
Abstract
Glutamine synthetase is a key enzyme which has a regulatory role in the brain glutamate pool. According to previously published proteomic analysis, it was shown that the expression level of this enzyme is affected by morphine administration. In our study, we examined the activity of glutamine synthetase in various structures of rat brain (cortex, striatum, hippocampus and spinal cord) that are biochemically and functionally involved in drug addiction and antinociception caused by morphine. We were not able to observe any significant changes in the enzyme activity between morphine-treated and control samples despite previously reported changes in the expression levels of this enzyme. These findings stressed the fact that changes observed in the expression of particular proteins during proteomic studies may not be correlated with its activity.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
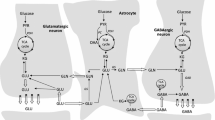
Glutamine synthetase is a key enzyme which has a regulatory role in the brain glutamate pool. Dysfunctions of this enzyme and perturbations in the glutamate–glutamine cycle seem to be involved in several neurological disorders. There is also a growing number of evidence that gutamatergic transmission is engaged in drug addiction [1].
After reviewing previously published morphinome studies (proteome in morphine dependence), we found that glutamine synthetase is one of the proteins whose concentration in the brain is connected with addiction processes. Morón et al. [2] found a downregulation of glutamine synthetase in hippocampal postsynaptic density (PSD) fraction after morphine administration. Upregulation of this enzyme was indicated by Prokai et al. [3] during proteomic studies of cerebral cortex synaptosomes. Kim et al. [4] discovered phosphorylation of its tyrosine residues. Additionally, we found publication considering changes in the level of glutamine synthetase in the spinal cord of morphine-tolerant mice [5] examined by Western blotting. Additional information about the mentioned studies is collected in Table 1.

Glutamine synthetase activity (μM min −1 mg−1) in different structures from the CNS. Striatum C: 19.5 ± 4.1, n = 6; M: 21.7 ± 2.6, n = 6, p = 0.293. Hippocampus C: 27.8 ± 3.5, n = 9; M: 26.8 ± 3.8, n = 8, p = 0.607. Cortex C: 30.8 ± 7.0, n = 9; M: 31.2 ± 4.8, n = 9, p = 0.902. Spinal cord C: 10.9 ± 1.8, n = 8; M: 11.2 ± 1.1, n = 9, p = 0.710
The fact that changes in glutamine synthetase expression were linked to morphine administration by three different methods (isotope-coded affinity tags, (ICAT), 2D gel electrophoresis and Western blot) in four distinct laboratories seems very appealing: It indicates that quantitative changes occur in this enzyme during morphine uptake. Discrepancies between studies and the fact that in our study [6, 7] we did not observe any changes in the expression of this enzyme also encouraged us to examine the problem. Proteomic strategy allows the determination of quantitative changes in protein expression, i.e. the relative amount of a given protein is measured. In the case of enzymes, such as glutamine synthetase, the most important property is their activity, not the amount of protein. Therefore, in our study, we decided to examine the activity of glutamine synthetase in various structures from rat CNS that are involved in drug addiction (cortex, striatum, hippocampus) and antinociception (spinal cord) caused by morphine. In the cell, glutamine synthetase catalyzes the following reaction:
If we replaced NH +4 with NH2OH, as a result, we obtained L-γ-glutamyl-hydroxamate.
This compound in the presence of FeCl3 gives a characteristic colour reaction, which may be measured colorimetically and enables measuring the enzyme activity under certain circumstances [8].
Experimental
Animals and treatment
Twenty-five male Wistar rats, weighing between 150 and 200 g, were addicted to morphine via subcutaneous implantation of pellets containing 75 mg of morphine base. Twenty control animals received pellets with 0.9% NaCl instead of morphine. Pellets were implanted under light ether anaesthesia in the animals’ neck. Animals were housed in groups of five per cage, under 12:12-h light/dark cycle. Water and standard food were freely available. Acute morphine withdrawal, emerging after the administration of an opiate antagonist, is considered to be the physical manifestation of dependence [9]. Therefore, the progress of addiction was tested with the injection of naloxone. A dose of 10 nmol naloxone was administrated to five morphine-treated rats 72 h after pellet implantation (total volume injected was 5 μl per rat, naloxone concentration 2 nmol/μl). Since they show immediate signs of withdrawal, like wet dog shakes, grooming, washing, etc., they confirmed that the rest of animals receiving the drug were morphine-dependent. A detailed description of the procedure may be found here [10]. At the same time, animals destined for experiments were decapitated.
Ethical requirements
All experiments were performed in agreement with the respective Polish and European Communities Council Directives (86/609/EEC) and were approved by the local ethics committee (permission no. 367/2002).
Tissue excision
Animals were killed by decapitation 72 h after pellet implantation. Brains were removed from ten control and ten morphine-dependent rats, and cerebral cortices, striata and hippocampi were isolated. Due to the prolonged isolation of brain structures, spinal cords were removed from another set of ten control and ten morphine-dependent animals. Tissues were immediately placed separately in Eppendorf tubes, frozen on dry ice and stored at −80°C prior to analysis. Structure isolation was not always successful; therefore, in the final experiments, less than ten replicates were usually used.
Glutamine synthetase assay
The activity of glutamine synthetase in different structures from the CNS was measured according to Dennis et al. [11] with minor modifications [12]. Prior to the final analyses, the tissue samples were carried out on ice to prevent protein degradation. Before the experiment, all structures were weighed. Homogenates of 10% (tissue weight/volume) were prepared by tissue disruption in water for about 30 s each using a rotor-stator homogenizer (Pro200, PRO Scientific Inc., Oxford, CT, USA). Homogenates were centrifuged and the supernatant collected. The protein concentration in supernatant was measured by the Bradford method [13] using Sigma Aldrich kit. Finally, samples at a volume of 0.5 ml homogenate at a protein concentration of 1 mg/ml were prepared. Each sample was mixed with 0.5 ml of the reaction buffer. Final concentrations of the components in the assay were as follows: 44 mM Tris–HCl, 44 mM MgCl2⋅6H2O, 20 mM mercaptoethanol, 1 mM DTT, 10 mM ATP, 50 mM glutamate, 0.16% Triton X-100, freshly prepared 100 mM hydroxylamine, 5 μg/ml oligomycin, 1 mM ouabain and an ADP trap consisting of 10 mM phosphocreatine and 30 U creatinephosphokinase.
The samples were incubated in water bath at 37°C for 30 min. In the case of the spinal cord, the incubation lasted for 60 min. At the end of the reaction, 0.5 ml of ferric chloride stop solution was added to each sample. The samples were centrifuged (15,000×g, 2 min., ambient temperature) and the amount of the complex of L-γ-glutamylhydroxamate and iron(III) chloride was measured spectrophotometrically at 510 nm (Spectro2000, Labomed Inc., Los Angeles, CA, USA). The volume of disposable cuvettes was 0.5 ml.
Synthetic L-γ-glutamylhydroxamate water solutions (0.5 ml), at desired concentrations, mixed with 0.5 ml reaction buffer and 0.5 ml of ferric chloride stop solution, were used for the construction of calibration curves. Different tissue concentrations and different times of incubations were used to measure the linearity of the assay along the timescale and with various protein concentrations in homogenates.
Results
The activity of glutamine synthetase was measured in striatum, cortex, hippocampus and spinal cord of the morphine-dependent and control rats.
To estimate the background of the measurements, we introduced “blank” samples. Blanks were the samples that were incubated without homogenates. In such samples, there was no significant absorbance at 510 nm. This ensures that during further experiments, only changes in absorbance caused by the enzyme activity were observed, and they were not caused by any kind of reaction between the components of the reaction buffer. Under our assay conditions, the enzyme activity increased linearly with time and with protein concentration (data not shown).
Enzyme activity was expressed as micromolars of L-γ-glutamylhydroxamate produced in time (minutes) with protein concentration of 1 mg/ml. The mean values for each group of the samples with their standard deviations are given in Fig. 1. We were not able to observe any significant changes in the enzyme activity between morphine-dependent and control samples from any of the structures examined.
Therefore, we decided to check whether changing the sample concentration and incubation time influenced the results. Five control and five morphine-treated samples from hippocampus, at a protein concentration of 2 mg/ml, were taken and incubated for 45 min. In this case, we did not observe any significant differences either (data not shown).
Conclusions
In this paper, we have proven that despite the fact that several previous proteome/morphinome studies have indicated changes in glutamine synthetase expression or quantity in cells/tissues/organisms, we did not observe any significant changes in enzyme activity during our experiments. In the case of proteomic studies, we have to be aware of the fact that changes in protein expressions may not be of biological importance. For example, elevated levels of the enzyme may not be linked with its increased activity. In fact, the enzyme may be present in the sample in active as well as in inactive forms, which cannot be distinguished using typical, proteomic tools. Moreover, statistically significant changes in protein quantity are not always connected to changes in an organism’s actual state. Thus, proteomics may indicate proteins whose expression levels were affected by morphine administration, but their role in the examined occurrence have to be validated and finally confirmed by other (biochemical and behavioural) experiments.
Abbreviations
- GS:
-
Glutamine synthetase
- ICAT:
-
Isotope-coded affinity tags
- LC MS/MS:
-
Liquid chromatography mass spectrometry
- MALDI-TOF MS:
-
Matrix-assisted laser desorption ionisation time-of-flight mass spectrometry
- PSD:
-
Postsynaptic density
References
Gass JT, Olive MF (2008) Glutamatergic substrates of drug addiction and alcoholism. Biochem Pharmacol 75:218–265
Morón JA, Abul-Husn NS, Rozenfeld R, Dolios G, Wang R, Devi LA (2007) Morphine administration alters the profile of hippocampal postsynaptic density-associated proteins: a proteomics study focusing on endocytic proteins. Mol Cell Proteomics 6:29–42
Prokai L, Zharikova AD, Stevens SM Jr (2005) Effect of chronic morphine exposure on the synaptic plasma-membrane subproteome of rats: a quantitative protein profiling study based on isotope-coded affinity tags and liquid chromatography/mass spectrometry. J Mass Spectrom 40:169–175
Kim S, Chudapongse N, Lee S, Levin MC, Oh J, Park H, Ho IK (2005) Proteomic analysis of phosphotyrosyl proteins in morphine-dependent rat brains. Brain Res Mol Brain Res 133:58–70
Wu G, Wen Z, Chen W, Chang Y, Cherng C, Wong C (2007) The effect of dexamethasone on spinal glutamine synthetase and glutamate dehydrogenase expression in morphine-tolerant rats. Anesth Analg 104:726–730
Bodzon-Kulakowska A, Suder P, Mak P, Bierczynska-Krzysik A, Lubec G, Walczak B, Kotlinska J, Silberring J (2009) Proteomic analysis of striatal neuronal cell cultures after morphine administration. J Sep Sci 32(8):1200–1210
Suder P, Bodzon-Kulakowska A, Mak P, Bierczynska-Krzysik A, Daszykowski M, Walczak B, Lubec G, Kotlinska JH, Silberring J (2009) The proteomic analysis of primary cortical astrocyte cell culture after morphine administration. J Proteome Res 8(10):4633–4640
Minet R, Villie F, Marcollet M, Meynial-Denis D, Cynober L (1997) Measurement of glutamine synthetase activity in rat muscle by a colorimetric assay. Clin Chim Acta 268:121–132
Broseta I, Rodríguez-Arias M, Stinus L, Miñarro J (2002) Ethological analysis of morphine withdrawal with different dependence programs in male mice. Prog Neuropsychopharmacol Biol Psychiatry 26:335–347
Dylag T, Pachuta A, Raoof H, Kotlinska J, Silberring J (2008) A novel cryptic peptide derived from the rat neuropeptide FF precursor reverses antinociception and conditioned place preference induced by morphine. Peptides 29:473–478
Dennis SC, Lai JC, Clark JB (1980) The distribution of glutamine synthetase in subcellular fractions of rat brain. Brain Res 197:469–475
Steffens M, Huppertz H, Zentner J, Chauzit E, Feuerstein TJ (2005) Unchanged glutamine synthetase activity and increased nmda receptor density in epileptic human neocortex: implications for the pathophysiology of epilepsy. Neurochem Int 47:379–384
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254
Acknowledgements
This work was supported by grant no. 3048/B/H03/2009/37 from the Ministry of Science and Higher Education.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Bodzon-Kulakowska, A., Suder, P., Drabik, A. et al. Constant activity of glutamine synthetase after morphine administration versus proteomic results. Anal Bioanal Chem 398, 2939–2942 (2010). https://doi.org/10.1007/s00216-010-4244-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-010-4244-0