amount of analyte in the original sample, whereas the apparent recovery is the value corrected with the internal standard [20]. Both were calculated for the developed method. Twenty grams of sediment was spiked with 0.5 mL of a solution containing the target analytes (0.1 μg/mL) and with 0.5 mL of a solution containing the internal standards (0.05 μg/mL). Due to the lack of reference materials for these analytes, trueness could not be evaluated.
Precision, measured as repeatability (RSDr), was assessed on sediments spiked at 2.5 and 0.5 ng/g in five replicates at each level.
Three quality control criteria were used to ensure the correct identification of the target compounds: (a) The GC retention times matched those of the standard compounds within ±0.1 min. (b) The signal-to-noise ratio was >3:1. (c) The isotopic ratios for the selected ion pairs were within ±15% of the theoretical values. A procedural blank was run with each batch.
Results
Optimization of the method
Instrumental analysis: GC-(ECNI)MS
The first step of the instrumental optimization comprised the identification of mass-to-charge ratios that might be used as quantifier and qualifier responses for every analyte. Individual stock solutions in toluene (0.5 μg/mL) were injected in both EI and ECNI full-scan modes to obtain spectra information. The most relevant mass-to-charge ratios for each compound are shown in decreasing order of abundance in Table 2.
The PBCCH standard was not a pure compound. Four chromatographic peaks were registered, with the penta congener—congener A—being the most abundant peak (around 50%). The other peaks—congener B (31%), C (16%) and D (3%)—might be a mixture of tetra- and tri-derivates, although the molecular ion could not be confirmed by any of the ionization modes. The molecular ion of TBoCT was detected in both EI and ECNI modes, with the bromine ion being the most abundant and sensitive in ECNI. ECNI was also more sensitive than EI for pTBX and PBT, but there the only detected fragment was the bromine ion. Their respective molecular ions were the most abundant fragments in EI mass spectra, though. The mass spectrum of TBPhA in ECNI revealed that the principal ion was the molecular ion, followed by the fragment with a loss of bromine. In the EI spectrum, the molecular ion was the second most abundant one. As for TDBPP in ECNI mode, the principal ion was the bromine followed by the molecular ion with the loss of a 2,3-dibromopropyl group, which is more specific. Their molecular ion could not be confirmed by EI. BTBPE produced two ions in ECNI corresponding to the 2,4,6-tribromophenoxy (m/z 331) group and the loss of one bromine from this group (m/z 252), which can be used for identification purposes. The EI spectrum showed the most abundant ion at m/z 357, corresponding to a fragment ion with three bromine atoms. For DBDPE, the dominating ion in ECNI was the bromine too. The molecular ion with a loss of two bromines was detected, but with a very low sensitivity. In EI, the most abundant fragment centred at m/z 485, which corresponded to the cleavage of the ethyl bond.
The fragmentation pattern of the 13C-labelled compounds of BTBPE and DBDPE was also studied to assess their feasibility as internal standards (surrogates). As expected, in the ECNI mass spectrum of 13C-BTBPE, the bromine ion was the most abundant one. The m/z 256 fragment gave a signal that was sensitive enough to be used for quantification purposes. The EI spectrum centred at m/z 363. As for 13C-DBDPE, the bromine ion was the most abundant in ECNI. The m/z 824 fragment gave very low signal, thus preventing 13C-DBDPE from being applied as an appropriate internal standard for quantification.
Due to its higher sensitivity and selectivity to bromine and with less fragmentation, ECNI was selected as the primary ionization mode to quantify these new flame retardants. Chromatographic conditions were set up taking into consideration the analysis of these new BFRs together with PBDEs. Therefore, the analysis was carried out in two steps: PBCCH, TBoCT, pTBX, PBT, TBPhA, TBDPP and BTBPE were analysed together with tri-, tetra-, penta-, hexa- and hepta-PBDEs on a 60-m column, whereas DBDPE was analysed in the same run as octa-, nona-BDEs and BDE209 on a short column to avoid on-column degradation [21]. As internal standards (IS), BDE58 and 13C-BTBPE were used on the 60-m column and 13C-BDE209 was used on the 15-m column, even though 13C-BDE209 was reported not to be the most accurate internal standard for DBDPE because of their different response factors [22].
As far as the analysis of DBDPE is concerned, most of the difficulties encountered in the analysis of deca-BDE are also expected for this compound. DBDPE is poorly soluble in most organic solvents, even less than BDE209 [12]. It has a higher boiling point than BDE209, so it elutes even later from a non-polar GC column, thus being exposed to elevated temperatures for a longer time. DBDPE thermally degrades to (mainly) bromotoluenes [23] and photolytically debrominates, i.e. to nona- and octa-brominated congeners (tentatively identified) [24]. As this chemical has the same field of application as deca-BDE and is also present in the indoor environment [25], the same precautions to minimize background contamination should be taken [26]. Sulphur removal with tetrabutylammonium also caused debromination of DBDPE in standard solutions, but this problem was less pronounced in real samples, which was attributed to a protective effect of the matrix [24]. In the present study, sulphur was removed with a combination of GPC and a reaction with acid copper, which was reasonably effective as long as toluene was not used as solvent [22].
Sample treatment
The sample treatment for BFRs in sediments usually comprises an extraction procedure and at least one (or more) cleanup steps.
Extraction
Soxhlet extraction is the conventional method for PBDEs and other contaminants from environmental samples. Other extraction techniques, such as ultrasonic-assisted extraction, microwave-assisted extraction, supercritical fluid extraction and PLE, which significantly reduce the extraction time and the solvent consumption, are nowadays gaining wider acceptance.
In the present study, PLE was used as the extraction technique. As demonstrated in the study carried out by Houtman et al. [27], PLE and Soxhlet extraction provided comparable results in terms of recoveries for a large variety of environmental contaminants. The operational settings for PLE are described in “Analytical methodology”.
Cleanup procedure
Although PLE is often more selective than Soxhlet extraction, the crude extract is also likely to contain many co-extracted compounds (lipids, humic acids and sulphur) that may hinder the final analysis.
Sulphuric acid treatment, either directly or via impregnated silica columns [28], is the most commonly applied destructive matrix removal method. PBDEs are resistant to sulfuric acid treatment [18], but the resistance of these new BFRs under such conditions was unknown. The assessment of their degradation was conducted by passing through 5 g of acidified silica (44%, w/w) column 0.5 mL of a standard mixture (0.5 μg/mL) of the target analytes in toluene. The analytes were eluted with 35 mL of hexane/DCM (70:30, v/v). The eluate was concentrated under a nitrogen stream to 0.5 mL and then injected in the GC-(ECNI)MS. The recoveries ranged between 95% and 115% (data not shown) for most of the compounds, except for TBPhA (completely degraded), TDBPP and BTBPE. The degradation of TBPhA was expected due to the high chemical instability of the anhydrides. The almost complete hydrolysis of the phosphate might explain the low recovery of TBDPP, whereas this hydrolysis might only partially occur for the oxygen belonging to the phenoxy group of the BTBPE. Although higher recoveries of BTBPE would be expected if 13C-BTBPE were used as internal standard, the acidic treatment is not advisable for this compound and other alternative cleanup procedures were sought.
GPC is a non-destructive cleanup technique. The suitability of GPC for the removal of high-molecular matrix components from sediments and SPM was evaluated by the assessment of the elution profile and the recovery of the BFRs of interest. A matrix extract of 0.5 mL spiked with the mixture of the target analytes at 0.5 μg/mL was injected in the GPC system with a flow of 10 mL/min. Fractions of 10 mL were collected, concentrated to 0.5 mL and analysed by GC-(ECNI)MS. The first eluted BFR was TDBPP at 15 min (data not shown). PBCCH, TBPhA, BTBPE, PBDEs and DBDPE were completely eluted after 24 min, whereas TBoCT, pTBX and PBT needed three more minutes. Therefore, the collected fraction in the procedure was from 15 to 27 min. The absolute recoveries ranged from 81% to 114%. This allowed discarding the first 150 mL without losses of target compounds. However, it was observed that a dark-coloured fraction came out between 11 and 19 min (110–190 mL), which might contain part of the sediment matrix. This implied a partial co-elution of matrix with some of the BFRs. Elemental sulphur eluted between 25 and 28 min [27]. In summary, GPC removes a wide range of compounds that would interfere with the chemical analysis, but some of the remaining matrix components need to be eliminated by applying additional cleanup techniques.
The GPC extract (0.5 mL in toluene) was further purified by SPE. This is a fast and efficient technique, with very low solvent consumption and cross-contamination and often a high selectivity, but it needs a thorough optimization. The following aspects of the SPE procedure were optimized: (a) nature of the sorbent, (b) nature of the elution solvent, (c) elution volume, (d) conditioning step and (e) washing step.
Six different cartridges were tested: two polymeric phases (Lichrolut® EN based on polystyrene divinylbenzene and Oasis™ HLB based on divinlybenzene/N-vinylpirrolidone), a carbon-based phase (Supelclean ENVI-Carb), an amino-silica phase (NH −2 ), an octadecyl phase (C18-) and a weak cation exchange phase (Oasis™ WCX). Briefly, the cartridges were cleaned with 5 mL of DCM and conditioned with 5 mL of n-hexane. A sediment extract, which had been previously spiked with the analytes of interest and with the IS, was loaded on the cartridge. The compounds were eluted with 9 mL of DCM, which were then concentrated to 0.5 mL and analysed by GC-(ECNI)MS. The recoveries of analytes relative to the ISs are given in Table 3. The best results, in terms of relative recoveries and RSD values, were obtained with Oasis™ HLB. This is probably due to a higher hydrophobic character (coming from the divinylbenzene polymer) associated with an increased retention capability through hydrophilic interactions (due to the N-vinylpirrolidone polymer) between the sorbent and analytes. Unlike TBDDP, the high recovery for BDE209 and DBDBE could not be attributed to a different behaviour between the target analyte and its IS, but to the confirmed presence of natural contamination from the sediment sample. Lichrolut® EN also provided acceptable recoveries, but higher RSD values. Carbon phases, such as ENVI-Carb, are designed to extract polar compounds; hence, no complete recoveries (data not shown) were obtained either for TBoCT, pTBX, PBT, BTBPE, DBDPE, 13C-BTBPE and 13C-BDE209 on those phases. The high RSD values with Oasis™ WCX were due to the low recoveries of both the analytes and the ISs.
From these results, Oasis™ HLB was concluded to provide the most promising cleanup performance, so the optimization experiments for the stages described above were conducted using this stationary phase. Oasis™ HLB has already been proposed to extract PDBEs from biological fluids [29, 30], and it was the first time, to the best of our knowledge, that it was applied to sediments and SPM samples.
As far as the optimization of the conditioning step was concerned, the use of 5 mL of methanol instead of n-hexane was demonstrated to provide higher recoveries of the more polar analytes (data not shown).
After that, both the nature of the elution solvent and its elution volume were studied and optimized. The tested elution solvents were: DCM, DCM/n-hexane (3:1, v/v), DCM/toluene (3:1, v/v) and DCM/iso-octane (3:1, 1:1 and 1:3). As shown in Fig. 3, the recoveries increased when decreasing the polarity of the solvent (iso-octane < n-hexane < toluene). All the test mixtures with iso-octane gave similar high recoveries, but DCM/iso-octane (3:1, v/v) showed the lowest RSD. It was noted that the addition of toluene to DCM had a negative effect on the recovery of the low-molecular-weight BFRs (PBCCH, TBoCT, pTBX and PBT). The minimum solvent volume was estimated from the cumulative elution profile of the target analytes. Fractions of 1 mL of eluate were collected and analysed by GC-(ECNI)MS. Recoveries higher than 99% were obtained after the elution of 5 mL (data not shown).

Selection of the elution solvent on OasisTM HLB (y-axis: % of relative areas A analyte/A IS). Error bars showed the standard deviation (n − 3)
The compounds that will interfere in the GC/MS analysis are removed during the washing step. Although an aqueous solution of 5% methanol is suggested by the manufacturer as the washing solvent, other solvents were tested due to the insolubility of the iso-octane in water. The optimization of this step was conducted by loading 0.07 g of matrix spiked with the target analytes (300 ng/mL) in toluene onto the SPE cartridges. The Oasis™ HLB cartridges were washed with 0, 0.5, 1.0, 1.5 and 2.0 mL of n-hexane and 0.5 mL of methanol in two sets by triplicate. After the washing step, the cartridges were dried under a nitrogen stream and eluted with 5 mL of DCM/iso-octane (3:1, v/v). One set of extracts was evaporated until dryness and the residue was weighed. The other set was injected in the GC-(ECNI)MS and the recoveries were estimated. As expected, the larger the washing volume, the more interferences removed and the lower the recovery. By washing with 1 mL of n-hexane, only 1% of interferences were removed and 71% of recovery was achieved, whereas by washing with 2.5 mL of n-hexane, 62% of the interferences were removed but the recovery only yielded 32%. Methanol was shown to be more effective than n-hexane, thus separating 35% of the interferences with hardly any loss of the target analytes (95% recovery).
Summarizing, the optimized SPE procedure with Oasis™ HLB cartridge consisted of:
-
1.
Conditioning step: 5 mL of DCM, 5 mL of methanol
-
2.
Sample loading step: 0.5 mL extract in toluene
-
3.
Washing step: 0.5 mL of methanol
-
4.
Drying of the cartridge
-
5.
Elution step: 5 mL of DCM/iso-octane (3:1, v/v)
The final extract was concentrated to 0.5 mL under a nitrogen stream.
In principle, the extract was ready to be injected and analysed by GC-(ECNI)MS. However, some tests revealed the presence of matrix, which interfered with the analysis by decreasing the sensitivity of the heavier analytes (nona-BDEs, deca-BDE and DBDPE) after a few injections. DBDPE, like BDE209, is demonstrated to be a very sensitive compound that can be degraded in the GC system due to high temperatures or to the adsorption on some parts of the injection port. The degradation in the injection port is favoured when the sample is not cleaned up enough since the matrix particles, which remain in the system, act as active sites. A standard of the calibration curve was injected every five sample injections to check the stability of the signal.
Therefore, an extra cleanup step with silica deactivated with 3% (w/w) water was included after the SPE on Oasis™ HLB. The elution was assessed and optimized. PBCCH, TBoCT, PBT and pTBX eluted in the first fraction of 12 mL 100% iso-octane; whilst BTBPE, BDE209 and DBDPE eluted with the second fraction of 24 mL of 15% (v/v) of DEE in iso-octane. For TDBPP and TBPhA, a third more polar fraction of 16 mL of DEE was needed, which resulted in good recoveries of both analytes. The three fractions were mixed and concentrated to 0.5 mL under a nitrogen stream. This final extract was injected in the GC-(ECNI)MS. The recovery of the whole elution process ranged from 85% to 110% with RSD values around 5%. No signs of degradation of either DBDPE or BDE209 were observed during the sequence analysis.
An overview of the optimized method is shown in Fig. 2.
Method performance
The method performance was assessed in terms of linearity, LOD, LOQ, recovery and repeatability (see Table 4).
In general, good linearity was demonstrated for all compounds over three orders of magnitude. To validate the regression data, an ANOVA was performed at the 95% of confidence level. Since p values for lack-of-fit test were ≥0.05, the linear first-order models were suitable for the experimental data.
LOD and LOQ values were estimated on matrix; hence, TBPhA gave the highest value due to its higher instability. The lowest LOD and LOQ values corresponded to the most volatile BFRs.
The recovery of all the target analytes ranged from 50% to 60%, whereas the apparent recovery was 85–100%. The fact that the apparent recovery was not 100% for all the target compounds is due to the differences in behaviour compared with the internal standards (surrogates). Only BDE209 and BTBPE, for which isotopic labelled standards were used, had apparent recoveries close to 100%.
Method precision was evaluated by testing at two concentration levels. As expected, the highest RSDr% values corresponded to the lowest concentration. The repeatability at 0.5 ng/g ranged from 10% to 26%, whereas at 2.5 ng/g, it ranged from 2% to 14%. TBPhA showed the highest values.
Sample analysis
Sediments (20 g) and SPM (5 g) from different locations in the Western Scheldt estuary (see Fig. 1) were processed according to the method scheme in Fig. 2. The sampling locations were chosen based on the presence of a BFR production plant in Terneuzen (site 3), the high level of industrialization of the nearby harbour of Antwerp and the presence of the textile industry, which is known to use relatively high amounts of BFRs, further upstream of the river [23, 31–35]. The samples were quantified based on an eight-point linear calibration curve.
Table 5 shows that most of the “new” BFRs are present in the Western Scheldt, although some of them at relatively low concentrations as compared to PBDEs. TBPhA and TDBPP were not detected. PBT and BTBPE have maximum concentrations around 0.3 ng/g dry weight. PBCCH D is present at concentrations up to 1.2 ng/g. The relative pattern for the congeners of PBCCH differs from the standard and also differs within locations, which could be due to a debromination process. DBDPE shows a maximum of 10 ng/g dry weight. The presence of BTBPE and DBDPE was also verified by EI.
As far as the concentration of PBDEs is concerned, BDE209 was the predominant congener both in sediments and the SPME. After BDE209, the most predominant congeners were the nona-congeners (BDE206 and BDE207), which are presumably degradation products. The concentration of BDE209 was similar to the values reported in the literature [31, 36]. The next most abundant PBDE congeners in sediments were BDE47, BDE71, BDE 49 and BDE99. Their abundance depended on the location. As for the SPM sample, the detected levels of BDE153 and BDE154 were comparable to BDE47 and BDE99.
Wielingen, the most westerly location in the Scheldt estuary, in fact in coastal water, is the location with the lowest BFR levels, whilst Hansweert (east from Terneuzen, close to a BFR production site) and Ouden Doel (at the Belgium border) compete for the highest degree of BFR contamination. The input from Antwerp harbour and/or further upstream appears to be more important than a supposed contribution from the bromine industry at Terneuzen. Although there is a considerable tidal movement, which could move a possible BFR plume from Terneuzen to the east, it is unlikely that the higher level of brominated compounds in the eastern part of the Western Scheldt would only have been caused by such tidal influence [31]. At Terneuzen, both sediment and a SPM sample have been taken. On a dry weight basis, almost all PBDE and other BFR concentrations are higher in the SPM sample. Although peak values can occur in SPM, another explanation could be an increase in concentration with time of these compounds: The sediment sample is a more time-integrated sample, even though only the top layer was sampled, whilst the SPM reflects the actual situation at the moment of sampling.
To the best of our knowledge, this is the first time that the occurrence of BTBPE, PBCCH, TBoCT, PBT and pTBX in sediments and SPM from the Western Scheldt estuary is reported. BTBPE has been already identified and quantified in several environmental compartments from Asia and the USA [37, 38]. However, no documentation about the occurrence of PBCCH, TBoCT or pTBX in the environment was found.
DBDPE was introduced in the early 1990s as a replacement of deca-BDE [12]. Few studies have reported the presence of DBDPE in the environment: It has been detected in sediment from sewage treatment plants in Sweden [12], in tree bark from North America [39], in house dust from the USA [25] and in air near the Great Lakes [40]. DBDPE has also been found in fishes, birds and marine mammals [41]. With increasing regulation and the phasing out of production of the commercial usage of PBDEs, it is expected that the production and usage of DBDPE will mount in the near future [42]. Although the evidence for the environmental presence of DBDPE is still limited, its detection in a wide range of matrices warns of a potential environmental concern.
Conclusion
The developed methods allow the determination of a number of new BFRs together with PBDEs. These methods were applied to samples from the Western Scheldt estuary (the Netherlands) to screen their environmental occurrence, which resulted in the first identification ever of PBCCH, pTBX and TBoCT in SPM and sediments. Their concentration ranged from 0.05 to 3.0 μg/kg dry weight. PBT, BTBPE and DBDPE were also found.
References
Hyotylainen T, Hartonen K (2002) Determination of brominated flame retardants in environmental samples. Trac-Trends Anal Chem 21:13–29
Birnbaum LS, Staskal DF (2004) Brominated flame retardants: cause for concern? Environ Health Perspect 112:9–17
Viberg H, Fredriksson A, Eriksson P (2004) Neonatal exposure to the brominated flame-retardant 2,2′,4,4′,5-pentabromodiphenyl ether decreases cholinergic nicotinic receptors in hippocampus and affects spontaneous behaviour in the adult mouse. Environ Toxicol Pharmacol 17:61–65
Hamers T, Kamstra JH, Sonneveld E, Murk AJ, Kester MHA, Andersson PL, Legler J, Brouwer A (2006) In vitro profiling of the endocrine-disrupting potency of brominated flame retardants. Toxicol Sci 92:157–173
de Wit CA (2002) An overview of brominated flame retardants in the environment. Chemosphere 46:583–624
Sjodin A, Dg P, Bergman A (2003) A review on human exposure to borminated flame retardants. Environ Int 29:829–839
Hites RA (2004) Polybrominated diphenyl ethers in the environment and in people: a meta-analysis of concentrations. Environ Sci Technol 38:945–956
Inoue K, Harada K, Takenaka K, Uehara S, Kono M, Shimizu T, Takasuga T, Senthilkumar K, Yamashita F, Koizumi A (2006) Levels of concentration ratios of polychlorinated biphenyls and polybrominated diphenyl ethers in serum and breast milk in Japanese mothers. Environ Health Perspect 114:1179–1185
Brominated Science and Environmental Forum BSEF. www.bsef.com
Brominated Science and Environmental Forum BSEF. http://www.bsef.com/regulation/
European Court of Justice. 2008-04-01, Case C-14/06. Brussels, Belgium
Kierkegaard A, Bjorklund J, Friden U (2004) Identification of the flame retardant decabromodiphenyl ethane in the environment. Environ Sci Technol 38:3247–3253
Kajiwara N, Noma Y, Takigami H (2008) Photolysis studies of technical decabromodiphenyl ether (decaBDE) and ethane (DeBDethane) in plastics under natural sunlight. Environ Sci Technol 42:4404–4409
Arias PA (2001) 2nd International Workshop on Brominated Flame Retardants BFR 2001, Stockholm, Sweden, p 17
Tullo AH (2003) Resting easier. Chem Eng News 81:43–44
Hoh E, Zhu LY, Hites RA (2005) Novel flame retardants 1,2-bis(2,4,6-tribromophenoxy)ethane and 2,3,4,5,6-pentabromoethylbenzene in United States’ environmental samples. Environ Sci Technol 39:2472–2477
Schlummer M, Brandl F, Mäurer A, van Eldik R (2005) Analysis of flame retardants additives in polymer fractions of waste of electric and electronic equipment (WEEE) by means of HPLC-UV/MS and GPC-HPLC-UV. J Chromatog A 1064:39–51
Bjorklund J, Tollback P, Ostman C (2003) Mass spectrometric characteristics of decabromodiphenyl ether and the application of isotopic dilution in the electron capture negative ionization mode for the analysis of polybrominated diphenyl ethers. J Mass Spectrom 38:394–400
EUROCHEM Guidelines (1998) The fitness for purpose of analytical methods: a laboratory guide to method validation and related topics. LGC (Teddington) Ltd. http://www.eurachem.ul.pt/
Burns DT, Danzer K, Townshend A (2002) Use of terms “recovery” and “apparent recovery” in analytical procedures. Pure Appl Chem 74:2201–2205
de Boer J, Allchin C, Law R, Zegers B, Boon JP (2001) Method for the analysis of polybrominated diphenylethers in sediments and biota. TrAC Trends Anal Chem 20:591–599
Konstantinov A, Arsenault G, Chittim B, Kolic T, MacPherson K, McAlees A, McCrindle R, Potter D, Reiner EJ, Tashiro C, Yeo B (2006) Characterization of mass-labelled [C-13 (14)]-decabromodiphenylethane and its use as a surrogate standard in the analysis of sewage sludge samples. Chemosphere 64:245–249
Jakab E, Uddin MA, Bhaskar T, Sakata Y (2003) Thermal decomposition of flame-retarded high-impact polystyrene. J Anal Appl Pyrol 68–9:83–99
Kierkegaard A, Sellstrom U, McLachlan MS (2009) Environmental analysis of higher brominated diphenyl ethers and decabromodiphenyl ethane. J Chromatog A 1216:364–375
Stapleton HM, Allen JG, Kelly SM, Konstantinov A, Klosterhaus S, Watkins D, McClean MD, Webster TF (2008) Alternate and new brominated flame retardants detected in US house dust. Environ Sci Technol 42:6910–6916
de Boer J, Wells DE (2006) Pitfalls in the analysis of brominated flame retardants in environmental human and food samples—including results of three interlaboratory studies. Trac-Trends Anal Chem 25:364–372
Houtman CJ, Leonards PEG, Kapiteijn W, Bakker JF, Brouwer A, Lamoree MH, Legler J, Klamer HJC (2007) Sample preparation method for the ER-CALUX bioassay screening of (xeno-)estrogenic activity in sediment extracts. Sci Total Environ 386:134–144
Covaci A, Voorspoels S, de Boer J (2003) Determination of brominated flame retardants with emphasis on polybrominated diphenyl ethers (PBDEs) in environmental and human samples—a review. Environ Int 29:735–756
Covaci A, Voorspoels S (2005) Optimization of the determination of polybrominated diphenyl ethers in human serum using solid-phase extraction and gas chromatography–electron capture negative ionization mass spectrometry. J Chromatog B 827:216–223
Thomsen C, Liane VH, Becher G (2007) Automated solid-phase extraction for the determination of polybrominated diphenyl ethers and polychlorinated biphenyls in serum-application on archived Norwegian samples from 1977 to 2003. J Chromatog B 846:252–263
de Boer J, Wester PG, van der Horst A, Leonards PEG (2003) Polybrominated diphenyl ethers in influents, suspended particulate matter, sediments, sewage treatment plant and effluents and biota from the Netherlands. Environ Pol 122:63–74
Covaci A, Gheorghe A, Voorspoels S, Maervoet J, Steen Redeker E, Blust R, Schepens P (2005) Polybrominated diphenyl ethers, polychlorinated biphenyls and organochlorine pesticides in sediment cores from the Western Scheldt river (Belgium): analytical aspects and depth profiles. Environ Int 31:367–375
Klamer HJC, Leonards PEG, Lamoree MH, Villerius LA, Akerman JE, Bakker JF (2005) A chemical and toxicological profile of Dutch North Sea surface sediments. Chemosphere 58:1579–1587
Verslycke TA, Vethaak AD, Arijs K, Janssen CR (2005) Flame retardants, surfactants and organotins in sediments and mysid shrimp of the Scheldt estuary (the Netherlands). Environ Pol 136:19–31
Lopez P, Brandsma SA, Leonards PEG, de Boer J (2009) Methods for the determination of phenolic brominated flame retardants and by-products, formulation intermediates and decomposition products of brominated flame retardants in water. J Chromatog A 1216:334–345
Voorspoels S, Covaci A, Maervoet J, Schepens P (2004) PBDEs in marine and freshwater sediments from Belgium: levels, profiles and relations with biota. J Environ Monit 6:914–918
Gauthier LT, Hebert CE, Weseloh DVC, Letcher RJ (2007) Current-use flame retardants in eggs of herring gulls (Larus argentatus) from the Laurentian Great Lakes. Environ Sci Technol 41:4561–4567
Shi T, Chen SJ, Luo XJ, Zhang XL, Tang CM, Luo Y, Ma YJ, Wu JP, Peng XZ, Mai BX (2009) Occurrence of brominated flame retardants other than polybrominated diphenyl ether in environmental and biota samples from southern China. Chemosphere 74:910–916
Zhu LY, Hites RA (2006) Brominated flame retardants in tree bark from North America. Environ Sci Technol 40:3711–3716
Venier HRA (2008) Flame retardants in the atmosphere near Great Lakes. Environ Sci Technol 42:4745–4751
Luo XJ, Zhang XL, Liu J, Wu JP, Luo Y, Chen SJ, Mai BX, Yang ZY (2009) Persistent halogenated compounds in waterbirds from e-waste recycling region in south China. Environ Sci Technol 43:306–311
Ricklund N, Kierkegaard A, McLachlan MS (2008) An international survey of decabromodiphenyl ethane (deBDethane) and decabromodiphenyl ether (decaBDE) in sewage sludge samples. Chemosphere 73:1799–1804
Acknowledgements
P. López acknowledges Caja Madrid and CAI (Spain) for personal funding through their respective Fellowship Programs
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary materials
Below is the link to the electronic supplementary material.
ESM 1
(PDF 786 kb)
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
López, P., Brandsma, S.A., Leonards, P.E.G. et al. Optimization and development of analytical methods for the determination of new brominated flame retardants and polybrominated diphenyl ethers in sediments and suspended particulate matter. Anal Bioanal Chem 400, 871–883 (2011). https://doi.org/10.1007/s00216-011-4807-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-011-4807-8