
Abstract
In human bronchial epithelial (16HBE14o−) cells, CB1 and CB2 cannabinoid receptors are present, and their activation by the endocannabinoid virodhamine and the synthetic non-selective receptor agonist CP55,940 inhibits adenylyl cyclase and cellular interleukin-8 release. Here, we analyzed changes in intracellular calcium ([Ca2+]i) evoked by Δ9-tetrahydrocannabinol (Δ9-THC), CP55,940, and virodhamine in 16HBE14o− cells. Δ9-THC induced [Ca2+]i increase and a large transient [Ca2+]i mobilization, the latter probably reflecting store-depletion-driven capacitative Ca2+ entry (CCE). In contrast, CP55,940 induced a rather moderate Ca2+ influx and a sustained [Ca2+]i mobilization. CP55,940-induced Ca2+ influx was inhibited by Ni2+, indicating CCE, possibly mediated by transient receptor potential channel TRPC1, the mRNA of which is expressed in 16HBE14o− cells. CP55,940-induced calcium alterations were mimicked by virodhamine concentrations below 30 μM. Interestingly, higher virodhamine induced an additional Ca2+ entry, insensitive to Ni2+, but sensitive to the TRPV1 antagonist capsazepine, the TRPV1-TRPV4 inhibitor ruthenium red, and the non-CCE (NCCE) inhibitors La3+ and Gd3+. Such pharmacological profile is supported by the presence of TRPV1, TRPV4, and TRPC6 mRNAs as well as TRPV1 and TRPC6 proteins in 16HBE14o− cells. Cannabinoid receptor antagonists increased virodhamine-induced Ca2+ entry. Virodhamine also enhanced arachidonic acid release, which was insensitive to cannabinoid receptor antagonism, but sensitive to the phospholipase A2 inhibitor quinacrine, and to capsazepine. Arachidonic acid induced [Ca2+]i increase similar to virodhamine. Collectively, these observations suggest that [Ca2+]i alterations induced by Δ9-THC, CP55,940 and by low concentrations of virodhamine involve mobilization and subsequent CCE mechanisms, whereas such responses by high virodhamine concentrations involve NCCE pathways.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
In the last decades, two cannabinoid receptor (CBR) subtypes have been identified, cloned, expressed, and functionally investigated (Matsuda et al. 1990; Munro et al. 1993). The CB1R has primarily been observed in central and peripheral nervous structures (Tsou et al. 1998), whereas the CB2R is predominantly found in immune cells (Berdyshev 2000; Galiegue et al. 1995; Munro et al. 1993; Schatz et al. 1997). Interestingly, (endo)cannabinoid-mediated effects independent of the CB1R and the CB2R have been described (Oz 2006; Pertwee 2005). Such (endo)cannabinoid responses may involve orphan receptors GPR119 and GPR55; in addition, transient receptor potential (TRP) channels are also a matter of debate (Brown 2007; Rao and Kaminski 2006b; Clapham et al. 2005).
Both the CB1R and the CB2R belong to the G-protein-coupled receptor superfamily and primarily couple to pertussis toxin-sensitive and adenylyl cyclase-inhibitory Gi/o-proteins (Childers and Deadwyler 1996; Felder et al. 1995; Hofmann et al. 2000; Gkoumassi et al. 2007). Enhancement of cAMP production through linkage to Gs-proteins, however, has been reported as well (Bonhaus et al. 1998; Gkoumassi et al. 2007; Glass and Felder 1997). Recently, we have shown that both CBR subtypes are present in human bronchial epithelial (16HBE14o−) cells and are functionally coupled to adenylyl cyclase. We found that, in response to the endocannabinoid virodhamine and the synthetic cannabinoid CP55,940, the CB2R signaling pathway is predominant and inhibits adenylyl cyclase and the cellular release of interleukin-8. Following pretreatment with pertussis toxin, increased accumulation of cAMP became apparent, however, which was mediated by CB1R. Interestingly, the synthetic cannabinoid CP55,940 evoked such 16HBE14o− cell responses less efficiently compared to the endocannabinoid virodhamine (Gkoumassi et al. 2007). Indeed, CP55,940 acts as a non-selective receptor agonist (Burkey et al. 1997; Thomas et al. 2007), whereas virodhamine acts as a partial agonist on the CB1R and as a full agonist on the CB2R (Porter et al. 2002).
Next to the modulation of adenylyl cyclase, (endo)cannabinoid-induced changes in intracellular calcium ([Ca2+]i) have been investigated in various cell types. We reported that the phytocannabinoid Δ9-tetrahydrocannabinol (Δ9-THC) and the synthetic cannabinoid CP55,940 induced Ca2+ influx and [Ca2+]i mobilization in DDT1 MF-2 smooth muscle cells, processes being dependent on the CB1R (Filipeanu et al. 1997) and on the release of arachidonic acid (Begg et al. 2001; Demuth et al. 2005). Recent reports indicated that Δ9-THC, but not CP55,940, induced Ca2+ alterations in resting T cells independent of CB1R and CB2R (Rao et al. 2004; Rao and Kaminski 2006a), but dependent on Ca2+ influx through the TRP channel TRPC1 (Rao and Kaminski 2006b). Interestingly, the endocannabinoid anandamide has been shown to activate the TRP family member TRPV1 (Clapham et al. 2005).
In bronchial epithelial cells, changes in intracellular calcium are importantly involved in the regulation of mucus secretion, cytokine production, surfactant secretion, and ciliary beat frequency (Conway et al. 2003; Sakamoto et al. 2007; Haller et al. 1998; Lansley et al. 1992). Exposure of epithelial cells to cannabinoids may represent a result of marijuana smoke (Δ9-THC) or endogenously released cannabinoids like virodhamine and anandamide (Porter et al. 2002). However, the effects of (endo)cannabinoids on intracellular calcium alterations in bronchial epithelial cells have not been described yet. Therefore, we aimed in the current study to identify and to characterize alterations in intracellular calcium evoked by (endo)cannabinoids in human bronchial epithelial (16HBE14o−) cells. As it has been reported that the newly identified (endo)cannabinoid virodhamine is present in two to nine times higher concentrations in peripheral tissues than the classical (endo)cannabinoid anandamide (Porter et al. 2002), we studied the effect of virodhamine in comparison to Δ9-THC and CP55,940. We demonstrated that Δ9-THC, CP55,940, and low concentrations of virodhamine induced [Ca2+]i increases in 16HBE14o− cells involving mobilization and subsequent capacitive calcium entry (CCE) mechanisms, whereas high virodhamine concentrations induced [Ca2+]i increases predominantly through non-CCE (NCCE) pathways.
Materials and methods
Chemicals and drugs
All chemicals were of analytical grade and obtained from Sigma (Zwijndrecht, The Netherlands), unless stated otherwise. Virodhamine [O-(2-aminoethyl)-5Z,8Z,11Z,14Z-eicosatetraenoate], ruthenium red, capsazepine [N-[2-(4-chlorophenyl)ethyl]-1,3,4,5-tetrahydro-7,8-dihydroxy-2H-2-benzazepine-2-carbothioamide], and SR141716A (N-piperidino-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-3-pyrazole-carboxamide) were obtained from Tocris-Cookson (Avonmouth, UK). Δ9-Tetrahydrocannabinol (Δ9-THC) was a gift from Dr. H. W. Frijlink (University of Groningen). SR144528 [N-[(1S)-endo-1,3,3-trimethyl bicyclo [2.2.1] heptan-2-yl]-5-(4-chloro-3-methylphenyl)-1-(4-methylbenzyl)-pyrazole-3-carboxamide] was a gift from Sanofi (Montpellier, France). CP55,940 (5-(1,1-dimethylheptyl)-2-[5-hydroxy-2-(3-hydroxypropyl)cyclohexyl]phenol) was obtained from Sigma (Zwijndrecht, The Netherlands). Stock solutions of Δ9-THC, virodhamine and arachidonic acid, were prepared in ethanol, shielded from light, and stored at −20°C. SR141716A and SR144528 were dissolved in dimethyl sulfoxide (DMSO) and stored at room temperature. Lanthanum (III) chloride, gadolinium (III) chloride, and nickel (II) chloride solutions were prepared freshly in distilled water. Stock solution of quinacrine [6-chloro-9-(4-diethylamino-1-methylbutylamino)-2-methoxyacridine dihydrochloride] was prepared in DMSO and stored at 4°C. Thapsigargin was dissolved in DMSO and stored at −20°C. Capsazepine was dissolved in ethanol and stored at −20°C. Ruthenium red was dissolved in distilled water and stored at room temperature. The antibody directed to TRPC6 was a gift from Dr. Alexander Dietrich (Hofmann et al. 2000; Dietrich et al. 2003), and the antibody directed to TRPV1 was from Biomol (Exeter, UK).
Cell culture
The immortal human bronchial epithelial cell line 16HBE14o− (Cozens et al. 1994) was a kind gift from Dr. D. C. Gruenert (University of Vermont, Burlington VT, USA). Cells were grown at 37°C in minimum essential medium (MEM), supplemented with 10% fetal bovine serum, penicillin (50 µg/ml), streptomycin (50 µg/ml), and l-glutamine (2 mM) in fibronectin/collagen coated flasks to near confluency in an atmosphere of 5% CO2/95% O2. For arachidonic acid release studies, cells were plated into 9.6 cm2 six well plates. All culture media and supplements were obtained from Life Technologies (Breda, The Netherlands). Cell culture dishes were from Costar (Badhoevedorp, The Netherlands), and flasks were from Greiner (Alphen a/d Rijn, The Netherlands).
Intracellular Ca2+ measurements
Intracellular [Ca2+] was measured using Fura-2 fluorometry as reported previously (Demuth et al. 2005). Briefly, cells were trypsinized and loaded in suspension with 3 μM Fura-2 acetoxymethylester at 37°C for 45 min in the dark. Fluorescence was measured at 37°C. When used, receptor antagonists (SR141716A and SR144528 1 µM) and channel inhibitors (capsazepine 10 µM, ruthenium red 20 µM, Gd3+ 1 µM, La3+ 10 µM, Ni2+ 1 mM) were added 10 min prior to the agonist.
RNA isolation and RT-PCR
Total RNA was isolated using the RNeasy minikit (Qiagen, Venlo, The Netherlands) according to the manufacturer's instructions. RNA samples were purified with a RQ1 RNAse-free DNAse to remove genomic DNA contaminations. Subsequently, RNA (1 μg) was transcribed into cDNA using the Promega RT system kit. PCR reactions were performed using the Promega RT-PCR Core system kit (Leiden, The Netherlands), according to the manufacturer's instructions, using a Mastercycler Gradient (Eppendorf, Hamburg, Germany). For the detection TRP channels the following primer pairs were obtained from Invitrogen (Leek, The Netherlands): TRPC1 (forward): 5′-GTG CGG GAG GTG AAG GAG GAG AAT-3′; TRPC1 (reverse): 5′-CTG CAT GGG GCT TGG GTA GAG ATA-3′; TRPC6 (forward): 5′-ATG ATA TGG GCT GAA TGT AAA GA-3′; TRPC6 (reverse): 5′-TCA AAG TAG GAA AAC CAG AG-3′; TRPV1 (forward): 5′-CTA ACT CCA GGC CAC CTC CA-3′; TRPV1 (reverse): 5′-GCC ATG TTG CGT CTC TCG AT-3′; TRPV4 (forward): 5′-TAC TAT CGA GGT CAG ACA GC-3′; TRPV4 (reverse): 5′-CCG CAG CAG TTC ATT GAT GG-3′. With these primers, the amplified gene fragments were 463, 752, 562, and 678 bp, respectively. PCR reactions that comprised of a denaturing step at 94°C for 5 min was followed by amplification for 30 cycles at 94°C, 1 min; annealing temperature, 1 min; 72°C, 1 min; with final extension at 72°C for 10 min. The annealing temperature used for TRPC1 was 52°C, 60°C for TRPC6, and 58°C for TRPV1 and TRPV4. The amplified product (2 µl) was separated on a 2% agarose gel, and the DNA was visualized by ethidium bromide. A 100 bp ladder (MBI Fermentas, St. Leon-Rot, Germany) was used as DNA marker. Restriction analysis was performed with EcoRI for TRPC1 (expected fragments 335 and 128), with HindIII for TRPC6 (expected fragments 342 and 410), with PstI for TRPV1 (expected fragments 328 and 234), and HincII for TRPV4 (expected fragments 477 and 201; Sigma, Zwijndrecht, The Netherlands) for 2 h at 37°C according to the manufacturer's protocol.
Immunoblot analysis
Western blot analysis was performed as described before (Gkoumassi et al. 2007; Hassock et al. 2002; Hofmann et al. 2000). Briefly, cell lysate (corresponding to 10–15 µg protein per lane) was separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis, using 10% gel at 100 V. Proteins in the gel were transferred onto nitrocellulose membranes, which were subsequently blocked with blocking buffer (Tris–HCl 50 mM, NaCl 150 mM, Tween-20 0.1%, dried milk powder 5%) for 90 min. Membranes were incubated overnight at 4°C using 1:1,000 dilution of rabbit polyclonal antiserum raised against the C-terminal peptide VGHNKQPSIRSSEDFHLNSFNNPP (amino acids 829–852 of hTRPC6); the rabbit polyclonal antiserum against the TRPV1 was used at a dilution of 1:200. After three washes of 10 min each, membranes were incubated with horseradish peroxidase-labeled secondary antibodies (1:3,000 in blocking buffer) for 90 min at room temperature, followed by another three washes. Antibodies were visualized by enhanced chemiluminescence.
Arachidonic acid release measurements
[3H]Arachidonic acid release was measured as described previously (Demuth et al. 2005; Van der Zee et al. 1995). In short, cells were grown to confluency in 9.6 cm2 six well plates and labeled with 0.25 µCi of [3H]arachidonic acid (specific activity, 60–100 Ci (2.22–3.70 TBq)/mmol) in serum-free MEM (1 ml), for 3 h at 37°C. Cells were washed once in extracellular salt solution (ECS) containing (mM): NaCl, 125; KCl, 6; MgCl2, 2.5; NaH2PO4, 1.2; Hepes, 20; glucose, 11; sucrose, 67; CaCl2, 1.2; pH 7.4, twice with ECS containing 1% BSA (fatty acid-free) and once again with ECS to eliminate unincorporated radioactivity; subsequently, cells were allowed to equilibrate for 15 min, in the absence or presence of the inhibitors. Cells were stimulated with the agonist for 5 min, and the incubation was stopped by removing the ECS. [3H]Arachidonic acid release into the ECS was quantified using liquid-scintillation counting.
Data analysis
Values are expressed as means ± standard error of the mean (SEM). Comparison of pairs of treatments was made using Student's t test. P < 0.05 was considered significant. All curves were fitted using a logistic fit four-parameter model (SigmaPlot 10, SYSTAT Software, Chicago IL, USA).
Results
To assess the effects of (endo)cannabinoids on alterations in intracellular calcium ([Ca2+]i) in 16HBE14o− cells, we incubated the cells with different concentrations of Δ9-THC, CP55,940, and virodhamine. The phytocannabinoid Δ9-THC evoked a large dose-dependent [Ca2+]i increase (Fig. 1b and c), consisting of a relatively fast peak (transient) and a sustained plateau phase (Fig. 1a). In the absence of extracellular Ca2+, a depressed transient response remained, whereas the sustained plateau phase was abrogated (Fig. 1b and c). Analysis of the concentration-effect curves indicated pEC50 values of 4.57 ± 0.05 (n = 6) and of 4.56 ± 0.08 (n = 5) for Δ9-THC peak responses in the presence and absence of extracellular Ca2+, respectively; maximal Δ[Ca2+]i following Δ9-THC stimulation were 472 ± 36 and 176 ± 29 nM Ca2+, in the presence and absence of extracellular Ca2+, respectively. For the plateau phase a pEC50 value of 4.59 ± 0.73 and a maximal Δ[Ca2+]i of 199 ± 28 nM were obtained in the presence of extracellular Ca2+, which changed into 5.00 ± 0.23 and a maximal [Ca2+]i of 21 ± 8 nM under Ca2+-free conditions. The similarities between the pEC50 values suggest that [Ca2+]i mobilization and influx following Δ9-THC administration share a common origin.

Changes in [Ca2+]i evoked by Δ9-tetrahydrocannabinol (Δ9-THC) in 16HBE14o− cells. a Representative traces obtained in the presence and absence of extracellular Ca2+ induced by 100 µM Δ9-THC administered at t = 60 s. b Concentration-response curves of the maximal Δ[Ca2+]i for peak values induced by Δ9-THC in the presence (closed circles) and absence (open circles) of extracellular Ca2+. c Concentration-response curves of the maximal Δ[Ca2+]i for the plateau values induced by Δ9-THC in the presence (closed circles) or absence (open circles) of extracellular Ca2+. Data in b and c are presented as means ± SEM of five to eight experiments
The synthetic cannabinoid CP55,940 induced a moderate and rather sustained dose-dependent [Ca2+]i increase (Fig. 2). The maximal Δ[Ca2+]i, in the presence of extracellular Ca2+ was 70.3 ± 2.5 nM Ca2+ with a pEC50 value of 5.32 ± 0.03 for this agonist. Of note, higher concentrations of CP55,940 did not further increase [Ca2+]i (data not shown). In the absence of extracellular Ca2+, the value for the maximal Δ[Ca2+]i was 27.6 ± 0.6 nM with a pEC50 value of 5.76 ± 0.18 (Fig. 2b), indicating that CP55,940-induced [Ca2+]i increase involves continuous [Ca2+]i mobilization and [Ca2+]i influx.

Changes in [Ca2+]i evoked by CP55,940 in 16HBE14o− cells. a Representative traces obtained in the presence and absence of extracellular Ca2+ induced by 100 µM CP55,940 administered at t = 60 s. b Concentration-response curves of the maximal Δ[Ca2+]i values induced by CP55,940 in the presence (closed circles) and absence (open circles) of extracellular Ca2+. Data in b are presented as means ± SEM of four experiments
To gain insight into the impact of capacitative Ca2+ entry (CCE) in cannabinoid-driven calcium alterations in 16HBE14o− cells, we first estimated CCE following intracellular store depletion (Fig. 3). After emptying Ca2+ stores with thapsigargin under Ca2+-free conditions, reintroduction of extracellular Ca2+ induced a large CCE-dependent Ca2+ influx, which could be abolished concentration dependently by the CCE inhibitor Ni2+ (Sweeney et al. 2002a), but not by the NCCE inhibitors Gd3+ or La3+ (Clapham et al. 2005; Demuth et al. 2005; Fig 3a and b). The CCE-dependent Ca2+ entry was inhibited by Ni2+ ions with a pIC50 of 2.99 ± 0.25 (Fig. 3c). Importantly, the CP55,940-induced Ca2+ influx was also completely blocked by the CCE inhibitor Ni2+, strongly pointing to the involvement of CCE in CP55,940 driven Ca2+ influx (Fig. 4a). Previous studies have implicated that TRPC1 is involved in CCE (Clapham et al. 2005; Sweeney et al. 2002b) and that cannabinoids induce Ca2+ influx in part via these channels (Rao and Kaminski 2006b). Indeed, PCR analysis revealed that mRNA for the TRP channel TRPC1 was expressed by 16HBE14o− cells (Fig. 4b), which is in agreement with previous findings in primary HBE cells (Corteling et al. 2004).

Effects of Ni2+, La3+, and Gd3+ on thapsigargin-evoked capacitative Ca2+ entry (CCE) in 16HBE14o− cells. Internal Ca2+ stores were depleted with 1 µM thapsigargin in Ca2+-deficient medium. Reintroduction of extracellular Ca2+ (1 mM) increased intracellular [Ca2+] reflecting activation of CCE. a Sample trace of the effect of cumulatively administered concentrations of Gd3+ (0.01, 0.1, 1, 10, and 100 µM) on CCE-dependent increases in [Ca2+]i starting at *a, followed by addition of Ni2+ (*b 1 mM; *c 3 mM; *d 10 mM). b Sample trace of the effect of La3+. Cumulative concentrations were 0.01, 0.1, 1, 10, and 100 µM starting at *a, followed by addition of Ni2+ (*b 1 mM; *c 3 mM; *d 10 mM). In contrast to Ni2+, no effects on CCE-dependent increases in [Ca2+]i were observed with Gd3+ or La3+ (n = 3). c Concentration-response curve of the effect induced by Ni2+ alone. Data in c are presented as means ± SEM of three experiments

Effect of CCE blockade on CP55,940-induced [Ca2+]i increase. a CP55,940 (10 µM)-induced changes in [Ca2+]i in the presence and absence of extracellular Ca2+ and in the presence of Ni2+ (10 mM, added after reaching the sustained phase in the presence of extracellular Ca2+). Data in a are presented as means ± SEM of four experiments. *P < 0.05 vs. control. N.S. not significantly different. b mRNA expression of TRPC1 in 16HBE14o− cells. Total RNA was reverse-transcribed into cDNA, and specific fragments were amplified by PCR, separated on 2% agarose gel, and visualized with ethidium bromide. Restriction analysis, performed with EcoRI, is presented in the right lane
Virodhamine (O-arachidonoyl ethanolamine), a novel endocannabinoid structurally closely related to anandamide, also induced [Ca2+]i increase in a concentration-dependent fashion (Fig. 5). Interestingly, however, we observed two distinctive patterns, consisting of a slow and moderate [Ca2+]i increase at virodhamine concentrations below 30 µM and a large, fast, and almost linear [Ca2+]i increase at high virodhamine (30 μM) and higher (not shown) concentrations. The vehicle ethanol induced only a negligible [Ca2+]i increase (data not shown). Under Ca2+-free conditions, higher concentrations of virodhamine did not further enhance [Ca2+]i increase, indicating that this cellular response was fully dependent on influx of extracellular Ca2+ (Fig. 5b and c). Under Ca2+-free conditions, a plateau in the maximal Δ[Ca2+]i was reached with a maximal Δ[Ca2+]i of 50.0 ± 10.1 nM and a pEC50 value of 5.10 ± 0.19 (Fig. 5c). Surprisingly, the CB1R antagonist SR141716A further enhanced the virodhamine (30 µM)-induced [Ca2+]i increase, whereas the NCCE-inhibitor Gd3+ and the TRPV1-TRPV4 inhibitor ruthenium red strongly reduced the large Ca2+ influx component (Fig. 5b). To further assess the potential role of CBR subtypes and Ca2+ channels, we quantified the virodhamine-induced calcium alterations (Fig. 6). Both the CB1R antagonist SR141716A and the CB2R antagonist SR144528 augmented [Ca2+]i increase in response to 10 and 30 μM virodhamine, suggesting an inhibitory effect of the CBRs on virodhamine-induced Ca2+ influx. The CBR antagonists did not alter the control level of [Ca2+]i (data not shown). As (endo)cannabinoids have been implicated in the activation of the vanilloid receptor/Ca2+ channel TRPV1 (Clapham et al. 2005), we studied the effect of the TRPV1 antagonist capsazepine (CPZ) on the virodhamine-induced Ca2+ influx as well. At low (10 μM) virodhamine concentrations, CPZ did not alter the virodhamine-induced [Ca2+]i increase (Fig. 6a). At high (30 μM) virodhamine concentrations, however, the virodhamine-induced [Ca2+]i increase was sensitive to CPZ (Fig. 6b), implying the involvement of TRPV1 channels in this response. The classical vanilloid-TRPV receptor agonist capsaicin (1–100 µM) by itself did not affect intracellular calcium concentrations (data not shown), and pretreatment with capsaicin did not significantly increase virodhamine-induced calcium alterations (P = 0.14; Fig 6a). Interestingly, the TRPV1–TRPV4 inhibitor ruthenium red was able to reduce virodhamine (30 µM)-induced [Ca2+]i increase as well (Fig. 6b). As we observed the presence of TRPV1 and TRPV4 mRNAs and TRPV1 protein in 16HBE14o− cells (Fig. 6c and d) and a previous study has reported the presence of the TRPV4 protein in differentiated primary HBE cells (Sidhaye et al. 2008), a potential role for such Ca2+ channels in virodhamine-induced Ca2+ influx should be envisaged as well. Almost complete inhibition of virodhamine-induced Ca2+ influx was achieved by the NCCE inhibitors Gd3+ and La3+ used at low concentrations of 1 and 10 µM, respectively (Fig. 6b). In contrast, the CCE inhibitor Ni2+ did not inhibit virodhamine-induced Ca2+ influx, even at a concentration of 1 mM (Fig. 6b). Collectively, these observations clearly point to a role NCCE mechanisms in the calcium response induced by high virodhamine concentrations. Currently, TRPC6 channels are the only channels known to be sensitive to low concentrations of Gd3+ and La3+ (Clapham et al. 2005). Indeed, in agreement with findings in primary HBE cells (Corteling et al. 2004), we detected both mRNA and protein of TRPC6 in 16HBE14o− cells. Next to the 100 kDa immunoreactive positive TRPC6 band, an additional band of 73 kDa was visible, similar to the band reported before in HEK293 cells (Dietrich et al. 2003).

Virodhamine-induced changes in [Ca2+]i. a Representative traces showing the effect of virodhamine 10, 20, and 30 µM. b Effect of 30 µM virodhamine (control) on [Ca2+]i in the absence and presence of 1 µM SR141716A (SR1), 20 µM ruthenium red (RR), and 1 µM Gd3+ (Gd3+) and in the absence of extracellular Ca2+ (Ca2+-free). The different inhibitors were added 10 min prior to virodhamine application at t = 60 s. c Concentration-response curves of the maximal sustained increases of [Ca2+]i induced by virodhamine in the presence (closed circles) and absence (open circles) of extracellular Ca2+. Data in c are presented as means ± SEM of four experiments

Quantification of the effects of various inhibitors on virodhamine-induced changes in [Ca2+]i and detection of TRP channels. a Maximal sustained increases in intracellular [Ca2+] induced by 10 µM virodhamine in the absence or presence of SR141716A (1 µM, SR1), SR144528 (1 µM, SR2), capsaicin (10 μM, CPS), or capsazepine (10 µM, CPZ). The different antagonists were added 10 min prior to virodhamine application at t = 60 s. Data in a are presented as means ± SEM of four to seven experiments. b Ca2+-influx rate induced by 30 µM virodhamine in the absence or presence of SR141716A, SR144528, capsazepine, ruthenium red (20 µM, RR), Gd3+ (1 µM), La3+ (10 µM), or Ni2+ (1 mM). Ca2+-influx rate represents the maximal slope and is expressed as percent of the control rate (30 μM virodhamine). Data in b are presented as means ± SEM of eight experiments. *P < 0.05 vs. control. c mRNA expression of TRPC6, TRPV1, and TRPV4 in 16HBE14o− cells. Total RNA was reverse-transcribed into cDNA, and specific fragments were amplified by PCR, separated on 2% agarose gel, and visualized with ethidium bromide. Restriction analysis, performed with HindIII (TRPC6), PstI (TRPV1), and HincII (TRPV4) are presented in the right lanes. d Western analysis of the TRPC6 channel in 16HBE14oֿ cells compared to HEK293 cells used as positive control
Since arachidonic acid has been shown to be an important mediator in NCCE-dependent Ca2+ entry (Demuth et al. 2005;Van der Zee et al. 1995; Shuttleworth and Thompson 1999), we also investigated the release of this lipid mediator by 16HBE14o− cells upon stimulation with virodhamine. A concentration-dependent release of arachidonic acid was observed (pEC50 = 5.19 ± 0.16), which did not increase further at concentrations above 10 μM, indicating that virodhamine-induced arachidonic acid release may be involved in NCCE (Fig. 7a). This release was sensitive to the phospholipase A2 inhibitor quinacrine, but insensitive to CBR antagonists (Fig. 7b). Virodhamine-induced arachidonic acid release was, however, sensitive to capsazepine treatment, and a trend towards a decrease in arachidonic acid release was observed for ruthenium red, indicating that TRPV1 channels may be involved in these responses. Finally, to assess whether arachidonic acid could be involved in the activation of TRPV1 by virodhamine, we investigated the effects of arachidonic acid on [Ca2+]i increase. Concentrations of arachidonic acid between 1 and 30 μM induced a concentration-dependent [Ca2+]i increase, with a pEC50 = 5.17 ± 0.63 and a maximal Δ[Ca2+]i of 97.3 ± 7.5 nM (Fig. 8a and b). Higher concentrations of arachidonic acid (≥100 μM) induced a large, fast, and almost linear increase at high concentrations (Fig 8a), very similar to the effects observed for the high virodhamine concentrations.

Effects of virodhamine on arachidonic acid release. a Concentration-response curve of virodhamine-induced arachidonic acid release. Data in a are presented as means ± SEM of eight to 12 experiments. b Effect of SR141716A (1 µM, SR1), SR144528 (1 µM, SR2), quinacrine (0.1 mM, Quin), ruthenium red (20 µM, RR), and capsazepine (10 µM, CPZ) on the arachidonic acid release induced by 30 µM virodhamine. *P < 0.05 vs. control (virodhamine). Data in b are presented as means ± SEM of eight experiments

Changes in [Ca2+]i evoked by arachidonic acid in 16HBE14o− cells. a Representative traces obtained with different concentrations (1–100 μM) of arachidonic acid administered at t = 60 s. b Concentration-response curves of the maximal Δ[Ca2+]i values induced by arachidonic acid. Data in b are presented as means ± SEM of four experiments
Discussion
In the present study, we report for the first time that the structurally distinct (endo)cannabinoids Δ9-THC, CP55,940, and virodhamine induce intracellular [Ca2+]i increase driven by different mechanisms in human bronchial epithelial 16HBE14o− cells. All three compounds induce an [Ca2+]i increase by Ca2+ entry from the extracellular space, but for Δ9-THC, this Ca2+ influx is subsequent to a large transient [Ca2+]i mobilization and may therefore reflect a store-depletion-driven CCE mechanism. Indeed, we reported in previous studies in DDT1 MF-2 smooth muscle cells that the Δ9-THC-induced Ca2+ release from thapsigargin-sensitive intracellular stores involved the CB1R and subsequent CCE mechanisms (Filipeanu et al. 1997). Interestingly, Δ9-THC induced an additional Ca2+ influx in these cells independent of CB1R and CB2R. Recently, a robust Δ9-THC-induced Ca2+ influx was reported in resting T cells, also independent of CB1R and CB2R (Rao and Kaminski 2006a) and at least partly mediated by TRPC1 channels (Rao and Kaminski 2006b). While thapsigargin-sensitive CCE mechanisms are operative in 16HBE14o− cells and cellular gene transcription of TRPC1 channels was present and the involvement of TRPC1 channels in CCE mechanisms being well established (Clapham et al. 2005; Sweeney et al. 2002a), it is reasonable to assume that subsequent to Ca2+ release from intracellular, Δ9-THC-induced Ca2+ entry may be gated through TRPC1 channels in 16HBE14o− cells indeed.
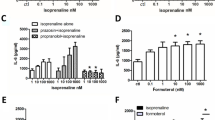
The synthetic non-selective CBR agonist CP55,940 induced only a modest and a sustained [Ca2+]i mobilization. We reported earlier that CP55,940 is also less efficacious compared to the endocannabinoid virodhamine in inhibiting cAMP accumulation and release of interleukin-8 from 16HBE14o− cells (Gkoumassi et al. 2007). The Ca2+ influx induced by CP55,940 was inhibited by Ni2+, indicating the involvement of a CCE mechanism, possibly mediated by TRPC1 channels. Previous studies, however, showed that CP55,940 induced [Ca2+]i increase independent of CB1R and predominantly by NCCE mechanisms in DDT1 MF-2 smooth muscle cells (Demuth et al. 2005), while in renal tubular epithelial cells, CP55,940 increased [Ca2+]i by Ca2+ entry following Ca2+ release from thapsigargin-sensitive pools in a CBR-independent manner (Chou et al. 2001). Collectively, these studies indicate that mechanisms distinct from those in other cell types are operative in CP55,940-induced [Ca2+]i increase in 16HBE14o− cells.
Remarkably, the (endo)cannabinoid virodhamine induced Ca2+ increase at concentrations which were 150 times higher than we described previously for CBR-mediated inhibition of cAMP accumulation and interleukin-8 release from 16HBE14o− cells (Gkoumassi et al. 2007). This observation suggests Ca2+ entry via CBR-independent channel activation. In support of this, changes in [Ca2+]i evoked by both low and high concentrations of virodhamine were increased rather than decreased in the presence of both CB1R and CB2R antagonists, which would suggest inhibitory effects of CBRs on [Ca2+]i alterations under these conditions or even aspecific actions of the CBR antagonists on cation channels (Rao et al. 2004; Rao and Kaminski 2006a; White and Hiley 1998).
Virodhamine-induced arachidonic acid release and Ca2+ entry showed the same concentration-dependence, suggesting that such cellular responses are driven by the same NCCE mechanisms. Receptor-independent effects by (endo)cannabinoids may be dependent on their lipophilic nature (Oz 2006). Such processes, however, do not exclude potential physiologically important effects with regard to the activation of TRP channels, as concentrations of arachidonic acid-derivatives may reach concentrations up to 5 mM in for example human platelets, and concentrations as used in our current study are physiologically not uncommon (Brash 2001; Oz 2006). Recent studies even indicated that higher concentrations of virodhamine are necessary for attaining the full agonist effect (Porter et al. 2002; Steffens et al. 2005; Ho and Hiley 2004).
Physiological functions driven by TRPV1 and TRPV4, including detection of heat, H+ ions, and extracellular osmolarity (Clapham et al. 2005) may play an important role in [Ca2+]i regulation in bronchial epithelial cells and may be influenced by endocannabinoids (Jia and Lee 2007). Indeed, [Ca2+]i changes may affect the regulation of mucus secretion, cytokine production, surfactant secretion, and ciliary beat frequency of epithelial cells (Conway et al. 2003; Sakamoto et al. 2007; Haller et al. 1998; Lansley et al. 1992). Ca2+ entry induced by high virodhamine concentrations in 16HBE14o− cells was inhibited by the TRPV1-TRPV4 inhibitors capsazepine and ruthenium red. Presence of TRPV1 and TRPV4 mRNAs and protein (TRPV1, present study; TRPV4 (Sidhaye et al. 2008)) suggest their possible involvement in the observed responses for virodhamine. Surprisingly, the TRPV1 agonist capsaicin did not affect [Ca2+]i or virodhamine-induced changes in [Ca2+]i. These findings might be explained by the existence of different splice variants of the TRPV1 channel, which could not be detected by our PCR primers. Of these splice variants, TRPV1b has been shown to be activated by heat but not by capsaicin or protons (Lu et al. 2005), indicating that this variant may be involved in the observed effects. It is also feasible to assume that the signaling properties from capsaicin differ from that of virodhamine. In addition to TRPV1 and TRPV4, TRPC6 is also likely to be involved in this Ca2+ entry process since this channel mediates NCCE (Grant et al. 2007) and is highly sensitive to low concentrations of La3+ and Gd3+ (Clapham et al. 2005), properties fitting well with our results. Accordingly, both mRNA and protein for TRPC6 were expressed in 16HBE14o− cells.
Structural differences between Δ9-THC, CP55,940, and virodhamine may be important for the distinct effects on [Ca2+]i increase in 16HBE14o− cells. CP55,940 shares much structural similarity with Δ9-THC, whereas virodhamine, which is an ester of arachidonic acid and ethanolamine, has much more similarity to arachidonic acid. Indeed, arachidonic acid-induced [Ca2+]i changes were very similar to the effects induced by virodhamine. These effects and the effects seen with capsazepine and ruthenium red on virodhamine-induced Ca2+ influx and arachidonic acid release indicate that virodhamine, directly or indirectly, activates TRPV1 and TRPV4 in an arachidonic acid-dependent fashion.
In conclusion, we have shown that the (endo)cannabinoids Δ9-tetrahydrocannabinol, CP55,940, and virodhamine use different mechanisms of action to elevate [Ca2+]i in 16HBE14o− cells. Δ9-THC, CP55,940, and low concentrations of virodhamine increase [Ca2+]i through mobilization and subsequent CCE mechanisms, whereas higher concentrations of virodhamine induce [Ca2+]i increases involving non-CB1R, non-CB2R-mediated NCCE pathways.
References
Begg M, Baydoun A, Parsons ME, Molleman A (2001) Signal transduction of cannabinoid CB1 receptors in a smooth muscle cell line. J Physiol 531:95–104
Berdyshev EV (2000) Cannabinoid receptors and the regulation of immune response. Chem Phys Lipids 108:169–190
Bonhaus DW, Chang LK, Kwan J, Martin GR (1998) Dual activation and inhibition of adenylyl cyclase by cannabinoid receptor agonists: evidence for agonist-specific trafficking of intracellular responses. J Pharmacol Exp Ther 287:884–888
Brash AR (2001) Arachidonic acid as a bioactive molecule. J Clin Invest 107:1339–1345
Brown AJ (2007) Novel cannabinoid receptors. Br J Pharmacol 152:567–575
Burkey TH, Quock RM, Consroe P, Ehlert FJ, Hosohata Y, Roeske WR, Yamamura HI (1997) Relative efficacies of cannabinoid CB1 receptor agonists in the mouse brain. Eur J Pharmacol 336:295–298
Childers SR, Deadwyler SA (1996) Role of cyclic AMP in the actions of cannabinoid receptors. Biochem Pharmacol 52:819–827
Chou KJ, Tseng LL, Cheng JS, Wang JL, Fang HC, Lee KC, Su W, Law YP, Jan CR (2001) CP55, 940 increases intracellular Ca2+ levels in Madin-Darby canine kidney cells. Life Sci 69:1541–1548
Clapham DE, Julius D, Montell C, Schultz G (2005) International union of pharmacology. XLIX. Nomenclature and structure-function relationships of transient receptor potential channels. Pharmacol Rev 57:427–450
Conway JD, Bartolotta T, Abdullah LH, Davis CW (2003) Regulation of mucin secretion from human bronchial epithelial cells grown in murine hosted xenografts. Am J Physiol Lung Cell Mol Physiol 284:L945–L954
Corteling RL, Li S, Giddings J, Westwick J, Poll C, Hall IP (2004) Expression of transient receptor potential C6 and related transient receptor potential family members in human airway smooth muscle and lung tissue. Am J Respir Cell Mol Biol 30:145–154
Cozens AL, Yezzi MJ, Kunzelmann K, Ohrui T, Chin L, Eng K, Finkbeiner WE, Widdicombe JH, Gruenert DC (1994) CFTR expression and chloride secretion in polarized immortal human bronchial epithelial cells. Am J Respir Cell Mol Biol 10:38–47
Demuth DG, Gkoumassi E, Droge MJ, Dekkers BG, Esselink HJ, van Ree RM, Parsons ME, Zaagsma J, Molleman A, Nelemans SA (2005) Arachidonic acid mediates non-capacitative calcium entry evoked by CB1-cannabinoid receptor activation in DDT1 MF-2 smooth muscle cells. J Cell Physiol 205:58–67
Dietrich A, Schnitzler M, Emmel J, Kalwa H, Hofmann T, Gudermann T (2003) N-linked protein glycosylation is a major determinant for basal TRPC3 and TRPC6 channel activity. J Biol Chem 278:47842–47852
Felder CC, Joyce KE, Briley EM, Mansouri J, Mackie K, Blond O, Lai Y, Ma AL, Mitchell RL (1995) Comparison of the pharmacology and signal transduction of the human cannabinoid CB1 and CB2 receptors. Mol Pharmacol 48:443–450
Filipeanu CM, de ZD, Nelemans SA (1997) Delta9-tetrahydrocannabinol activates [Ca2+]i increases partly sensitive to capacitative store refilling. Eur J Pharmacol 336:R1–R3
Galiegue S, Mary S, Marchand J, Dussossoy D, Carriere D, Carayon P, Bouaboula M, Shire D, Le FG, Casellas P (1995) Expression of central and peripheral cannabinoid receptors in human immune tissues and leukocyte subpopulations. Eur J Biochem 232:54–61
Gkoumassi E, Dekkers BG, Droge MJ, Elzinga CR, Schmidt M, Meurs H, Zaagsma J, Nelemans SA (2007) Virodhamine and CP55, 940 modulate cAMP production and IL-8 release in human bronchial epithelial cells. Br J Pharmacol 151:1041–1048
Glass M, Felder CC (1997) Concurrent stimulation of cannabinoid CB1 and dopamine D2 receptors augments cAMP accumulation in striatal neurons: evidence for a Gs linkage to the CB1 receptor. J Neurosci 17:5327–5333
Grant AD, Cottrell GS, Amadesi S, Trevisani M, Nicoletti P, Materazzi S, Altier C, Cenac N, Zamponi GW, Bautista-Cruz F, Lopez CB, Joseph EK, Levine JD, Liedtke W, Vanner S, Vergnolle N, Geppetti P, Bunnett NW (2007) Protease-activated receptor 2 sensitizes the transient receptor potential vanilloid 4 ion channel to cause mechanical hyperalgesia in mice. J Physiol 578:715–733
Haller T, Ortmayr J, Friedrich F, Volkl H, Dietl P (1998) Dynamics of surfactant release in alveolar type II cells. Proc Natl Acad Sci U S A 95:1579–1584
Hassock SR, Zhu MX, Trost C, Flockerzi V, Authi KS (2002) Expression and role of TRPC proteins in human platelets: evidence that TRPC6 forms the store-independent calcium entry channel. Blood 100:2801–2811
Ho WS, Hiley CR (2004) Vasorelaxant activities of the putative endocannabinoid virodhamine in rat isolated small mesenteric artery. J Pharm Pharmacol 56:869–875
Hofmann T, Schaefer M, Schultz G, Gudermann T (2000) Cloning, expression and subcellular localization of two novel splice variants of mouse transient receptor potential channel 2. Biochem J 351:115–122
Jia Y, Lee LY (2007) Role of TRPV receptors in respiratory diseases. Biochim Biophys Acta 1772:915–927
Lansley AB, Sanderson MJ, Dirksen ER (1992) Control of the beat cycle of respiratory tract cilia by Ca2+ and cAMP. Am J Physiol 263:L232–L242
Lu G, Henderson D, Liu L, Reinhart PH, Simon SA (2005) TRPV1b, a functional human vanilloid receptor splice variant. Mol Pharmacol 67:1119–1127
Matsuda LA, Lolait SJ, Brownstein MJ, Young AC, Bonner TI (1990) Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature 346:561–564
Munro S, Thomas KL, bu-Shaar M (1993) Molecular characterization of a peripheral receptor for cannabinoids. Nature 365:61–65
Oz M (2006) Receptor-independent actions of cannabinoids on cell membranes: focus on endocannabinoids. Pharmacol Ther 111:114–144
Pertwee RG (2005) Pharmacological actions of cannabinoids. Handb Exp Pharmacol1-51
Porter AC, Sauer JM, Knierman MD, Becker GW, Berna MJ, Bao J, Nomikos GG, Carter P, Bymaster FP, Leese AB, Felder CC (2002) Characterization of a novel endocannabinoid, virodhamine, with antagonist activity at the CB1 receptor. J Pharmacol Exp Ther 301:1020–1024
Rao GK, Kaminski NE (2006a) Cannabinoid-mediated elevation of intracellular calcium: a structure-activity relationship. J Pharmacol Exp Ther 317:820–829
Rao GK, Kaminski NE (2006b) Induction of intracellular calcium elevation by Delta9-tetrahydrocannabinol in T cells involves TRPC1 channels. J Leukoc Biol 79:202–213
Rao GK, Zhang W, Kaminski NE (2004) Cannabinoid receptor-mediated regulation of intracellular calcium by delta(9)-tetrahydrocannabinol in resting T cells. J Leukoc Biol 75:884–892
Sakamoto N, Hayashi S, Gosselink J, Ishii H, Ishimatsu Y, Mukae H, Hogg JC, van Eeden SF (2007) Calcium dependent and independent cytokine synthesis by air pollution particle-exposed human bronchial epithelial cells. Toxicol Appl Pharmacol 225:134–141
Schatz AR, Lee M, Condie RB, Pulaski JT, Kaminski NE (1997) Cannabinoid receptors CB1 and CB2: a characterization of expression and adenylate cyclase modulation within the immune system. Toxicol Appl Pharmacol 142:278–287
Shuttleworth TJ, Thompson JL (1999) Discriminating between capacitative and arachidonate-activated Ca(2+) entry pathways in HEK293 cells. J Biol Chem 274:31174–31178
Sidhaye VK, Schweitzer KS, Caterina MJ, Shimoda L, King LS (2008) Shear stress regulates aquaporin-5 and airway epithelial barrier function. Proc Natl Acad Sci U S A 105:3345–3350
Steffens M, Zentner J, Honegger J, Feuerstein TJ (2005) Binding affinity and agonist activity of putative endogenous cannabinoids at the human neocortical CB1 receptor. Biochem Pharmacol 69:169–178
Sweeney M, McDaniel SS, Platoshyn O, Zhang S, Yu Y, Lapp BR, Zhao Y, Thistlethwaite PA, Yuan JX (2002a) Role of capacitative Ca2+ entry in bronchial contraction and remodeling. J Appl Physiol 92:1594–1602
Sweeney M, Yu Y, Platoshyn O, Zhang S, McDaniel SS, Yuan JX (2002b) Inhibition of endogenous TRP1 decreases capacitative Ca2+ entry and attenuates pulmonary artery smooth muscle cell proliferation. Am J Physiol Lung Cell Mol Physiol 283:L144–L155
Thomas A, Baillie GL, Phillips AM, Razdan RK, Ross RA, Pertwee RG (2007) Cannabidiol displays unexpectedly high potency as an antagonist of CB1 and CB2 receptor agonists in vitro. Br J Pharmacol 150:613–623
Tsou K, Brown S, Sanudo-Pena MC, Mackie K, Walker JM (1998) Immunohistochemical distribution of cannabinoid CB1 receptors in the rat central nervous system. Neuroscience 83:393–411
Van der Zee L, Nelemans A, den HA (1995) Arachidonic acid is functioning as a second messenger in activating the Ca2+ entry process on H1-histaminoceptor stimulation in DDT1 MF-2 cells. Biochem J 305(Pt 3):859–864
White R, Hiley CR (1998) The actions of the cannabinoid receptor antagonist, SR 141716A, in the rat isolated mesenteric artery. Br J Pharmacol 125:689–696
Acknowledgments
Effimia Gkoumassi is a recipient of an Ubbo Emmius Fellowship from the School of Behavioral and Cognitive Neuroscience (BCN), University of Groningen, and Martina Schmidt is a recipient of a Rosalind Franklin Fellowship from the University of Groningen. Bart Dekkers is supported by a grant from the Netherlands Asthma Foundation (03.36). We thank Dr. Alexander Dietrich for the immunoblot detection of TRPC6 in 16HBE14o− cells.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Effimia Gkoumassi and Bart G. J. Dekkers contributed equally to this work.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Gkoumassi, E., Dekkers, B.G.J., Dröge, M.J. et al. (Endo)cannabinoids mediate different Ca2+ entry mechanisms in human bronchial epithelial cells. Naunyn-Schmied Arch Pharmacol 380, 67–77 (2009). https://doi.org/10.1007/s00210-009-0406-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00210-009-0406-z