
Abstract
This review provides an overview of direct and indirect technologies to screen protein–ligand interactions with mass spectrometry. These technologies have as a key feature the selection or affinity purification of ligands in mixtures prior to detection. Specific fields of interest for these technologies are metabolic profiling of bioactive metabolites, natural extract screening, and the screening of libraries for bioactives, such as parallel synthesis libraries and small combichem libraries. The review addresses the principles of each of the methods discussed, with a focus on developments in recent years, and the applicability of the methods to lead generation and development in drug discovery.

Schematic view of the principle of filtration based 96-well affinity selection MS binding assays
Similar content being viewed by others

Introduction
The development of new lead compounds in drug discovery has been of continuous importance over the past few decades. However, high attrition rates and the decreasing number of new drug approvals in the past few years have increased the necessity for new development tools. Protein affinity selection methods utilizing mass spectrometry (MS) are among the more recently developed methods that can be a valuable addition to traditional drug discovery techniques. They distinguish themselves by utilizing the very high sensitivity and selectivity that is inherent to mass-spectrometric detection, while retaining the biological specificity that is typical for commonly used plate reader assays.
The use of MS has a number of consequences when MS is used as analytical tool for readout of bioassays. A positive implication is the fact that no labels are required in MS. This widens the application area for these methods, including target proteins for which no label is available or can be developed. A second advantage is that the mass spectrometer measures the actual compounds that have affinity for the protein, instead of a labeled competitor. Since protein selection methods provide simultaneous biological and structural data on the compound(s) to be analyzed, the detection of false positives due to library impurities or degradation of compounds in a bioactive mixture is very improbable. A common drawback of using MS is that bioassay conditions (e.g., buffer used, pH, blocking reagents, detergents) have to be adjusted to create MS compatibility. Therefore, usually a compromise is made, resulting in less favorable MS and bioassay conditions.
In this review, a comprehensive overview of recent developments in the field of protein affinity selection methods that utilize MS is provided. Methods such as frontal affinity chromatography (FAC) [1], size-exclusion chromatography (SEC) [2], (pulsed) ultrafiltration [3], immobilized or dynamic protein affinity selection [4], and surface plasmon resonance (SPR) coupled with MS [5] can be named in this regard. The scope is limited to methods that involve a step that separates bound and unbound protein before detection of bioactives (ligands). All these methods consist of the same four steps: (1) complexation of the protein and test ligand, (2) separation of nonbound compounds from the protein–ligand mixture, (3) elution/dissociation, resulting in the release of the free ligand, and (4) detection of the eluted ligand by MS.
Affinity chromatography
Affinity chromatography is a process in which a protein (e.g., an antibody), small molecule, or other bioactive agent is immobilized on a solid support. When a mixture is injected onto the column containing this solid support, analytes that show affinity are retained. The method originates from the late 1960s, when it was used to extract and purify enzymes [6] or antibodies [7]. The method was based upon solid supports consisting of mainly sepharose or agarose. These materials were known for their low nonspecific adsorptive properties, and their relative ease of modification, allowing easy immobilization of the desired target. However, when the method was developed toward affinity chromatography with the aim of not only extracting and purifying the analytes, but also ranking them according to their affinity, it became a necessity to use support materials that were better equipped for the flow rates and capillary pressures that are common in high-performance liquid chromatography (HPLC). The use of pressure-resistant solid supports is often referred to as the defining advantage of high-performance affinity chromatography over conventional affinity chromatography, and was a starting point for rapid expansion of the number of applications reported in this field [8]. Two main approaches are in current use [9]. Firstly, FAC, in which a known concentration of analyte is infused onto an immobilized protein column, and the affinity is calculated on the basis of the saturation time and the shape of the breakthrough curve. The second is zonal elution, in which the analyte is injected onto the column together with a competitor. By varying the concentration of the competitor and measuring the retention time of the analyte, one can calculate its affinity.
Frontal affinity chromatography
The concept of FAC is more complicated than it appears at first sight. The target, often a receptor, is (covalently) immobilized on a column. An analyte is then infused at a known concentration, and the concentration of the analyte exiting the column is measured. At the beginning this amount will be very low, because there is a large amount of free receptor for the analyte to bind to. However, as a larger fraction of the immobilized receptor is bound, the amount of analyte being measured will slowly increase. At some point, the column will be saturated and the signal in the detector will equal that of the infused analyte concentration. The speed at which this happens is dependent on the association (k on) and dissociation (k off) constants of the receptor–analyte interaction, which can be calculated by varying the concentration of the analyte. A schematic representation of the chromatographic process and the analytical results is given in Fig. 1.

Frontal affinity chromatography (FAC) 1 a compound or mixture is continuously infused onto an affinity column (2). The column (backbone) material that is immobilized with target protein, such as a receptor, retains compounds with affinity. 3 detection of breakthrough of ligands with mass spectrometry (MS). 4a–7a snapshots of the chromatographic breakthrough process of the ligands at different time points during a FAC analysis. 4b–7b corresponding detection of breakthrough events with MS per ligand. At full breakthrough, the infused ligand concentration equals the eluted concentration
When developing a FAC method, the handbook of affinity chromatography by Hage [8] and the paper on practical protocol development by Ng et al. [10] should provide all the theoretical and practical background required. Current developments focus on solid support improvement to facilitate protein immobilization, reduce nonspecific binding, and stabilize certain classes of target proteins, and the miniaturization of the protein affinity column. Prime examples are the use of immobilized artificial membranes (IAMs) and monolithic affinity columns.
A significant development in the field of solid supports is the use of IAMs to form immobilized receptor columns. Ever since the introduction of FAC in conventional HPLC systems, target proteins have been immobilized mainly on silica, glass, or polystyrene, often resulting in the loss of a major part of their activity. To widen the applicability of FAC, a solid support contributing to protein stability was required. IAMs consisting of silica beads covered with membrane lipids to be used as a stationary phase were developed by Pidgeon and Venkataram [11] in 1989. In recent years Beigi et al. [12, 13] have shown the immobilization of G-protein-coupled receptors (GPCRs), whereas Temporini et al. [14] have immobilized the orphan receptor GPR17 on an IAM. Later, this IAM was applied by Calleri et al. [15] for characterization of ligands. For the study of multidrug transporters, IAMs were applied to the determination of known ligands, for which the enantioselectivity and IC50 values could be determined [16].
Moaddel and Wainer [17] provided a comprehensive guide on IAM development and preparation, allowing fast implementation by groups with little experience in the field. In the case of ligand-gated ion channels, one study describes use of immobilized nicotinic acetylcholine receptors (nAChRs) on IAMs [18]. Similar techniques have been used that allow the efficient screening of bioactive mixtures, in some cases allowing hit ranking [19–21]. A good review describes the potential of FAC-MS for screening mixtures against immobilized target proteins and illustrates this with nice examples of pharmaceutical interest [22]. Xuan et al. [23] looked at the interactions of the drug carbamazepine with α1-acid glycoprotein to study relevant drug–plasma protein binding. By changing the temperature, the authors could elucidate that enthalpy was the main driving force in the binding interactions. Similar work studied human serum albumin (HSA) for plasma protein–drug binding [24], where equilibrium constants could be estimated for several drugs tested. For other (sulfonylurea) drugs, similar studies were performed recently in frontal and zonal modes to obtain information regarding protein–drug binding characteristics [25]. The use of affinity microcolumns for the rapid analysis of warfarin and l-tryptophan for binding of the drug to HSA yielded data of a kind similar to that in the previous two conventional bore column examples [26].
Columns in microfluidic chips or in nano liquid chromatography (LC) mode have very low protein consumption owing to their dimensions. Because these small columns are hard to pack with conventional bead-type materials, they have been a prominent target for the development of monolithic affinity columns. A very elegant method was reported by Sharma et al. [19], who immobilized membrane-bound nAChRs from Torpedo californica in a monolithic column from diglycerylsilane polymers. By adding the right amount of poly(ethylene glycol), they wholly incorporated the membrane and the active receptor in a silica monolith. Okanda and El Rassi [27] reported a monolithic silica column with incorporated lectins for affinity ranking in both nano-LC and capillary electrophoresis (CE).
Zonal affinity chromatography
Zonal elution is a straightforward method of performing affinity chromatography. Basically it is a classic competition experiment, implemented in a HPLC system. A known amount of analyte (or a mixture) is injected onto the affinity column, in the addition of a competitor. A schematic representation of the chromatographic process and the analytical results is given in Fig. 2. As depicted in the figure, relatively low affinity compounds are eluted as they are already eluted without the infusion of a competitor. When a series of experiments is performed using a differing concentration of competitor, the retention time of the analyte will vary. Higher concentrations of competitor result in lower analyte retention times since the competitor also binds to the affinity column. An advantage of this method is the low consumption of analyte. However, developments in affinity chromatography are focused on frontal analysis as it provides more information per experiment. Zonal elution is nowadays more often used as a means to acquire additional data on an interaction observed in frontal analysis, such as the location of a binding site [28]. One of the recently published methods based solely on zonal elution was developed by Bertucci et al. [29], who investigated the dynamics of the binding of HIV protease inhibitors to HSA. In another study using a special kind of column-switching approach, zonal affinity chromatography was coupled via heart cutting and used to screen extracts of Coptidis rhizoma for β2-adrenoceptor affinity [30].

Zonal affinity chromatography (ZAC) 1 a compound or mixture is injected onto an affinity column (2). The column (backbone) material that is immobilized with target protein, such as a receptor, retains compounds with affinity. 3 detection of eluted ligands with MS as peaks in the MS trace. 4a–7a snapshots of eluted ligands at different time points during a ZAC analysis. 4b–7b corresponding detection of ligand elution events with MS per ligand. When high-affinity ligands are to be analyzed, continuous infusion of a competitor is necessary to prevent extremely long retention times
In conclusion, FAC is a prominent technique in pharmaceutical research because of the high data density and variety of targets that can be immobilized, such as GPCRs. Owing to the fast development of frontal analysis, zonal elution is used more for measurement of additional binding information. When looking at frontal analysis, one must address three drawbacks. First, extended controls must be performed to validate the functionality of the protein since immobilization influences the freedom of movement and accessibility of binding sites. Second, immobilized protein columns have a limited stability and memory effects from high-affinity ligands or reactive compounds might compromise column performance. Third, it can be challenging to produce functionally equal columns.
Affinity selection—binding to immobilized targets
Affinity selection methods, also known as affinity capture or affinity trapping methods, are similar to affinity chromatography in the sense that their mechanism of action depends on possible binding of analytes to immobilized proteins on a solid support. However, in affinity selection no chromatographic separation of ligands is involved. The conditions in which the binding occurs are optimized to stabilize the protein–analyte complex, wash off nonbinders, and then dissociate the complex to detect the binding analytes. To identify binders, most of these methods employ mass-spectrometric detection. Because the compounds are not separated on the basis of their affinity for the receptor, the resulting analytes cannot be ranked, but can only be grouped in binders and nonbinders. Optimizing the threshold to distinguish these two groups, to eliminate false negatives and limit false positives, is one of the most challenging aspects of affinity selection development.
Although affinity selection in one form or another has been around for decades [31–33], it became more prominent in bioanalysis upon the introduction of the lectin-based affinity materials, used to extract bacteria and carbohydrates with a specific binding motif for matrix-assisted laser desorption ionization (MALDI) time-of-flight (TOF) analysis [34, 35]. These methods allowed detection of target proteins that are difficult to analyze in any conventional way owing to the complex matrices they are in. Since that time, a number of specific affinity materials have been developed with considerable success. Krugman et al. [36] presented their work in which a novel family of proteins found in leukocytes bound specifically to phosphoinositide affinity material. Kong at al. [37] described a diamond-based affinity material with the property of extracting proteins from complex mixtures with MALDI-TOF analysis. Recently, Ferrance [4] has shown the feasibility of immobilizing proteins on gellan (a polysaccharide) beads. The optical transparency of these beads allows direct measurement of the amount of protein successfully immobilized on the material.
Immobilized metal affinity chromatography (IMAC) comprises a large number of methods similar to protein affinity chromatography, but with the difference that it separates proteins, small organic ligands, or phosphopeptides on the basis of their affinity. Instead of protein–ligand affinity, metal–protein (or metal–ligand) affinity is determined. For proteins, the affinity mainly depends on the number and location of histidine residues in the proteins to be analyzed, and to a lesser extent on cysteine and tryptophan residues [38]. When a protein is His-tagged, a very high affinity between the His-tag and the immobilized metal ion efficiently traps any His-tagged protein. Jonker et al. [39] utilized this principle by trapping an in-solution-formed His-tagged protein–ligand complex on a small nickel-loaded column. This allows ligand–protein complex formation in solution, using the immobilized metal column merely to separate the bound and unbound fractions, followed by release of the ligands from the trapped proteins for mass-spectrometric detection. Figure 3 shows a schematic view of the analytical setup. Another variant of affinity capture methods is ligand fishing using magnetic particles. For this so-called magnetic bead dynamic protein affinity selection, IMAC coated magnetic beads are used to trap a His-tagged protein–ligand complex from a sample for further processing and final MS [40]. Figure 4 shows a schematic representation of the ligand trapping process. In other examples, Marsza et al. [41] employed magnetic beads coated with heat shock protein for ligand fishing, whereas Hu et al. [42] reported an enzyme inhibition screening assay utilizing enzymes immobilized on magnetic beads.

Dynamic protein affinity chromatographic system. The upper-left part comprises an online Ni2+ stripping pump with EDTA and a Ni2+ regeneration pump, both connected via a switch valve. The Ni2+-iminodiacetic acid (Ni-IDA) trapping column is able to retain His-tagged proteins. The lower-left part consists of a switch valve for selecting wash, equilibration, and disruption (of ligands bound to immobilized target protein) procedures via the liquid chromatography (LC) pumps connected. The solid-phase extraction (SPE) column traps disrupted ligands from the Ni-IDA column prior to gradient LC-MS

The dynamic trapping process of magnetic beads in high-performance LC [poly(ether ether ketone)] tubing. 1 injected plug of a bioactive compound mixture incubated with target protein and (para)magnetic beads flows through the LC tubing and arrives at the external magnet placed near the tubing. 2 beads with immobilized target protein and ligands (bound to the target protein) are retained in the magnetic field. Nonbinders are eluted to waste. 3 An eluent switch disrupts the target protein–ligand complex and allows ligands to be eluted. A post-magnetic field switch valve directs ligands to SPE for further analysis. 4 automated repositioning of the magnet away from the LC tubing allows the beads to go to waste (after switching the switch valve again)
In conclusion, the advantage of affinity selection methods is the wide applicability, the easy compensation that can be made for nonspecific binding, and their relatively high sensitivity. The major drawback is the fact that affinity ranking is not possible using these methods. The analytical results of these methods will be a group of binders and a group of nonbinders. The biggest challenge is to eliminate false negatives, and limit false positives as much as possible.
Affinity selection—binding to targets in solution
Separation by ultrafiltration
Traditional ultrafiltration
In the early 1980s, ultrafiltration was developed as a means to measure protein–ligand interactions in solution-based complexes [43, 44]. The concept of the technique is rather straightforward. A certain amount of pressure is applied to an amount of liquid containing protein–ligand complexes as well as free ligand and protein. This can be achieved by applying pressure using a pump, vacuum, or centrifugal force. The ultrafiltration unit contains a molecular weight cutoff membrane, allowing solvents and nonbound small molecules to pass through, but retaining proteins and protein–ligand complexes. This achieves effective separation of the bound and nonbound fractions of the analyte, allowing detection of the bound ligand by any available detection method. The huge advantage of this method over other existing methods is that complex formation takes place in solution, allowing the protein the same degree of freedom it would have in vivo.
A number of groups have developed methods using continuous ultrafiltration [45, 46]. In this technique, a fixed amount of protein is injected into an ultrafiltration chamber and the analyte is then pumped through the chamber. Fung et al. [47] were among the first to automate ultrafiltration for high-throughput analysis. Some other recent publications include the work of Comess et al. [48], who developed a high-throughput serial ultrafiltration method to screen a library of compounds for affinity for a pharmacologically relevant streptococcal enzyme. Finally, Li et al. [49] developed an online-coupled ultrafiltration–LC-MS method that was used to screen natural extracts for α-glucosidase inhibitors.
Some very recent examples include the quantification of unbound prednisolone, prednisone, cortisol, and cortisone in human plasma by ultrafiltration–LC-MS [50] and the development of an ultrafiltration–LC-MS-based ligand-binding assay for Mycobacterium tuberculosis shikimate kinase [51], to be used for the development of antimicrobial agents. For screening of potential lead compounds in malaria drug discovery, work was performed for the drug targets Plasmodium falciparum thioredoxin and glutathione reductases [52]. Another study used ultrafiltration-based affinity selection MS for the kinase Chk1 involved in DNA damage [53]. A slightly different example employed hollow-fiber membranes coupled online to a MS system for continuous affinity selection [54].
Pulsed ultrafiltration
The development of pulsed ultrafiltration by van Breemen et al. [55] significantly increased the applicability of ultrafiltration. Instead of filtration of an entire sample, a small amount of sample was injected into the ultrafiltration unit. The liquid flow pushes the nonbound fraction through the molecular weight cutoff membrane, to waste. Afterwards, the bound ligand is dissociated from the protein by buffer adjustment. The formerly bound fraction is then pushed through the membrane, and immediately measured and identified by MS. Figure 5 illustrates this process. The two main advantages over traditional ultrafiltration are that (1) less analyte is needed and (2) target proteins can be reused if a nondestructive dissociation buffer is applied. The method could be applied to complex mixtures as well as combinatorial libraries, and it functioned online, was automated, and was suitable for high-throughput screening. Since the publication of this method, most ultrafiltration applications have used some or all of the characteristics of pulsed ultrafiltration, very often without identifying them as such. As a result, there no longer exists a strict division between traditional and pulsed ultrafiltration. The technology and terminology involved are very often used incorrectly. However, a significant number of interesting advancements has been reported since. Some successful applications of pulsed ultrafiltration include its use by the group of van Breemen to study metabolic stability [56], inhibitors of protein aggregation [57], and ligands for human retinoid X receptor α [58]. In the field of ultrafiltration, most developments are focused on new applications, and not on improving the technology. The latest development was also by the group of van Breemen and involved the discovery of cyclooxygenase inhibitors from medicinal plants used to treat inflammation [59].

Pulsed ultrafiltration chromatography. 1 mixture with ligands and nonbinders injected into an ultrafiltration chamber. Ligands (blue) bind to target protein (2) that is retained in the upper part of the chamber (3) separated from the bottom part (4) by a membrane that is not permeable for large proteins. Small-molecule nonbinders can diffuse through the membrane and be eluted to the waste (W) via continuously infused (MS-compatible) buffer (5). When the mixture is injected and during the washing of nonbinders, a buffer is infused that allows target protein–ligand binding (6). During the ligand-disruption step for ligand elution to MS (7), the bottom part of the chamber is switched from the waste to MS and a disruption buffer is infused via 6 into the upper chamber
When reviewing ultrafiltration, it is apparent that two advantages of ultrafiltration-based affinity selection methods are (1) in-solution complex formation and (2) the possibility to reuse the target protein.
Separation by size-exclusion chromatography
The concept of separating compounds by size was developed in the 1960s in the form of gel permeation chromatography, mainly applied to the analysis of high molecular weight polymers [60]. The concept is as simple as it is elegant. A cross-linked dextran gel is used as the column material. The porosity of the gel determines its separation properties, resulting in less retention for larger molecules, and more retention for smaller molecules. This is caused by diffusion of the smaller molecules into pores in the polymeric gel. Because small molecules can enter these pores and the larger ones (proteins) can not, their retention time is longer. With the development of more advanced gels and other size-exclusion materials, the separation power of size-exclusion-based methods has steadily increased since their inception. Size-exclusion methods are easy to automate and very suitable for implementation in screening assays. While the columns used can be placed online or used off-line in high-throughput fashion (e.g., adapted to 96-well plates), other variants, such as spin-column-based methods, have also been used [61].
A direct successor to classic gel permeation chromatography is the SpeedScreen technology [2]. It consists of a double 96-well plate format. The upper part contains a size-exclusion gel, and has holes in the bottom of the plate. An in-solution protein–ligand incubation mixture is pipetted on top of the gel, and this is placed upon the second plate, a collection plate. The incubate is separated owing to centrifugal force, and bound and unbound ligands are separated. The 96-well plate is then analyzed using a LC-MS system. The system screens one 96-well plate within 10 min, has routinely been applied by Novartis, and has resulted in a number of lead compounds [62].
On the basis of the same concept, size-exclusion columns are being used in almost all branches of protein analysis. A much larger variety of materials is available in SEC than in gel permeation chromatography (which traditionally uses Sephadex gels), and the reusability of these materials allows the use of such columns in online LC methods. In 2004, Neogenesis developed an automated high-throughput-screening assay using online SEC coupled with MS for the assessment of protein–ligand interactions [63]. This method, named automated ligand identification system (ALIS), uses a standardized SEC-LC-MS setup, but contains an in-house-developed resin as the size-exclusion material. The fast affinity screening results included the discovery of a lipid phosphatase inhibitor involved in type 2 diabetes [64]. Characterization of orthosteric and allosteric ligands for the muscarinic M-2 acetylcholine receptor was also feasible with the system [65]. Furthermore, Whitehurst and Annis [66] described affinity selection methods and their emerging role for screening GPCR ligands.
Following the success of ALIS, a number of groups developed innovative applications for size-exclusion affinity measurements. Flarakos et al. [67] developed a method able to assess and rank ligand binding to HSA based on automated SEC coupled online to a two-dimensional LC-MS system. Binding of ligands to Akt-1 and Zap-70 kinases, the muscarinic acetylcholine receptor M2 [68], or the histamine H2 receptor [69] followed by SEC-based purification of the receptor–ligands and disruption for release of ligands to MS was performed by Annis et al. [68] and Derks et al. [69], respectively.
Separation by filtration or centrifugation
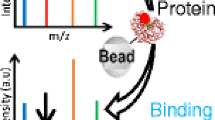
A method similar to that used in the work by Annis et al. [68] and Derks et al. [69] used a native marker ligand for, e.g., the dopamine D2 receptor, thereby allowing the analysis of ligand binding to GPCRs after a rapid solid-phase extraction MS step [70, 71]. A followup technology used MALDI as an ionization source. This adaptation allowed a large throughput increase owing to rapid analysis of the eventually spotted 96-sample MALDI plates [72]. Slightly different approaches use a rapid filtration step (in 96-filtration well plates) instead of SEC to obtain the same assay principle. Here, a native marker ligand is measured with MS after the filtration step [73]. A typical workflow of this setup is depicted in Fig. 6. The technology is actually more based on traditional radioligand binding assays in which the bound marker ligand is now not analyzed by the radioactivity of the marker, but by its molecular mass. The advantages here are the universal applicability (for most receptors the native ligand is available) and elimination of the need for a radiolabeled ligand. A slight variation on this theme is the use of short reversed-phase C18 columns using 8–9-s retention times to speed up LC-MS quantification in MS binding assays [74].

The principle of MS binding assays (e.g., SpeedScreen). Ligands, target protein, and tracer/target ligand in a 96-well plate (1) are aspirated (2) and subsequently dispensed onto a 96-well filtration plate (3). After washing of the filtration plate (4) to remove nonbinders, methanol is added (5) to allow disruption and elution of binders (6) to another 96-well plate (7) for measurement by LC-MS (8). Quantification of the tracer/target ligand by triple-quadrupole MS allows determination of the percentage of tracer/target ligand displacement by unknown ligand binders
Concluding, SEC coupled with MS is a very powerful method with wide applicability, easy automation, and proven results in both fundamental research and screening applications. Its disadvantages are nonspecific binding to the size-exclusion material, which can result in false positives or negatives, and that reusability of the columns is significantly reduced when more complex protein matrices are researched.
Affinity capillary electrophoresis
CE has been used for the separation of proteins and peptides for several decades; however, in the 1990s several groups developed methods to assess protein–ligand interactions [75], peptide–antibiotic binding [76], or protein–DNA binding [77] using capillary zone electrophoresis. Since then, the field of affinity CE (ACE) has gradually expanded and increased its applicability [78, 79]. Two major technological breakthroughs have contributed to the development of modern-day ACE: coupling with MS and the use of microfluidic chip technology. When focusing on protein–drug interactions, one needs to separate ACE methods into two categories: mobility shift methods and immobilized protein methods.
Mobility shift methods
In mobility shift methods, the protein and ligand form a noncovalent complex in solution. For compounds with a very slow k off, a small amount of this incubation mixture is injected into a standard CE-MS system, and the constituents of the mixture are separated on the basis of the fact that the noncovalent protein–ligand complex has an electrophoretic mobility distinctly different from that of the nonbound protein and ligand. The concentration of the bound and nonbound fractions can be quantified using MS, and protein–ligand affinity can be calculated from these results. An interesting example was reported by Groesll et al. [80] coupling CE with inductively coupled plasma MS. This enabled the researchers to assess binding between gallium(III)-based anticancer drugs and serum proteins.
In the case of protein–ligand complexes with a fast k off, the complex will dissociate quickly upon injection into the CE-MS system. In these cases, either the ligand or the protein will be added to the electrophoresis buffer, thus changing the mobility of its counterpart in the capillary. Many varieties of mobility shift methods have been developed and a frequently used variant is frontal analysis CE (FACE), distinctly different from FAC in the fact that the protein is not immobilized on a solid support, but is incubated with a ligand before injection. Recently, Fermas et al. [81] developed a method aimed at the study of complexation between antithrombin and heparin pentasaccharide. They combined a continuous infusion of a preincubated protein–ligand complex solution with mass-spectrometric detection. Since the development of FACE, it has become apparent it can be used to determine protein–ligand affinity for a great variety of targets and libraries. A small number of examples published in 2009 and 2010 are the interaction between lidocaine and hyaluronic acid [82], the interaction between a large number of drugs and the human liposome [83], and affinity studies performed on a large test set of drugs and HSA [84]. Many similar drug–protein binding studies have been performed, all providing precise and sensitive binding data [85]. In some cases, this technology allows albumin to act as a chiral selector owing to its many drug–protein binding characteristics [86]. Other recent examples allow the receptor affinity analysis of bioactive inflammatory cytokines present in skin biopsies in a chip-based CE system [87].
Immobilized protein methods
Immobilized protein methods involve any method in which the target protein is immobilized on a solid support, be it silica, (magnetic) beads, microfluidic chips, or anything else. When a sample containing one or more possible ligands to the target protein is injected into the system, the ligand(s) will bind to the immobilized target protein, thus facilitating sample cleanup, preconcentration, or performing a buffer change before detection. In the past couple of years, these methods have increased in relevance owing to the relative ease of immobilizing protein on microfluidic chips and magnetic beads. Yang et al. [88] reported an immunoaffinity CE method incorporating an anti-α-fetoprotein column inside a microfluidic chip allowing automated detection of early stage cancer diagnostic α-fetoprotein. Xiao et al. [89] have provided a proof-of-principle using a microfluidic chip with immobilized strands of DNA on it, able to selectively trap insulin and insulin-like growth factor 2, with subsequent MALDI-TOF-MS detection. The recent move toward the use of magnetic beads in affinity measurements combined with CE can also lead to interesting results and allows in-capillary affinity preconcentrations [90, 91]. When assessing the merits of ACE, it is apparent the field has evolved to a point where it can compete with other affinity methods described in this article. It is a relatively simple and straightforward method to implement and use routinely. However, ACE has hurdles to overcome such as nonspecific adsorption, Joule heating, and the implementation of mass-spectrometric detection.
Surface plasmon resonance–mass spectrometry
SPR differs fundamentally from all other methods presented in this review, because protein binding and detection take place at the same location. A SPR biosensor consists of a prism positioned against a thin metal layer (often gold) and a solution at the opposite side of the metal layer. Upon directing a beam of light onto the surface under different angles, surface plasmons occur at the interface between the metal and the solution, which implies that photons excite electrons in the metal film. At a specific angle of incidence, the wave vector of these excited electrons is equal to that of the surface plasmons, resulting in a total energy transfer and the light beam is no longer reflected. This is the principle of SPR and when it happens, the light beam is no longer reflected. SPR is used as a means of detection as it is dependent on the analytes present in the solution near the surface. When a ligand binds to a protein immobilized on the metal sensor, this changes the wave vector of the surface plasmons, and the angle of incidence at which SPR occurs, which is detected. However, as the technology only allows analysis of binding events and no identification occurs, SPR is nowadays often coupled to a mass spectrometer to identify the binders.
In the field of small-molecule research, significantly fewer methods have been reported, presumably owing to the dependency of the SPR signal on molecular weight and as a result, its improved performance for analytes above 15 kDa. However, recently Jecklin et al. [92] developed a SPR method for seven analytes under 500 Da. Utilizing human carbonanhydrase immobilized on a SPR chip, they managed to calculate accurate dissociation constants for the test set, and compared them with those from two reference methods. Also, Marchesini et al. [93] successfully measured the binding and dissociation kinetics of a test set of seven low molecular weight paralytic shellfish poisons binding to immobilized saxitoxin monoclonal antibody. Furthermore, recent publications by Wang et al. [94] and Miura et al. [95] show the development of indirect SPR affinity methods, in which the affinity of a small molecule is determined by its competition with a large ligand already bound to the immobilized target protein. Concluding, SPR is most efficient for compounds with a high molecular weight, but developments that target small molecules are upcoming.
Conclusion
Reviewing the wide spectrum of methods discussed in this paper for protein–ligand affinity interactions, we can separate all of them into two general categories. The first group consists of methods that immobilize the target protein on a solid support, whereas the second group of these methods allow in-solution complexation. Ultrafiltration and size exclusion are solution-based methods. The complex is formed in solution, approaching in vitro conditions as closely as possible. The same applies for dynamic protein affinity selection and a number of CE methods. All other techniques, frontal affinity, zonal elution, affinity capture, and most CE-based methods, use immobilized proteins. When assessing the progress made using immobilized proteins, we can draw a number of conclusions. FAC will remain relevant, because it is a cheap and easy-to-implement method to assess protein–ligand binding. The immobilized target proteins are becoming more and more complex in terms of their instability and the environment needed to maintain their activity, and none of the in-solution-based methods rival the data resulting from FAC methods.
The solution-based methods might not provide the same amount of affinity data, but they do not suffer from impaired binding properties due to protein immobilization, and each of them has advantages of its own. Ultrafiltration benefits from the reusability of the receptor, and the speed of measurement. CE affinity selection can target proteins unsuitable for any of the other methods, and will stay important despite issues with nonspecific absorption of the proteins to the capillary walls and MS compatibility. The size-exclusion methods are the ones most commonly applied in modern drug discovery for two simple reasons. SEC is very easy to implement and automate using standard 96-well plates and equipment, and it is significantly faster than all the other techniques discussed. SPR-MS is limited by its inability to measure small ligands conveniently, but advances in life sciences allow more and more target proteins to be used to study them for small-molecule binding interactions. Furthermore, a limited number of target proteins have been successfully immobilized on the SPR chip, but this number is steadily increasing. Finally, care must be taken that SPR coupled with MS is not used just as an expensive affinity selection device since the main strength of SPR is analysis of binding kinetics, which is only possible when studying single known compounds, whereas the strength of (LC-)MS is the analysis of unknown compounds in mixtures.
Abbreviations
- ACE:
-
affinity capillary electrophoresis
- ALIS:
-
automated ligand identification system
- CE:
-
capillary electrophoresis
- FAC:
-
frontal affinity chromatography
- FACE:
-
frontal analysis capillary electrophoresis
- GPCR:
-
G-protein-coupled receptor
- HPLC:
-
high-performance liquid chromatography
- HSA:
-
human serum albumin
- IAM:
-
immobilized artificial membrane
- IMAC:
-
immobilized metal affinity chromatography
- LC:
-
liquid chromatography
- MS:
-
mass spectrometry
- MALDI:
-
matrix-assisted laser desorption ionization
- nAChR:
-
nicotinic acetylcholine receptor
- SEC:
-
size-exclusion chromatography
- SPR:
-
surface plasmon resonance
- TOF:
-
time of flight
References
Ng W, Dai JR, Slon-Usakiewicz JJ, Redden PR, Pasternak A, Reid N (2007) J Biomol Screen 12(2):167–174
Muckenschnabel I, Falchetto R, Mayr LM, Filipuzzi I (2004) Anal Biochem 324(2):241–249
Sun Y, Gu C, Liu X, Liang W, Yao P, Bolton JL, van Breemen RB (2005) J Am Soc Mass Spectrom 16(2):271–279
Ferrance JP (2007) J Chromatogr A 1165(1–2):86–92
Nedelkov D (2010) Methods Mol Biol 627:261–268
Cuatreca P, Wilchek M, Anfinsen CB (1968) Proc Natl Acad Sci USA 61(2):636–643
Wofsy L, Burr B (1969) J Immunol 103(2):380–382
Hage DS (2006) Handbook of affinity chromatography, 2nd edn. Taylor and Francis, New York
Hage DS (2002) J Chromatogr B Anal Technol Biomed Life Sci 768(1):3–30
Ng ESM, Chan NWC, Lewis DF, Hindsgaul O, Schriemer DC (2007) Nat Protoc 2(8):1907–1917
Pidgeon C, Venkataram UV (1989) Anal Biochem 176(1):36–47
Beigi F, Wainer IW (2003) Anal Chem 75(17):4480–4485
Beigi F, Chakir K, Xiao RP, Wainer IW (2004) Anal Chem 76(24):7187–7193
Temporini C, Ceruti S, Calleri E, Ferrario S, Moaddel R, Abbracchio MP, Massolini G (2009) Anal Biochem 384(1):123–129
Calleri E, Ceruti S, Cristalli G, Martini C, Temporini C, Parravicini C, Volpini R, Daniele S, Caccialanza G, Lecca D, Lambertucci C, Trincavelli ML, Marucci G, Wainer IW, Ranghino G, Fantucci P, Abbracchio MP, Massolini G (2010) J Med Chem 53(9):3489–3501
Bhatia PA, Moaddel R, Wainer IW (2010) Talanta 81(4–5):1477–1481
Moaddel R, Wainer IW (2009) Nat Protoc 4(2):197–205
Zhang Y, Xiao Y, Kellar KJ, Wainer IW (1998) Anal Biochem 264(1):22–25
Sharma J, Besanger TR, Brennan JD (2008) Anal Chem 80(9):3213–3220
Jozwiak K, Haginaka J, Moaddel R, Wainer IW (2002) Anal Chem 74(18):4618–4624
Jozwiak K, Moaddel R, Yamaguchi R, Ravichandran S, Collins JR, Wainer IW (2005) J Chromatogr B Anal Technol Biomed Life Sci 819(1):169–174
Calleri E, Temporini C, Caccialanza G, Massolini G (2009) ChemMedChem 4(6):905–916
Xuan H, Joseph KS, Wa C, Hage DS (2010) J Sep Sci 33(15):2294–2301
Mallik R, Yoo MJ, Briscoe CJ, Hage DS (2010) J Chromatogr A 1217(17):2796–2803
Joseph KS, Hage DS (2010) J Chromatogr B Anal Technol Biomed Life Sci 878(19):1590–1598
Yoo MJ, Schiel JE, Hage DS (2010) J Chromatogr B Anal Technol Biomed Life Sci 878(20):1707–1713
Okanda FM, El Rassi Z (2006) Electrophoresis 27(5-6):1020–1030
Zhaoa XF, Zheng XH, Wei YM, Bian LJ, Wang SX, Zheng JB, Zhang YY, Li ZJ, Zang WJ (2009) J Chromatogr B Anal Technol Biomed Life Sci 877(10):911–918
Bertucci C, Pistolozzi M, Felix G, Danielson UH (2009) J Sep Sci 32(10):1625–1631
Zhao X, Nan Y, Xiao C, Zheng J, Zheng X, Wei Y, Zhang Y (2010) J Chromatogr B Anal Technol Biomed Life Sci 878(22):2029–2034
Huyer G, Kelly J, Moffat J, Zamboni R, Jia ZC, Gresser MJ, Ramachandran C (1998) Anal Biochem 258(1):19–30
Manalan AS, Klee CB (1987) Biochemistry 26(5):1382–1390
Kataoka K (1991) ACS Symp Ser 464:159–174
Bundy J, Fenselau C (1999) Anal Chem 71(7):1460–1463
Kaji H, Saito H, Yamauchi Y, Shinkawa T, Taoka M, Hirabayashi J, Kasai K, Takahashi N, Isobe T (2003) Nat Biotechnol 21(6):667–672
Krugmann S, Anderson KE, Ridley SH, Risso N, McGregor A, Coadwell J, Davidson K, Eguinoa A, Ellson CD, Lipp P, Manifava M, Ktistakis N, Painter G, Thuring JW, Cooper MA, Lim ZY, Holmes AB, Dove SK, Michell RH, Grewal A, Nazarian A, Erdjument-Bromage H, Tempst P, Stephens LR, Hawkins PT (2002) Mol Cell 9(1):95–108
Kong XL, Huang LCL, Hsu CM, Chen WH, Han CC, Chang HC (2005) Anal Chem 77(1):259–265
Gutierrez R, del Valle EMM, Galan MA (2007) Sep Purif Rev 36(1):71–111
Jonker N, Kool J, Krabbe JG, Retra K, Lingeman H, Irth H (2008) J Chromatogr A 1205(1–2):71–77
Jonker N, Kretschmer A, Kool J, Fernandez A, Kloos D, Krabbe JG, Lingeman H, Irth H (2009) Anal Chem 81(11):4263–4270
Marsza PM, Moaddel R, Kole S, Gandhari M, Bernier M, Wainer IW (2008) Anal Chem 80(19):7571–7575
Hu F, Deng C, Zhang X (2008) J Chromatogr B 871(1):67–71
Whitlam JB, Brown KF (1981) J Pharm Sci 70(2):146–150
Zhirkov YA, Piotrovskii VK (1984) J Pharm Pharmacol 36(12):844–845
Zlotos G, Oehlmann M, Nickel P, Holzgrabe U (1998) J Pharm Biomed Anal 18(4–5):847–858
Kinawi A, Teller C (1979) Arzneimittelforschung/Drug Res 29–2(10):1495–1500
Fung EN, Chen YH, Lau YY (2003) J Chromatogr B Anal Technol Biomed Life Sci 795(2):187–194
Comess KM, Schurdak ME, Voorbach MJ, Coen M, Trumbull JD, Yang H, Gao L, Tang H, Cheng X, Lerner CG, McCall O, Burns DJ, Beutel BA (2006) J Biomol Screen 11(7):743–754
Li HL, Song FR, Xing JP, Tsao R, Liu ZQ, Liu SY (2009) J Am Soc Mass Spectrom 20(8):1496–1503
Ionita IA, Akhlaghi F (2010) Ann Clin Biochem 47(4):350–357
Mulabagal V, Calderon AI (2010) Anal Chem 82(9):3616–3621
Mulabagal V, Calderon AI (2010) J Chromatogr B Anal Technol Biomed Life Sci 878(13–14):987–993
Comess KM, Trumbull JD, Park C, Chen ZH, Judge RA, Voorbach MJ, Coen M, Gao L, Tang H, Kovar P, Cheng XH, Schurdak ME, Zhang HY, Sowin T, Burns DJ (2006) J Biomol Screen 1(7):755–764
Jiang Y, Lee CS (2001) J Mass Spectrom 36(6):664–669
van Breemen RB, Huang CR, Nikolic D, Woodbury CP, Zhao YZ, Venton DL (1997) Anal Chem 69(11):2159–2164
Geun Shin Y, Bolton JL, van Breemen RB (2002) Comb Chem High Throughput Screen 5(1):59–64
Cheng X, van Breemen RB (2005) Anal Chem 7(21):7012–7015
Liu DT, Guo J, Luo Y, Broderick DJ, Schimerlik MI, Pezzuto JM, van Breemen RB (2007) Anal Chem 79(24):9398–9402
Cao H, Yu R, Choi Y, Ma ZZ, Zhang H, Xiang W, Lee DY, Berman BM, Moudgil KD, Fong HH, van Breemen RB (2010) Pharmacol Res 61(6):519–524
Moore JC (1964) J Polym Sci Part A Gen Pap 2(2PA):835–843
Siegel MM, Tabei K, Bebernitz GA, Baum EZ (1998) J Mass Spectrom 33(3):264–273
Brown N, Zehender H, Azzaoui K, Schuffenhauer A, Mayr LM, Jacoby E (2006) J Biomol Screen 11(2):123–130
Annis DA, Athanasopoulos J, Curran PJ, Felsch JS, Kalghatgi K, Lee WH, Nash HM, Orminati JPA, Rosner KE, Shipps GW, Thaddupathy GRA, Tyler AN, Vilenchik L, Wagner CR, Wintner EA (2004) Int J Mass Spectrom 238(2):77–83
Annis DA, Cheng CC, Chuang CC, McCarter JD, Nash HM, Nazef N, Rowe T, Kurzeja RJ, Shipps GW Jr (2009) Comb Chem High Throughput Screen 12(8):760–771
Whitehurst CE, Nazef N, Annis DA, Hou YM, Murphy DM, Spacciapoli P, Yao ZP, Ziebell MR, Cheng CC, Shipps GW, Flesch JS, Lau D, Nash HM (2006) J Biomol Screen 11(2):194–207
Whitehurst CE, Annis DA (2008) Comb Chem High Throughput Screen 11(6):427–438
Flarakos J, Morand KL, Vouros P (2005) Anal Chem 77(5):1345–1353
Annis DA, Nazef N, Chuang CC, Scott MP, Nash HM (2004) J Am Chem Soc 126(47):15495–15503
Derks RJE, Letzel T, de Jong CF, van Marie A, Lingeman H, Leurs R, Irth H (2006) Chromatographia 64(7–8):379–385
Hofner G, Wanner KT (2003) Angew Chem Int Ed 42(42):5235–5237
Niessen KV, Hofner G, Wanner KT (2005) Chembiochem 6(10):1769–1775
Hofner G, Merkel D, Wanner KT (2009) ChemMedChem 4(9):1523–1528
Zepperitz C, Hofner G, Wanner KT (2006) ChemMedChem 1(2):208–217
Hofner G, Wanner KT (2010) J Chromatogr B Anal Technol Biomed Life Sci 878(17–18):1356–1364
Kraak JC, Busch S, Poppe H (1992) 608(1-2):257-264
Dunayevskiy YM, Lyubarskaya YV, Chu YH, Vouros P, Karger BL (1998) J Med Chem 41(7):1201–1204
Baba Y, Tsuhako M, Sawa T, Akashi M, Yashima E (1992) Anal Chem 64(17):1920–1925
Chen Z, Weber SG (2008) Trends Anal Chem 27(9):738–748
Liu XJ, Dahdouh F, Salgado M, Gomez FA (2009) J Pharm Sci 98(2):394–410
Groessl M, Bytzek A, Hartinger CG (2009) Electrophoresis 30(15):2720–2727
Fermas S, Gonnet F, Varenne A, Gareil P, Daniel R (2007) Anal Chem 79(13):4987–4993
Mrestani Y, Hammitzch M, Neubert RHH (2009) Chromatographia 69(11–12):1321–1324
Franzen U, Jorgensen L, Larsen C, Heegaard NHH, Ostergaard J (2009) Electrophoresis 30(15):2711–2719
Liu XM, Li SY, Zhang JS, Chen XG (2009) J Chrom B Anal Technolog Biomed Life Sci 877(27):3144–3150
El-Hady D, Kuhne S, El-Maali N, Watzig H (2010) J Pharm Biomed Anal 52(2):232–241
Ye H, Yu L, Xu X, Zheng C, Lin W, Liu X, Chen G (2010) Electrophoresis 31(12):2049–2054
Phillips TM, Kalish H, Wellner E (2009) Electrophoresis 30(22):3947–3954
Yang WC, Sun XH, Wang HY, Woolley AT (2009) Anal Chem 81(19):8230–8235
Xiao JF, Carter JA, Frederick KA, McGown LB (2009) J Sep Sci 32(10):1654–1664
Adachi K, Yamaguchi M, Nakashige M, Kanagawa T, Torimura M, Tsuneda S, Sekiguchi Y, Noda N (2009) J Biosci Bioeng 107(6):662–667
Liu XJ, Gomez FA (2009) Electrophoresis 30(7):1194–1197
Jecklin MC, Schauer S, Dumelin CE, Zenobi R (2009) J Mol Recognit 22(4):319–329
Marchesini GR, Hooijerink H, Haasnoot W, Buijs J, Campbell K, Elliott CT, Nielen MWF (2009) Trends Anal Chem 28(6):792–803
Wang J, Munir A, Zhou HS (2009) Talanta 79(1):72–76
Miura N, Shankaran DR, Gobi KV, Kawaguchi T, Kim SJ (2008) Sens Lett 6(6):891–902
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
Published in the special issue on Advances in Analytical Mass Spectrometry with Guest Editor Maria Careri.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Jonker, N., Kool, J., Irth, H. et al. Recent developments in protein–ligand affinity mass spectrometry. Anal Bioanal Chem 399, 2669–2681 (2011). https://doi.org/10.1007/s00216-010-4350-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-010-4350-z