
Abstract
Tumours, which are initially sensitive to cytotoxic agents, often develop resistance to a broad spectrum of structurally unrelated drugs. The 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitors have been shown to inhibit ATP-binding cassette (ABC) transporters but have also impact on glycosylation of such proteins. Doxorubicin is a substrate for ABC transporters like P-glycoprotein (ABCB1) which is present in human RD rhabdomyosarcoma cells. It was therefore the aim of this study to identify the compartmentalisation and action of doxorubicin in simvastatin-treated RD cells. Due to autofluorescence of doxorubicin, intracellular distribution was monitored by confocal microscopy. The biological effects were traced on the level of colony formation, caspase activation and DNA injury. Here we show that simvastatin treatment leads to ABCB1 inhibition and down-regulation of the transporter. Consequently, these cells accumulate significant amounts of doxorubicin, predominantly in the nucleus and lysosomes. While clearance of the anthracycline into lysosomes is not altered by simvastatin treatment, it significantly enhanced nuclear accumulation in a HMG-CoA reductase-independent manner. Thus, in such treated cells, topoisomerase II activity is significantly inhibited, which is further corroborated by augmented double-strand DNA breaks. Moreover, colony formation was synergistically inhibited by the combination of simvastatin and doxorubicin. Given the fact that ABCB1 expression correlates with an adverse prognosis in many tumours, adjuvant chemotherapy including statins might represent a novel therapeutic concept to overcome ABCB1-mediated multidrug resistance by direct inhibition and down-regulation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Rhabdomyosarcoma is the most common soft tissue sarcoma of childhood, accounting for more than half of such cases in paediatric patients (Raney et al. 1993). Initial treatment is often successful, but relapses due to multidrug resistance (MDR) remain a major obstacle to survival. Thus, the overall outcome in paediatric rhabdomyosarcoma is therefore still a therapeutic challenge. In part, MDR may be caused by changes in the expression level or activity of ATP-binding cassette (ABC) transporters (Endicott and Ling 1989; Liscovitch and Lavie 2002; Szakacs et al. 2006).
Ways of reversing MDR via ABC transporters have been investigated extensively in recent years, and three generations of inhibitors have been developed (Liscovitch and Lavie 2002; Szakacs et al. 2006). Among these, verapamil, cyclosporine A and tariquidar have emerged as lead compounds for characterisation in vitro. Nevertheless, due to severe side effects, such as nephrotoxicity for cyclosporin A or cardiac toxicity for verapamil, these MDR modulators failed to convince in clinical trials (Cocker et al. 2000; Szakacs et al. 2006). Although there are many new MDR-modulating agents available for in vitro usage, no effective and clinically applicable therapy has yet evolved (Liscovitch and Lavie 2002; Szakacs et al. 2006; Tamaki et al. 2011).
The 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitors, commonly referred to as statins, are widely used in hypercholesterolaemia and prevent cardiovascular events (Lennernas and Fager 1997; Corsini et al. 1999; Group 2000). In recent years, evidence emerged that anti-inflammatory and anti-proliferative actions of statins also contribute to beneficial cardiovascular outcomes. These effects have led to a series of in vitro studies which came to the conclusion that statins also exert anti-tumour activity which has been targeted to cell cycle arrest, depletion of cholesterol precursors like isoprenoids and oxidative stress (Demierre et al. 2005; Sleijfer et al. 2005). However, in clinical studies, the role of statins in oncology is still controversial and seems to be strongly dependent on the molecular identity of the tumour (Graaf et al. 2004; Poynter et al. 2005; Nielsen et al. 2012).
Statins are capable to inhibit the rhodamine 123 efflux from tumour cells via their lactone form but not in the hydrolysed acidic form (Bogman et al. 2001). Such inhibition of efflux activity could be attributed to ABCB1 interaction with statins (Wang et al. 2001; Sakaeda et al. 2002; Chen et al. 2005). In a previous work, we made similar observations and could show that glycosylation of ABCB1 is reduced in simvastatin-treated human neuroblastoma cells (Sieczkowski et al. 2010). The mutual enhancement of cytotoxicity between statins and doxorubicin has been shown in neuroblastoma cells and RD rhabdomyosarcoma cells to trigger the mitochondrial apoptotic machinery (Werner et al. 2004; Sieczkowski et al. 2010). Since statins are capable to trigger caspase activity per se, it is difficult to estimate the apoptotic potential of doxorubicin in drug combinations. Moreover, it is unclear whether enhanced intracellular doxorubicin accumulation is targeted to individual organelles and whether this translates into specific doxorubicin-mediated cytotoxicity. However, one would postulate that enhanced intracellular doxorubicin concentrations might translate into augmented topoisomerase II inhibition.
Thus, it was the aim of the present work to confirm synergism between simvastatin and doxorubicin. Moreover, intracellular compartmentalisation of doxorubicin and functional implications thereof were investigated.
Materials and methods
Materials
Dulbecco’s modified Eagle medium (DMEM) was purchased from Gibco® (Invitrogen®, Paisley, Scotland, UK). Doxorubicin, daunorubicin, rhodamine 123, protease inhibitors (pefablock, leupeptin and aprotinin), soft agar and other common chemicals were from Sigma Chemical Co.® (St. Louis, MO, USA). Simvastatin and verapamil were purchased from Merck® Research Laboratories (Rahway, NJ, USA). Concanamycin A and fluorescent substrates for caspases 3 and 8 were from Alexis Biochemicals® (San Diego, CA, USA). Lysotracker® Red was obtained from Molecular Probes® (Eugene, OR, USA).
Cell culture
Human rhabdomyosarcoma cells (RD cells) of the spindle cell type were obtained from ATCC® (Manassas, VA, USA) and kept in growth medium (DMEM, 10 % foetal calf serum, 50 U/ml penicillin G and 50 μg/ml streptomycin) at 37 °C under 5 % CO2 for 2–5 days prior to experiments. The CCRFvcr1000 cells were cultured as previously described (Chiba et al. 1996; Hiessbock et al. 1999) and used to confirm simvastatin as inhibitor of ABCB1.
Caspase assays
RD cells were incubated with simvastatin, doxorubicin and/or mevalonic acid for the indicated times and concentrations. Thereafter, the cells were lysed (lysis buffer—25 mM HEPES (pH 7.4), 5 mM EDTA, 1 mM EGTA, 5 mM MgCl2, 5 mM DTT, supplemented with protease inhibitors 1.4 μg/ml aprotinin, 10 μg/ml leupeptin and 100 μM pefablock) and centrifuged (35,000 × g, 30 min), and aliquots of the supernatant were used for a fluorescence-based caspase assay (reaction buffer—25 mM HEPES pH 7.4, 10 % sucrose, 1.4 mM CHAPS and 5 mM DTT and 50 μM AFC-conjugated caspase substrates) (Werner et al. 2004). The corresponding pellet is termed “membrane fraction”, resuspended in radio immunoprecipitation assay (RIPA) buffer and further used for Western blotting.
Topoisomerase II decatenation assay and histone H2AX phosphorylation
RD cells (5 × 107 cells) were treated with drugs according to the figure legends for 24 h. The cells were lysed and processed for nuclear extraction according to Dansk et al. (1988). Nuclear extracts (20 μg) were assayed for topoisomerase II activity (30 min at 37 °C) using a topoisomerase II decatenation kit (Inspiralis®, Norwich, UK). The reaction was terminated with 2.5 % SDS and the DNA separated on a 1 % agarose gel. Densiometric analysis was carried out using the ImageJ® software (http://rsbweb.nih.gov/ij/index.html).
Alternatively, the phosphorylation of histone H2AX was used as a sensitive measure for double-strand DNA breaks. A flow cytometry based histone H2AX phosphorylation kit was used (Millipore®, Temecula, CA, USA). RD cells were treated as depicted in the figure legend and further processed and stained with an FITC-conjugated antibody specific for serine139 phosphorylation of histone H2AX according to the instructions of the manual. The positive stained cells were quantified by flow cytometry. The fluorescence signal in the absence of the antibody was used as a negative control and RD cells treated with 10 μM etoposide as a positive control.
Western blot analysis
Aliquots of membrane fractions (15 μg) from RD cells resuspended in RIPA buffer were applied to an 8 % sodium dodecyl sulphate polyacrylamide gel. The separated proteins were transferred to nitrocellulose membranes (Watman®, Dassel, Germany) and exposed to an ABCB1-specific primary antibody (C219 mouse monoclonal P-gp antibody, Abcam®, Cambridge, UK; diluted at 1:300) for 90 min at room temperature. Specific bands were visualised with an appropriate horse radish peroxidase-conjugated secondary antibody (anti-mouse IgG, horseradish peroxidase-linked antibody, GE Healthcare®, Bucks, UK; diluted 1:15,000, 60 min at room temperature) using an ECL Plus detection system (GE Healthcare®, Bucks, UK). Immunodetection of α-tubulin (monoclonal anti-α-tubulin; Sigma®; St. Louis, USA; dilution of 1:7,500) was used to control protein loading.
Colony forming assay
Trypsinised RD cells were seeded in 6- and 12-well plates at a density of 500 cells per well, precoated with 0.5 % soft agar. After 24 h, cells were incubated in the absence and presence of doxorubicin, simvastatin or a combination of both at the given concentrations. After 15 days, the cells were fixed with 50 % ethanol and stained with 0.5 % methylene blue. Cell growth was evaluated by counting colonies with more than 50 cells.
Fluorescence-activated cell sorting (FACS) analysis
RD cells were treated with simvastatin or verapamil for 10 min and subsequently with doxorubicin for further 60 min. Cells were analysed for doxorubicin accumulation on a FACScan (Beckton Dickinson®, New York, USA) with an excitation of 488 nm and an emission filter of 600 nm.
For histone H2AX phosphorylation, RD cells were exposed to the indicated compounds for 18 h and treated according to the kit instructions. The FITC-conjugated antibody against histone H2AX phosphorylation was detected with an excitation at 488 nm and an emission at 515 nm. A total of 104 cells were counted per sample and the data were processed using WinMDI software® (http://www.cyto.purdue.edu/flowcyt/software/Winmdi.htm) or FACSDiva® (BD Biosciences®, San Jose, CA, USA).
Confocal fluorescence microscopy
RD cells were grown on glass cover slides and incubated for 18 h in the absence or presence of simvastatin, verapamil or concanamycin A for 1 h. Such treated cells were subsequently exposed to 1 μM doxorubicin for an additional hour. Cells were washed with PBS and supplied with fresh culture medium. Images were collected using a confocal microscope (Zeiss® LSM 410; Jena, Germany), an excitation of 488 nm and an emission of 543 nm for LysoTracker® Red (Molecular Probes®, Leiden, Netherlands) or 570 nm for doxorubicin. The digitised pictures from a single focal plane containing two to six cells were stored. The doxorubicin intensity was measured in the nucleus (55–69 cells) and lysosomes (91–296 cells) using MetaMorph® software (Universal Imaging Corporation, Downington, PA, USA) and ImageJ® software (http://rsweb.nuh.gov/ij).
For the doxorubicin co-localisation in the lysosomes, cells were pre-incubated in the culture medium with 10 μM simvastatin and 15 μM verapamil for 1 h and another hour together with 1 μM doxorubicin. Pictures of doxorubicin autofluorescence were obtained as described above. Subsequently, 20 nM LysoTracker® Red was added and the fluorescence signal collected.
The kinetic analysis of nuclear accumulation of doxorubicin was carried out under conditions described above with minor changes. RD cells were incubated for 20 min in the absence and presence of simvastatin or the combination of simvastatin plus mevalonic acid. Immediately after the doxorubicin addition, a series of pictures was taken for the indicated period with the settings described above.
Daunorubicin, rhodamine 123 and cacein flux measurements
CCRFvcr1000 cells were pre-treated with various concentrations of simvastatin or 15 μM verapamil for 20 min. Daunorubicin and rhodamine 123 were used as transporter substrates as previously reported (Chiba et al. 1996; Hiessbock et al. 1999). The IC50 values for simvastatin were determined in zero trans efflux and steady-state protocols. In the steady-state protocol, apparent IC50 values were corrected for differences in ABCB1 expression considering a pump-leak situation.
For calcein efflux measurements, RD cells were pre-treated with various concentrations of simvastatin or 15 μM verapamil for 20 min or 48 h. Cells were gently detached and 5 × 105 cells were resuspended in HEPES/Na-Puffer (20 mM HEPES, 132 mM NaCl, 3.5 mM KCl, 1 mM CaCl2, 0.5 mM MgCl2, 5 mM glucose) supplemented with the respective drugs from the 20-min pre-incubation. No drugs were supplemented to cell suspensions from the 48-h pre-treatment. The experiment was started with 1 μM calcein-AM uptake and reaching a plateau. After 30 min, the extracellular calcein was quenched by 1 mM of CoCl2. The fluorescence was monitored at an excitation wavelength at 485 nm and an emission at 535 nm using a fluorescence plate reader (VICTOR-3, Perkin-Elmer®, MA, USA) with sampling rates of 10 and 30 s or 3 min, depending on the phase of the experiment (calcein efflux, calcein-AM uptake or plateau phase, respectively).
Miscellaneous procedures
Experiments were carried out at least three times in duplicates and presented as mean ± SD, if not otherwise stated. The curves were subjected to non-linear least squares regression and fitted to the according equations (Hill equation) using the Sigmaplot® software (Jandl, Erkrath, Germany). Statistical analysis was carried out by Student’s t test or for multiple comparisons by ANOVA and post hoc Dunnett’s test (when compared to control) or Tukey’s test (for pairwise comparison). A value of p < 0.05 was considered statistically significant.
Results
Simvastatin and doxorubicin synergistically activate the mitochondrial pathway of apoptosis
In human RD cells, HMG-CoA reductase inhibitors like simvastatin readily activate the effector caspase 3 in a concentration-dependent manner (Fig. 1a). The EC50 of simvastatin-induced caspase 3 activation shifted by more than one order of magnitude from 9.9 ± 1.2 to 0.69 ± 0.2 μM (mean ± SD, n = 4; p < 0.001) upon co-application with doxorubicin at a clinically relevant concentration (Lee et al. 1980). Under these conditions, caspase 8 was not significantly activated, neither by simvastatin alone nor by a combination with doxorubicin (Fig. 1b). The simvastatin-induced caspase activation is strongly dependent on HMG-CoA reductase inhibition and thus prevented by co-application with mevalonic acid (Werner et al. 2004; Minichsdorfer and Hohenegger 2009; Sieczkowski et al. 2010). In contrast, doxorubicin-induced caspase 3 activation is insensitive to co-administration of mevalonic acid (Fig. 1c). However, a more than additive stimulation of caspase 3 is seen with the co-administration of doxorubicin and simvastatin. This is significantly reduced by mevalonic acid to the level of doxorubicin alone. Thus, the mutual sensitisation for apoptosis is only seen for the intrinsic pathway of apoptosis, which has been previously depicted for statins in more detail (Werner et al. 2004). These results confirm that simvastatin potentiates doxorubicin toxicity in RD cells at concentrations that have been well tolerated in patients receiving simvastatin (Lennernas and Fager 1997; Larner et al. 1998).

Potentiation of caspase 3 activity by simvastatin plus doxorubicin. RD cells were grown in the absence and presence of simvastatin (sim), doxorubicin (dox) and mevalonic acid (ma, 1 mM) at the given concentrations for 24 h. Aliquots of the supernatant of lysed cells were used for caspase 3 (a, c) and caspase 8 activity (b). The data are given as mean values ± SD (n = 4). Statistical significance was determined with ANOVA and post hoc Tukey’s test (asterisk denotes significance versus control *p = 0.039, **p < 0.001; number sign denotes significance versus sim ##p < 0.001; plus sign denotes significance versus sim+dox ++p < 0.001; n.s. not significant)
Synergistic inhibition of colony formation by simvastatin and doxorubicin
HMG-CoA reductase inhibitors have been shown to synchronise tumour cells by blocking the transition of G1–S in the cell cycle, which explains their anti-proliferative effects in many cell types (Rao et al. 1999; Dimitroulakos et al. 2001; Ukomadu and Dutta 2003). We have therefore investigated colony formation of RD cells in the presence of increasing concentrations of simvastatin and doxorubicin, exemplified in Fig. 2a. In fact, RD cells are highly susceptible to inhibition of colony formation with an IC50 of 3.42 ± 0.7 nM doxorubicin (mean ± SD; n = 3) (data not shown). Consistent with our previous observations on caspase 3 and 9 activation, the efficacy to suppress colony formation is more pronounced in the combination of simvastatin and doxorubicin as compared to the individual compounds (Fig. 2a) (Werner et al. 2004). The potency of the individual compounds to prevent colony formation is not influenced by the presence of the second compound as depicted in Fig. 2a (IC50 for simvastatin 3.0 ± 1.3 μM versus simvastatin plus 5 nM doxorubicin 2.8 ± 2.1 μM; mean ± SD, n = 3). The isobologram analysis revealed a clear synergistic interaction for the drug combination of simvastatin with doxorubicin (Fig. 2b).

Synergistic inhibition of colony formation by simvastatin and doxorubicin. Aliquots of 500 RD cells were seeded in 12-well plates, and after 24 h, cells were incubated with the indicated doxorubicin and simvastatin concentrations (a). After 15 days, colonies of more than 50 cells were counted. The data are given as mean ± SEM (n = 3). The fractional inhibitory concentrations (FIC) of various combinations of simvastatin and doxorubicin were blotted in an isobologram (b). The solid line indicates additivity. The experimental data points were calculated from experiments as described in (a) and confirm synergism
Inhibition and down-regulation of ABCB1 by simvastatin
Taking advantage of the fluorescence properties of doxorubicin, a short pre-treatment of RD cells with simvastatin resulted in a significant increase in doxorubicin fluorescence as monitored by FACS analysis (Fig. 3a, b). Similarly, the first generation ABCB1 inhibitor verapamil also enhanced the intracellular accumulation of doxorubicin. In a control experiment without doxorubicin, autofluorescence of the cells in the absence or presence of simvastatin was not observed (data not shown).

Enhanced intracellular doxorubicin content in the presence of simvastatin or verapamil. Doxorubicin accumulation was assessed with flow cytometry in RD cells treated for 1 h with 1 μM doxorubicin (ctl) (a). Preincubation of the cells for 10 min with 3 μM (sim3), 10 μM (sim10) simvastatin or 15 μM verapamil (ver) enhanced the doxorubicin fluorescence intensity. The experiment was repeated twice with similar results. The mean increase in doxorubicin fluorescence under conditions given in a is depicted in the bar diagram (b). The data were processed with WinMDI software and presented as geometrical mean ± SD (n = 3)
There are already reports that statins are capable of inhibiting ABC transporters (Bogman et al. 2001; Wang et al. 2001; Sakaeda et al. 2002; Sieczkowski et al. 2010). We have therefore investigated the effects of simvastatin on ABCB1 in greater detail in CCRFvcr1000 cells, which express a defined number of transporter molecules (Chiba et al. 1996; Hiessbock et al. 1999). Such an approach enables to normalise the transporter activity to the actual number of transporters in the cells. Using this experimental approach, first-order rate constants of daunorubicin and rhodamine 123 efflux show a clear inhibition by simvastatin with IC50 values of 4.6 ± 1.6 and 49.3 ± 4.7 μM (mean ± SD; n = 3), respectively (Fig. 4). The Hill coefficient between 1.1 and 1.2 in both cases is indicative for a non-cooperative interaction. Comparable results were obtained when verapamil was used as a P-glycoprotein inhibitor which gave IC50 values in the same concentration range (Chiba et al. 1996; Hiessbock et al. 1999). In order to further corroborate this assumption, calcein efflux was determined in RD cells (Fig. 5). The calcein efflux is best described by fitting the data points to a biexponential decay. Thus, the two half lives of calcein efflux were significantly prolonged already in the presence of 1 μM simvastatin (Fig. 5b, c).

Simvastatin inhibits ABCB1 in CCRFvcr100 cells. The ABCB1 expressing CCRFvcr100 cells were loaded with daunorubicin (a) or rhodamine 123 (b) and the kinetics of the decrease in fluorescence was determined in the presence of increasing concentrations of simvastatin. The calculated first-order rate constants (V max/K m) were blotted in these concentration–response relationships. The symbols represent the mean ± SD (n = 3)

Simvastatin inhibits ABC transporter-mediated calcein efflux in RD cells. RD cells were incubated in the absence (ctl) or presence of 3 μM simvastatin (sim) 20 min prior to 1 μM calcein-AM exposure (a). The fluorescence (excitation 485 nm, emission 535 nm) was monitored over time until a plateau was reached. Calcein efflux was visualised by quenching extracellular calcein by the addition of 1 mM CoCl2. The symbols and bars represent the mean ± SEM (n = 6). The calcein efflux was fitted to bi-exponential decays and the half lives are depicted from applications with 1 (1 sim) or 10 (10 sim) μM simvastatin or 15 μM verapamil (15 ver) (b, c). The bars and errors represent the mean ± SD (n = 8)
ABCB1 is present in RD cells and, importantly, is down-regulated upon incubation for 48 h with increasing concentrations of simvastatin (Fig. 6a, b). Thus, these observations corroborate and expand earlier observations of our laboratory in human neuroblastoma cells that deglycosylation might impair not only the function of ABCB1 but also protein expression (Sieczkowski et al. 2010). To confirm a functional consequence, such treated RD cells were washed and subjected to calcein efflux measurements, and again, a significant prolongation of the half lives was observed. Hence, a direct inhibition of ABCB1 by simvastatin is in accordance to earlier observations and is now expanded by a significant down-regulation of the transporter (Bogman et al. 2001; Wang et al. 2001; Sakaeda et al. 2002; Sieczkowski et al. 2010).

Down-regulation of ABCB1 by simvastatin. The presence of ABCB1 was confirmed by Western blot analysis in untreated and simvastatin-treated (sim) RD cells, incubated for 48 h (a). Protein loading is controlled with α-tubulin. The intensity of the 140- and 170-kDa ABCB1 protein band was corrected for protein loading and the normalised intensities are depicted as mean ± SD (n = 3) in b. The asterisk denotes statistical significance versus control (*p < 0.05). Similar to the experiments depicted in Fig. 5, calcein efflux was determined in RD cells after 48 h of incubation with 3 and 30 μM simvastatin (sim) versus control (ctl). Simvastatin was removed before calcein-AM loading. The half lives of the first (c) and second calcein release phase (d) were presented as mean ± SD (n = 6)
Mining intracellular doxorubicin accumulation under simvastatin treatment
In order to further trace intracellular doxorubicin accumulation, RD cells were scanned by confocal microscopy. Doxorubicin accumulates in the nuclei, but also in punctate-patterned organelles, which co-localise with lysosomes (Fig. 7a). A lysosomal sequestration of doxorubicin represents a possible clearance mechanism of RD cells. However, no changes were observed in lysosomal doxorubicin fluorescence intensity or lysosomal radius (Fig. 7b, c).

Doxorubicin is sequestered into lysosomes of RD cells. RD cells were incubated with 1 μM doxorubicin (Dox) and simultaneously with 20 nM Lysotracker® Red (Lyso) for 1 h (a). Cells were supplied with fresh medium and pictures were taken with a confocal laser microscope. The intensity of the lysosomal pattern outside the nuclear region was analysed for lysosomal radius and mean lysosomal fluorescence intensity in the absence (ctl) and presence of 10 μM simvastatin (sim) or 15 μM verapamil (ver) (b, c) after a drug exposure of 1 h. The bars represent the mean ± SD (n = 91–296; n.s. not significant)
The nucleus is the primary target organelle for doxorubicin. The anthracycline binds to DNA and inhibits topoisomerase II, which results in blockade of DNA replication and transcription (Larsen et al. 2000). Consequently, one can postulate that cytotoxicity of doxorubicin depends on the concentration in the nucleus. Under control conditions, doxorubicin accumulates readily in the nuclei (Fig. 8). Upon treatment with simvastatin or verapamil for 18 h, the nuclear doxorubicin fluorescence increased significantly (Fig. 8b). As a weak base, doxorubicin is sequestered in acidic compartments such as lysosomes as already shown in Fig. 7a. This sequestration is thought to represent one mechanism of multidrug resistance that may exist separately from the ABC transporters (Schindler et al. 1996). Treatment of the cells with concanamycin A, an inhibitor of the lysosomal proton pump, destroys these organelles and still leads to accumulation and nuclear deposition of doxorubicin (Fig. 7a, b). Importantly, 50 nM concanamycin A is below the concentration range which has been described to interfere with ABCB1 function (0.1–10 μM) (Sharom et al. 1995).

Enhanced nuclear deposition of doxorubicin by simvastatin, verapamil or concanamycin A treatment. RD cells were grown on glass cover slides and incubated for 18 h in the absence (ctl) or presence of 10 μM simvastatin (sim), 15 μM verapamil (ver) or 50 nM concanamycin A (con) for 1 h. Such treated cells were subsequently exposed to 1 μM doxorubicin for an additional hour (a). Representative experiments are depicted and the bar indicates 10 μm. The intensity of the nuclear doxorubicin accumulation is blotted for the conditions described under a (b). The bars represent the mean ± SD (n = 55–69 cells). To resolve the kinetics of the nuclear doxorubicin accumulation, RD cells were preincubated for 20 min in the absence (filled circles) or presence of 10 μM simvastatin (open triangle) or a combination of 10 μM simvastatin plus 1 mM mevalonic acid (filled square) (c). The experiment was started by the addition of doxorubicin (1 μM) and pictures were taken every 2 min. Images were analysed with Metafluor® imaging software. The symbols represent the mean ± SEM (n = 14–38)
To analyse the kinetics of nuclear doxorubicin uptake, time series were taken from doxorubicin-exposed cells using confocal microscopy. Simvastatin immediately accelerated the nuclear compartmentalisations of doxorubicin, which was not prevented by co-incubation with mevalonic acid (Fig. 8c). This is of utmost importance and indicates that an HMG-CoA reductase-independent mechanism is responsible for the nuclear doxorubicin accumulation, which is already observed within 1 h of co-administration.
Enhanced topoisomerase II inhibition and DNA double-strand breaks by simvastatin plus doxorubicin
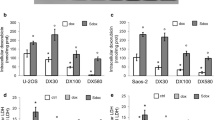
Simvastatin-induced inhibition of ABCB1 results in enhanced nuclear deposition of doxorubicin. Therefore, nuclear extracts from RD cells were probed for decatenation of kinetoplast DNA. This assay is specific for topoisomerase II activity, the target enzyme of anthracyclines. Expectedly, the topoisomerase II activity is inhibited to a greater extent by the drug combination of simvastatin plus doxorubicin (Fig. 9a, b). Complementary, topoisomerase II inhibition may result in less DNA repair and this should facilitate the detection of DNA double-strand breaks. Thus, the increment in histone H2AX phosphorylation on serine139 was used as a sensitive measure for DNA double-strand breaks (Watters et al. 2009). Again, a combination of simvastatin and doxorubicin significantly enhanced DNA injury compared to the individual compounds alone (Fig. 9c, d). Interestingly, simvastatin alone or in a combination with mevalonic acid failed to increase DNA double-strand breaks. This indicates that the induction of apoptosis by simvastatin alone has not reached DNA laddering within the experimental time frame of 18 h. Moreover, these findings corroborate the initial hypotheses that simvastatin-induced nuclear compartmentalisation of doxorubicin significantly enhances efficacy of the anthracycline.

Enhanced topoisomerase II inhibition and DNA double-strand breaks by co-application of simvastatin with doxorubicin. Topoisomerase II activity from nuclear extracts (20 μg) of RD cells is depicted in the absence (ctl) or presence of 0.1 μM doxorubicin (dox), 1 mM mevalonic acid (ma), 1 μM simvastatin (sim) or the combination of 0.1 μM doxorubicin with 1 mM mevalonic acid or 1 μM simvastatin (a). The standards for catenated kinetoplast DNA (kDNA) and decatenated (decat.) kDNA are indicated. Quantification of catenated and mini-circles kDNA is given in b (bars represent mean ± SD, n = 5). Asterisks indicate statistical significance versus control determined with ANOVA and post hoc Dunnett’s test (*p < 0.05; ***p < 0.001). In order to detect DNA double-strand breaks, RD cells were exposed to 0.1 μM doxorubicin (dox), 1 mM mevalonic acid (ma), 1 μM simvastatin (sim), 10 μM etoposide (etop) or the combination of doxorubicin with simvastatin or simvastatin plus mevalonic acid for 18 h. Thereafter, the cells were probed for histone H2AX phosphorylation by FACS analysis and a normalised histogram is shown (c). Quantification of histone H2AX phosphorylation is summarised in d (mean ± SD, n = 3–8). Indicated statistical significance was determined with ANOVA and post hoc Tukey’s test
Discussion
Statins, like simvastatin, are used for the treatment of hypercholesterolaemia and prevention of cardiovascular events (Corsini et al. 1999; Group 2000; Nielsen et al. 2012). They belong to the most widely prescribed drugs and are well tolerated, besides occasionally occurring skeletal muscle side effects, which are now understood as apoptotic events (Corsini et al. 1999; Sacher et al. 2005). In recent years, evidence has accumulated that statins also exert an anti-proliferative activity including cell cycle arrest, inhibition of angiogenesis, stimulation of anti-tumour immunity and impairment of metastatic potential (Sleijfer et al. 2005). In different tumour cell lines, it was shown that statins induce growth inhibition by blocking the transition of G1–S phase in the cell cycle (Lee et al. 1998; Rao et al. 1999; Dimitroulakos et al. 2001; Wachtershauser et al. 2001; Ukomadu and Dutta 2003) and by induction of apoptosis via the mitochondrial pathway in human RD cells (Werner et al. 2004), melanoma cells (Minichsdorfer and Hohenegger 2009), human lymphocytes, myeloma cells (Cafforio et al. 2005) and others (Dimitroulakos et al. 2001; Sieczkowski et al. 2010). However, apoptosis is seen in tumour cells only above 1–3 μM of the individual statins which represent a more than 10-fold higher concentration compared to pharmacological statin treatment (Corsini et al. 1999). This is hampered by co-application of the anthracycline doxorubicin which is capable to shift the concentration–response curve for simvastatin to the left by a factor of 10 (Fig. 1a).
In our study, we show that 100 nM doxorubicin is capable of shifting the EC50 of simvastatin-induced caspase 3 activation from 9.9 to 0.7 μM. This anthracycline concentration is found in the plasma of tumour patients treated with doxorubicin (Giaccone et al. 1997; Swenson et al. 2003). In contrast, the peak plasma concentrations reached in patients after oral simvastatin administration are approximately 0.14 μM (Lennernas and Fager 1997). Thus, induction of apoptosis with an EC50 of 0.7 μM simvastatin is clearly above this value. However, we speculate that lower doses of these drugs may also be effective in prolonged incubations, since the apoptotic effects of statins are time and dose dependent (Werner et al. 2004; Minichsdorfer and Hohenegger 2009). Indeed, low-dose statins given continuously seem to exhibit some anti-tumour activity (Sleijfer et al. 2005; Nielsen et al. 2012), which might explain the protective effect observed in case–control studies (Boudreau et al. 2004; Graaf et al. 2004; Poynter et al. 2005) and in a study performed in hepatocellular carcinoma (Kawata et al. 2001). The latter observation is of particular interest because the concentrations which statins might reach in the liver are significantly higher than those in the serum. The liver–plasma ratio of 10:2 after subcutaneous application of atorvastatin, simvastatin, lovastatin or pravastatin confirms this assumption (Chen et al. 2007). Importantly, liver injury is a rare statin side effect and often linked to concomitant drug administration or pre-existing liver diseases (Corsini et al. 1999).
Although doxorubicin represents a hallmark in many anti-cancer drug regimens, its role in children soft tissue sarcoma has been controversially discussed (Bisogno et al. 2005). However, our in vitro results are indicative of a rational with a potential role of statins as an emerging candidate for combinatory chemotherapy. Synergistic interactions between statins and various cytotoxic drugs were found in vitro and in vivo: (1) simvastatin was found to act synergistically with carmustine in vitro and in vivo (Soma et al. 1995). (2) Lovastatin potentiated the anti-tumour effects of cisplatin in MmB16 melanoma cells (Feleszko et al. 1998), of 5-fluorouracil and cisplatin in colon cancer cells (Agarwal et al. 1999), of doxorubicin in Colon-26 cells, v-Ha-ras-transformed NIH-3T3 sarcoma cells and Lewis lung carcinoma cells in vitro and in vivo (Feleszko et al. 2000). (3) Cerivastatin synergistically enhanced cytotoxicity of 5-fluorouracil in colorectal cancer cells (Wang et al. 2002) and of doxorubicin and cisplatin in human breast cancer cells (Kozar et al. 2004). Interestingly, with the exception of doxorubicin and in part carmustine, these chemotherapeutics are no substrates for ABCB1 (Szakacs et al. 2006). Cisplatin is a substrate for ABCC2, ABCC5 and ABCC6 and the thiopurine 5-fluorouracil for ABCC5 and ABCC11 (for review, see Szakacs et al. 2006). Accordingly, statins may impact also other ABC transporters, which has been confirmed for ABCC1 and ABCC2 (Wang et al. 2001; Sakaeda et al. 2002; Chen et al. 2005). One should have in mind that CYP3A4 substrates in many cases share also substrate specificity for ABCB1 (Gottesman et al. 2002; Szakacs et al. 2006). However, in the clinics, side effects due to interactions via ABC transporters are not described for statins (Corsini et al. 1999; Szakacs et al. 2006; Tamaki et al. 2011), although a polymorphism in the gene of the organic anion-transporting polypeptide (OATP1B1) has been attributable for elevated statin plasma levels and increased risk for myotoxicity (Link et al. 2008). Given the fact that tumour cells replicate faster than normal tissue, one can speculate that statins might have a greater impact on the expression of ABC transporters in tumour cells compared to normal cells.
In RD cells, simvastatin induced nuclear accumulation of doxorubicin, which is not related to inhibition of HMG-CoA reductase (Fig. 8). Thus, one has to postulate a novel drug target for simvastatin. In accordance with previous findings (Bogman et al. 2001; Wang et al. 2001; Sakaeda et al. 2002; Sieczkowski et al. 2010), we confirm that simvastatin is a potent inhibitor of ABCB1. This is corroborated by the following findings: (1) verapamil, a prototypical first generation ABCB1 inhibitor, also led to enhanced nuclear accumulation of doxorubicin. (2) Doxorubicin is a substrate of ABCB1 and confers doxorubicin resistance which is therefore of clinical relevance (Endicott and Ling 1989; Gottesman et al. 2002; Szakacs et al. 2006). Moreover, ABCB1 is expressed in RD cells and calcein efflux is inhibited by simvastatin (Fig. 6a). (3) Simvastatin is also efficient in another cellular system. In CCRFvcr1000 cells which stably express a defined number of ABCB1 transporters, daunorubicin and rhodamine123 fluxes were inhibited by simvastatin. (4) An alternative explanation might be provided by our recent observations on the glycosylation level of ABCB1 in the presence of simvastatin in human neuroblastoma cells (Sieczkowski et al. 2010). Simvastatin reduced the 170-kDa fully glycosylated species and thereby might impair the plasma membrane export of the transporter. A similar down-regulation is observed in RD cells upon simvastatin exposure times up to 48 h (Fig. 6a, b). Western blot analyses confirm a strong decline of the 170-kDa ABCB1 band, but also of the 140-kDa core-glycosylated species.
Interestingly, the lactone forms of statins are more potent to inhibit P-glycoprotein, compared to their corresponding acid forms (Bogman et al. 2001). For comparison, lovastatin in the acid form with the open lactone ring exerts a greater potency in pro-apoptotic action in cancer cell lines (Dimitroulakos et al. 2001). Pravastatin already exists in its acidic form and cannot inhibit ABCB1 (Bogman et al. 2001; Wang et al. 2001). Indirectly, these results imply that HMG-CoA reductase inhibition is not a prerequisite for ABCB1 inhibition. This is supported by our finding that the enhanced nuclear accumulation of doxorubicin is not abrogated by the co-application of a saturating concentration of mevalonic acid (Fig. 8c). Thus, ABCB1 and other ABC transporters like ABCC1 and ABCC2 are confirmed to be new targets for statins (Bogman et al. 2001; Wang et al. 2001; Chen et al. 2005; Sieczkowski et al. 2010).
Besides the efflux mechanism of ABC transporter, cellular drug clearance may also involve sequestration to acidic compartments such as lysosomes, recycling endosomes, the trans-Golgi network or secretory vesicles (Schindler et al. 1996; Larsen et al. 2000). Indeed, a punctuated staining of doxorubicin was detected in RD cells, which was attributed to lysosomes. In contrast to nuclear deposition, simvastatin did not modulate the sequestration of the weak base doxorubicin to this acidic compartment (Figs. 7 and 8). Conversely, destruction of lysosomes by the V-type ATPase inhibitor concanamycin A resulted in a significant elevation of nuclear doxorubicin (Fig. 7b). Remarkably, this finding highlights the importance of the lysosomal drug clearance machinery of tumour cells. Concanamycin A was also found to inhibit ABCB1, although in a concentration range between 0.1 and 10 μM (Sharom et al. 1995). Thus, in our experimental settings, 50 nM concanamycin A is assumed to have no effect on the ABC transporter.
Nevertheless, the combination of simvastatin and doxorubicin shows a significant increment in topoisomerase II inhibition and DNA double-strand breaks (Fig. 9). Importantly, incubation with simvastatin alone had no effect on topoisomerase II activity or the occurrence of DNA double-strand breaks. This is again indicative for a synergistic action of the two drugs and highlights the importance of doxorubicin guidance into the nucleus by simvastatin.
Taken together, these data lead to the conclusion that simvastatin enhances the nuclear deposition of doxorubicin by direct inhibition of ABCB1 and on long term by down-regulation of the transporter. Consequently, this effect translates into further inhibition of topoisomerase II and enhanced DNA double-strand breaks. Interestingly, ABCB1 expression assessed by immunohistochemistry was an adverse prognostic factor in paediatric rhabdomyosarcoma patients (Chan et al. 1990). It remains to be elucidated whether statins can partially overcome chemotherapy resistance in vivo, prevent relapses and improve the overall outcome in rhabdomyosarcoma.
Abbreviations
- ABC transporter:
-
ATP-binding cassette transporters
- ABCB1:
-
ABC transporter B1, P-glycoprotein
- AFC:
-
7-Amino-4-trifluoro-methylcoumarin
- ANOVA:
-
Analysis of variance
- DMEM:
-
Dulbecco’s modified Eagle medium
- FACS:
-
Fluorescence-activated cell sorting
- HMG-CoA reductase:
-
3-Hydroxy-3-methylglutaryl coenzyme A reductase
- MDR:
-
Multidrug resistance
- RIPA buffer:
-
Radio immunoprecipitation assay buffer
References
Agarwal B, Bhendwal S, Halmos B, Moss SF, Ramey WG, Holt PR (1999) Lovastatin augments apoptosis induced by chemotherapeutic agents in colon cancer cells. Clin Cancer Res 5:2223–2229
Bisogno G, Ferrari A, Bergeron C, Scagnellato A, Prete A, Alaggio R, Casanova M, D'Angelo P, Di Cataldo A, Carli M (2005) The IVADo regimen—a pilot study with ifosfamide, vincristine, actinomycin D, and doxorubicin in children with metastatic soft tissue sarcoma: a pilot study of behalf of the European Pediatric Soft Tissue Sarcoma Study Group. Cancer 103:1719–1724
Bogman K, Peyer AK, Torok M, Kusters E, Drewe J (2001) HMG-CoA reductase inhibitors and P-glycoprotein modulation. Br J Pharmacol 132:1183–1192
Boudreau DM, Gardner JS, Malone KE, Heckbert SR, Blough DK, Daling JR (2004) The association between 3-hydroxy-3-methylglutaryl conenzyme A inhibitor use and breast carcinoma risk among postmenopausal women: a case–control study. Cancer 100:2308–2316
Cafforio P, Dammacco F, Gernone A, Silvestris F (2005) Statins activate the mitochondrial pathway of apoptosis in human lymphoblasts and myeloma cells. Carcinogenesis 26:883–891
Chan HS, Thorner PS, Haddad G, Ling V (1990) Immunohistochemical detection of P-glycoprotein: prognostic correlation in soft tissue sarcoma of childhood. J Clin Oncol 8:689–704
Chen C, Mireles RJ, Campbell SD, Lin J, Mills JB, Xu JJ, Smolarek TA (2005) Differential interaction of 3-hydroxy-3-methylglutaryl-coa reductase inhibitors with ABCB1, ABCC2, and OATP1B1. Drug Metab Dispos 33:537–546
Chen C, Lin J, Smolarek T, Tremaine L (2007) P-glycoprotein has differential effects on the disposition of statin acid and lactone forms in mdr1a/b knockout and wild-type mice. Drug Metab Dispos 35:1725–1729
Chiba P, Ecker G, Schmid D, Drach J, Tell B, Goldenberg S, Gekeler V (1996) Structural requirements for activity of propafenone-type modulators in P-glycoprotein-mediated multidrug resistance. Mol Pharmacol 49:1122–1130
Cocker HA, Pinkerton CR, Kelland LR (2000) Characterization and modulation of drug resistance of human paediatric rhabdomyosarcoma cell lines. Br J Cancer 83:338–345
Corsini A, Bellosta S, Baetta R, Fumagalli R, Paoletti R, Bernini F (1999) New insights into the pharmacodynamic and pharmacokinetic properties of statins. Pharmacol Ther 84:413–428
Danks MK, Schmidt CA, Cirtain MC, Suttle DP, Beck WT (1988) Altered catalytic activity of and DNA cleavage by DNA topoisomerase II from human leukemic cells selected for resistance to VM-26. Biochemistry 27:8861–8869
Demierre MF, Higgins PD, Gruber SB, Hawk E, Lippman SM (2005) Statins and cancer prevention. Nat Rev Cancer 5:930–942
Dimitroulakos J, Ye LY, Benzaquen M, Moore MJ, Kamel-Reid S, Freedman MH, Yeger H, Penn LZ (2001) Differential sensitivity of various pediatric cancers and squamous cell carcinomas to lovastatin-induced apoptosis: therapeutic implications. Clin Cancer Res 7:158–167
Endicott JA, Ling V (1989) The biochemistry of P-glycoprotein-mediated multidrug resistance. Annu Rev Biochem 58:137–171
Feleszko W, Zagozdzon R, Golab J, Jakobisiak M (1998) Potentiated antitumour effects of cisplatin and lovastatin against MmB16 melanoma in mice. Eur J Cancer 34:406–411
Feleszko W, Mlynarczuk I, Balkowiec-Iskra EZ, Czajka A, Switaj T, Stoklosa T, Giermasz A, Jakobisiak M (2000) Lovastatin potentiates antitumor activity and attenuates cardiotoxicity of doxorubicin in three tumor models in mice. Clin Cancer Res 6:2044–2052
Giaccone G, Linn SC, Welink J, Catimel G, Stieltjes H, van der Vijgh WJ, Eeltink C, Vermorken JB, Pinedo HM (1997) A dose-finding and pharmacokinetic study of reversal of multidrug resistance with SDZ PSC 833 in combination with doxorubicin in patients with solid tumors. Clin Cancer Res 3:2005–2015
Gottesman MM, Fojo T, Bates SE (2002) Multidrug resistance in cancer: role of ATP-dependent transporters. Nat Rev Cancer 2:48–58
Graaf MR, Beiderbeck AB, Egberts AC, Richel DJ, Guchelaar HJ (2004) The risk of cancer in users of statins. J Clin Oncol 22:2388–2394
Group SSSSS (2000) Follow-up study of patients ransomised in the Scandinavian Simvastatin Survival Study (4S) of cholesterol lowering. Am J Cardiol 86:257–262
Hiessbock R, Wolf C, Richter E, Hitzler M, Chiba P, Kratzel M, Ecker G (1999) Synthesis and in vitro multidrug resistance modulating activity of a series of dihydrobenzopyrans and tetrahydroquinolines. J Med Chem 42:1921–1926
Kawata S, Yamasaki E, Nagase T, Inui Y, Ito N, Matsuda Y, Inada M, Tamura S, Noda S, Imai Y, Matsuzawa Y (2001) Effect of pravastatin on survival in patients with advanced hepatocellular carcinoma. A randomized controlled trial. Br J Cancer 84:886–891
Kozar K, Kaminski R, Legat M, Kopec M, Nowis D, Skierski JS, Koronkiewicz M, Jakobisiak M, Golab J (2004) Cerivastatin demonstrates enhanced antitumor activity against human breast cancer cell lines when used in combination with doxorubicin or cisplatin. Int J Oncol 24:1149–1157
Larner J, Jane J, Laws E, Packer R, Myers C, Shaffrey M (1998) A phase I–II trial of lovastatin for anaplastic astrocytoma and glioblastoma multiforme. Am J Clin Oncol 21:579–583
Larsen AK, Escargueil AE, Skladanowski A (2000) Resistance mechanisms associated with altered intracellular distribution of anticancer agents. Pharmacol Ther 85:217–229
Lee YT, Chan KK, Harris PA, Cohen JL (1980) Distribution of adriamycin in cancer patients: tissue uptakes, plasma concentration after IV and hepatic IA administration. Cancer 45:2231–2239
Lee SJ, Ha MJ, Lee J, Nguyen P, Choi YH, Pirnia F, Kang WK, Wang XF, Kim SJ, Trepel JB (1998) Inhibition of the 3-hydroxy-3-methylglutaryl-coenzyme A reductase pathway induces p53-independent transcriptional regulation of p21(WAF1/CIP1) in human prostate carcinoma cells. J Biol Chem 273:10618–10623
Lennernas H, Fager G (1997) Pharmacodynamics and pharmacokinetics of the HMG-CoA reductase inhibitors. Similarities and differences. Clin Pharmacokinet 32:403–425
Link E, Parish S, Armitage J, Bowman L, Heath S, Matsuda F, Gut I, Lathrop M, Collins R (2008) SLCO1B1 variants and statin-induced myopathy—a genomewide study. N Engl J Med 359:789–799
Liscovitch M, Lavie Y (2002) Cancer multidrug resistance: a review of recent drug discovery research. IDrugs 5:349–355
Minichsdorfer C, Hohenegger M (2009) Autocrine amplification loop in statin-induced apoptosis of human melanoma cells. Br J Pharmacol 157:1278–1290
Nielsen SF, Nordestgaard BG, Bojesen SE (2012) Statin use and reduced cancer-related mortality. N Engl J Med 367:1792–1802
Poynter JN, Gruber SB, Higgins PD, Almog R, Bonner JD, Rennert HS, Low M, Greenson JK, Rennert G (2005) Statins and the risk of colorectal cancer. N Engl J Med 352:2184–2192
Raney RB Jr, Hays DM, Tefft M (1993) Rhabdomyosarcoma and the undifferentiated sarcomas. In: Pizzo PA, Poplack DG (eds) Principles and practice of pediatric oncology, 2nd ed. Lippincott Williams & Wilkins, Philadelphia, pp 769–794
Rao S, Porter DC, Chen X, Herliczek T, Lowe M, Keyomarsi K (1999) Lovastatin-mediated G1 arrest is through inhibition of the proteasome, independent of hydroxymethyl glutaryl-CoA reductase. Proc Natl Acad Sci U S A 96:7797–7802
Sacher J, Weigl L, Werner M, Szegedi C, Hohenegger M (2005) Delineation of myotoxicity induced by 3-hydroxy-3-methylglutaryl CoA reductase inhibitors in human skeletal muscle cells. J Pharmacol Exp Ther 314:1032–1041
Sakaeda T, Takara K, Kakumoto M, Ohmoto N, Nakamura T, Iwaki K, Tanigawara Y, Okumura K (2002) Simvastatin and lovastatin, but not pravastatin, interact with MDR1. J Pharm Pharmacol 54:419–423
Schindler M, Grabski S, Hoff E, Simon SM (1996) Defective pH regulation of acidic compartments in human breast cancer cells (MCF-7) is normalized in adriamycin-resistant cells (MCF-7adr). Biochemistry 35:2811–2817
Sharom FJ, Yu X, Chu JW, Doige CA (1995) Characterization of the ATPase activity of P-glycoprotein from multidrug-resistant Chinese hamster ovary cells. Biochem J 308(Pt 2):381–390
Sieczkowski E, Lehner C, Ambros PF, Hohenegger M (2010) Double impact on p-glycoprotein by statins enhances doxorubicin cytotoxicity in human neuroblastoma cells. Int J Cancer 126:2025–2035
Sleijfer S, van der Gaast A, Planting AS, Stoter G, Verweij J (2005) The potential of statins as part of anti-cancer treatment. Eur J Cancer 41:516–522
Soma MR, Baetta R, De Renzis MR, Mazzini G, Davegna C, Magrassi L, Butti G, Pezzotta S, Paoletti R, Fumagalli R (1995) In vivo enhanced antitumor activity of carmustine [N, N′-bis(2-chloroethyl)-N-nitrosourea] by simvastatin. Cancer Res 55:597–602
Swenson CE, Bolcsak LE, Batist G, Guthrie TH Jr, Tkaczuk KH, Boxenbaum H, Welles L, Chow SC, Bhamra R, Chaikin P (2003) Pharmacokinetics of doxorubicin administered i.v. as Myocet (TLC D-99; liposome-encapsulated doxorubicin citrate) compared with conventional doxorubicin when given in combination with cyclophosphamide in patients with metastatic breast cancer. Anticancer Drugs 14:239–246
Szakacs G, Paterson JK, Ludwig JA, Booth-Genthe C, Gottesman MM (2006) Targeting multidrug resistance in cancer. Nat Rev Drug Discov 5:219–234
Tamaki A, Ierano C, Szakacs G, Robey RW, Bates SE (2011) The controversial role of ABC transporters in clinical oncology. Essays Biochem 50:209–232
Ukomadu C, Dutta A (2003) p21-dependent inhibition of colon cancer cell growth by mevastatin is independent of inhibition of G1 cyclin-dependent kinases. J Biol Chem 278:43586–43594
Wachtershauser A, Akoglu B, Stein J (2001) HMG-CoA reductase inhibitor mevastatin enhances the growth inhibitory effect of butyrate in the colorectal carcinoma cell line Caco-2. Carcinogenesis 22:1061–1067
Wang E, Casciano CN, Clement RP, Johnson WW (2001) HMG-CoA reductase inhibitors (statins) characterized as direct inhibitors of P-glycoprotein. Pharm Res 18:800–806
Wang W, Collie-Duguid E, Cassidy J (2002) Cerivastatin enhances the cytotoxicity of 5-fluorouracil on chemosensitive and resistant colorectal cancer cell lines. FEBS Lett 531:415–420
Watters GP, Smart DJ, Harvey JS, Austin CA (2009) H2AX phosphorylation as a genotoxicity endpoint. Mutat Res Genet Toxicol Environ Mutagen 679:50–58
Werner M, Sacher J, Hohenegger M (2004) Mutual amplification of apoptosis by statin-induced mitochondrial stress and doxorubicin toxicity in human rhabdomyosarcoma cells. Br J Pharmacol 143:715–724
Acknowledgments
The authors are indebted to Dr. Peter Ambros (St. Anna Children’s Cancer Research Institute, Vienna, Austria) for helpful discussion and critical reading of the manuscript and to Anton Karel for technical assistance. The work was supported by grants from the Herzfeldersche Familienstiftung and the Austrian Science foundation, FWF-Project P22385.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Werner, M., Atil, B., Sieczkowski, E. et al. Simvastatin-induced compartmentalisation of doxorubicin sharpens up nuclear topoisomerase II inhibition in human rhabdomyosarcoma cells. Naunyn-Schmiedeberg's Arch Pharmacol 386, 605–617 (2013). https://doi.org/10.1007/s00210-013-0859-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00210-013-0859-y