
Abstract
Human hypertension affects affects more than 20% of the adult population in industrialized countries, and it is implicated in millions of deaths worldwide each year from stroke, heart failure and ischemic heart disease. Available evidence suggests a major genetic impact on blood pressure regulation. Studies in monogenic hypertension revealed that renal salt and volume regulation systems are predominantly involved in the genesis of these disorders. Mutations here affect the synthesis of mineralocorticoids, the function of the mineralocorticoid receptor, epithelial sodium channels and their regulation by a new class of kinases, termed WNK kinases. It has been learned from monogenic hypotension that almost all ion transporters involved in the renal uptake of Na+ have a major impact on blood pressure regulation. For essential hypertension as a complex disease, many candidate genes have been analysed. These include components of the renin–angiotensin-aldosterone system, adducin, β-adrenoceptors, G protein subunits, regulators of G protein signalling (RGS) proteins, Rho kinases and G protein receptor kinases. At present, the individual impact of common polymorphisms in these genes on the observed blood pressure variation, on risk for stroke and as predictors of antihypertensive responses remains small and clinically irrelevant. Nevertheless, these studies have greatly augmented our knowledge on the regulation of renal functions, cellular signal transduction and the integration of both. Together, this provides the basis for the identification of novel drug targets and, hopefully, innovative antihypertensive drugs.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Before reviewing the genetics of hypertension we have to start with some facts on ‘hypertension’ itself. According to our current understanding, blood pressure is a highly regulated quantity, affected by a multitude of physiological systems that finally integrate and maintain blood pressure levels to secure an adequate blood perfusion of all tissues despite widely varying metabolic demands. Blood pressure values are distributed continuously in a unimodal fashion skewed to the ends of the distribution curves. Given this continuous distribution of blood pressure levels, the classification of ‘hypertension’ as a discrete entity requires the operational definition of blood pressure thresholds above which cardiovascular risk increases or therapeutic interventions are of clinical benefit. Within the last 2 decades, classification thresholds have gradually been reduced owing to improved therapeutic options and refined epidemiologic analyses. Accordingly, ‘hypertension’ prevalences may ultimately comprise a considerable part and eventually the majority of the adult population in industrialized and aging societies in the near future, for instance in Western Europe. Living long enough (and perhaps, becoming fat enough), many of us will develop hypertension, a situation similar to diseases like Alzheimer’s. Hence, we have to understand the pathogenesis of hypertension as a dynamic process potentially beginning in early childhood. On the other hand, some octogenarians or even centenarians still have normal or low blood pressure values. This lends interest to ‘hypotensive’ mechanisms (potentially genes) in addition to the delineation of their hypertensive counterparts.
Second, available evidence suggests that genetic mechanisms contribute to blood pressure regulation. There is a substantial correlation between blood pressure values of parents and children, documenting that the similarity of blood pressure values within families is greater than between families (Longini et al. 1984). Observational studies with monozygotic twins demonstrate greater concordance than with dizygotic twins (Feinleib et al. 1977). Adoption studies further indicate that familial aggregation of blood pressure levels is not simply attributable to shared environment, since biological siblings exhibit a higher concordance than adoptive siblings (Biron et al. 1976; Rice et al. 1989). An analogous situation is the observed variation in body height which is governed by hereditary (including gender as a hereditary factor) and environmental factors (differing between ethnicities). Likewise, environmental and nutritional factors, as well as different lifestyles, contribute to the pathogenesis of primary hypertension as important modifiable risk factors. Guesses (more or less educated) suggest that 30–60% of the observed variation in blood pressure is attributable to genetic factors with five or ten or more (....and even more, if we refer to negative results from recent genome-wide screens in affected families) different genes, each with only a limited impact. Environment (nurture) contributes to the remaining half in blood pressure variation. Together, we face a complex interplay of different blood pressure raising and lowering genes, many with pleiotropic effects, in concert with numerous exogenous factors. In addition, primary hypertension is tightly entangled with the complexities of other disorders including the metabolic syndrome, diabetes type II, preeclampsia or the progress of renal diseases.
Here, we will review the current knowledge on the genetics of hypertension. An innocent search in the Medline database for ‘hypertension and genetics’ will result in a tsunami of many thousands of references, addressing putative connections between the genesis of hypertension and apparently all bodily functions and biological systems. For reasons of space and the scope of a pharmacological journal, we will focus here primarily on hypertension candidate genes that have been characterised in terms of a potential function. Thus, we will skip most of the evidence from pure association or linkage studies, pertaining to anonymous loci. Furthermore, we will concentrate on genetic studies that were confirmed in independent human samples, and animal models of genetic hypertension are only presented if their results show close parallels to the situation in humans.
Citing the sometimes poetic ‘wisdom’ of a former US Secretary of Defence on strategy, there are ‘known knowns’, ‘unknown knowns’, ‘known unknowns’ and ‘unknown unknowns’. At present, genetics of hypertension belong to the ‘known unknowns’ and with ongoing scientific progress we will most likely face many surprising ‘unknown unknowns’. This review, however, addresses ‘known knowns’ which may belong to the ‘unknown knowns’ for some readers. Mendelian forms of hypertension are the most important ‘known knowns’ in the field of hypertension genetics, since they are paradigmatic examples for the pathophysiology of systems primarily involved in blood pressure control. Most rewarding, we can trace the pathomechanisms here from variations in genomic DNA to the phenotype ‘increased blood pressure’.
Monogenic or Mendelian forms of hypertension
Monogenic or Mendelian forms of hypertension are extremely rare disorders. The best described forms are autosomal diseases that are accompanied by a distinct phenotype in addition to elevated blood pressure. Insightful clinical observations allowed for generating pedigrees, performing classical linkage analyses, identifying disease loci and, ultimately, the unravelling of genetic ‘defects’, which are believed to be indicative for similar abnormalities in primary hypertension.
We have included in this review monogenic forms of hypertension syndroms for which hypertension is the leading symptom. Of course, there exist other complex genetic syndromes that are accompanied by a rise in blood pressure, e.g. familial pheochromocytoma, which are out of the scope of this survey. Interestingly, most mutations identified so far directly affect renal Na+ homoeostasis. Increased renal salt absorption is accompanied by increased water reabsorption, resulting in an augmented intravascular volume. In turn, the preload—i.e., the venous blood return to the heart—increases, raising cardiac output and blood pressure. If this situation continues, regulatory processes are activated that also contribute to the maintenance of hypertension.
Glucocorticoid-remediable aldosteronism (GRA)
GRA is a rare autosomal dominant early onset form of hypertension characterised by volume expansion, moderate metabolic alkalosis and hypokalemia (not in all cases) accompanied by low plasma renin activity similar to the situation in primary hyperaldosteronism despite normal or decreased aldosterone levels (Sutherland et al. 1966). In fact, treatment with aldosterone antagonists or other potassium-sparing diuretics attenuates the symptoms. Crucial for the further exploration of the disease was the discovery of large amounts of 18-hydroxycortisol and 18-oxocortisol, otherwise uncommon steroids in the urine that exhibit mineralocorticoid activity (Fig. 1). Hallmark of the disease is the observation that treatment with glucocorticoids, e.g. prednisone, blocks the production of the abnormal steroids and lowers blood pressure (Sutherland et al. 1966). Using the anomalous generation of steroids as an intermediate phenotype in a large pedigree, Lifton and colleagues linked this syndrome to a locus on chromosome 8q (Lifton et al. 1992a,b; Pascoe et al. 1992a). Two closely related genes for adrenal steroid biosynthesis are confined to this locus in a tandem arrangement, 11β-hydroxylase (CYP11B1) and aldosterone synthase (CYP11B2) (Fig. 2a). While CYP11B2 activity is the rate-limiting step in aldosterone biosynthesis in adrenal glomerulosa regulated by angiotensin II (AngII), CYP11B1 is essential for cortisol biosynthesis in adrenal fasciculata under tight transcriptional control of ACTH. Unequal meiotic crossing-over happens at that locus as rare events favoured by the high sequence homologies (95%) of both genes. This gives rise to a chimeric gene that harbours the promoter- and regulatory region of 11β-hydroxylase synthase, followed by the structural part of aldosterone synthase resulting in an ensemble where the original CYP11B1 and CYP11B2 genes flank the described additional chimeric gene.

Structure of the glucocorticoids cortisol and cortisone and the abnormal steroids 18-hydroxycortisol and 18-oxocortisol observed in glucocorticoid-remediable aldosteronism. Indicated is the site of conversion from cortisol to cortisone mediated by 11β hydroxysteroid dehydrogenase. Conversion to cortisone in mineralocorticoid-responsive tissues prevents cortisol from binding to the mineralocorticoid receptor. Loss of function mutations in 11β hydroxysteroid dehydrogenase causes apparent mineralocorticoid excess

a Unequal crossing over of aldosterone synthase and 11β hydroxylase genes, resulting in a hybrid gene that brings aldosterone synthase activity under control of 11β hydroxylase which results in aberrant expression of mineralocorticoid synthesis in the zona fasciculata under control of ACTH (b)
Now under control of the 11β hydroxylase promoter aldosterone synthase, activity of the chimeric gene is ectopically expressed in the fasciculata of the adrenal gland, where it mediates together with CYP17 (17-α hydroxylase) the synthesis of abnormal 18-hydroxy- and 18-oxycortisol from glucocorticoid precursors. In contrast to the wildtype gene, expression of the chimeric aldosterone synthase gene is no longer regulated by angiotensin II but becomes sensitive to ACTH, which links mineralocorticoid secretion inappropriately to glucocorticoid secretion (Fig. 2b). 18-oxo- and 18-hydroxycortisol bind to the mineralocorticoid receptor (MR) in the distal tubule and cortical collecting duct, and stimulate Na+ absorption coupled to H+ and K+ secretion. Ultimately, this causes plasma volume expansion and a decrease in plasma renin activity with normal or low aldosterone levels. Suppression of ACTH by exogenous glucocorticoids attenuates abnormal steroidogenesis and ameliorates hypertension (Lifton et al. 1992a,b). Women with GRA exhibit an increased risk for preeclampsia (Wyckoff et al. 2000). Following the initial characterization of the molecular mechanisms in GRA, similar unequal crossing over mutations have been identified in other families. In all cases, the breakpoints are located 5′ of intron 4. There is considerable genetic heterogeneity in carriers of the GRA hybrid gene, ranging from normotension, via mild (indistinguishable from primary hypertension) to severe hypertension. This heterogeneity may be attributable in part to different breakpoints, but other genetic and environmental factors appear to play a major role.
Apparent mineralocortiocid excess (AME)
The MR is a nuclear hormone receptor expressed in principal cells of the distal nephron that controls gene transcription events which ultimately result in increased salt reabsorption. One paradigmatic ‘unknown unknown’ in pharmacology was the discovery that the cloned MR binds cortisol and aldosterone with equal affinities, potentially pertaining to the considerable homology with the glucocorticoid receptor (Fig. 4; Ariza et al. 1987). Since circulating cortisol concentrations in plasma are several orders of magnitudes higher than those of aldosterone, it was puzzling how a specific mineralocorticoid effect could arise. The now well-known ‘known’ solution to this question was the identification of 11β hydroxysteroid dehydrogenase type 2 (HSD11B2; cortisol 11-beta-reductase), the expression of which is confined to aldosterone-sensitive tissues including the distal tubule and the cortical collecting duct. HSD11B2 converts cortisol to cortisone that has apparently no relevant affinity for the MR (Fig. 3; Funder et al. 1987). Inhibition of HSD11B2 by carbenoxolone or glycyrrhetinic acid (upon consumption of large amounts of licorice) results in the unregulated occupation of MR with cortisol and, ultimately, in sodium retention, volume expansion and increases in blood pressure, accompanied by low renin activity, low aldosterone concentrations and in part a metabolic alkalosis (Stewart et al. 1987).

Pathogenesis of apparent mineralocorticoid excess (AME). The mineralocorticoid receptor (MR; indicated in the lower panel) binds aldosterone and cortisol with similar affinities, which would result in a permanent mineralocorticoid response given the higher circulating cortisol levels. Cortisone, however, does not interact with the MR. 11β-hydroxysteroid-dehydrogenase is expressed in mineralocorticoid-responsive tissues, as shown here for the cortical collecting duct, and converts cortisol into cortisone, thus protecting the MR. Loss-of-function mutations of the 11β-dehydrogenase result in an uncontrolled stimulation of the MR. Depicted are target mechanisms of the mineralocorticoid response, including enhanced expression of Na+/K+ ATPase and epithelial sodium channel molecules
Apparent mineralocorticoid excess was described as a rare autosomal recessive syndrome that comprises severe childhood hypertension, strokes, volume expansion, hypokalemia, and metabolic alkalosis, all symptoms responsive to aldosterone antagonists, despite the virtual absence of aldosterone in blood plasma (New et al. 1977). Based on the obvious similarities with the phenotype of ‘licorice intoxication’ (phenocopy), Mune and co-workers followed a candidate gene approach, and identified homozygous loss-of-function mutations (Arg208Cys; Arg213Cys) in the renal isoform of HSD11B2 in eight of nine families with AME (Mune et al. 1995). Similarly to blocking with glycyrrhetinic acid, the mutated HSD11B2 fails to inactivate cortisol, leaving the MR unprotected. Subsequently, other inactivating mutations in HSD11B2 were identified in different pedigrees (Arg208His; Asp223Asn; Arg279Cys; Arg337Cys; Arg337His; Tyr299del) which explain in part the observed genetic heterogeneity (OMIM, Online-Medelian Inheritance in Man; http://www.ncbi.nlm.nih.gov; accessed November 2006). There is evidence that heterozygous carriers of such mutations may develop late-onset AME (Li et al. 1997). Since AME is accompanied with low renin activity, and 40% of patients with primary hypertension demonstrate low renin levels, speculation was raised that mutations in the HSD11B2 gene on chromosome 16q with moderately decreased enzymatic activity could contribute to the pathogenesis of (salt-sensitive) hypertension and preeclampsia. In two different (inbred) families, HSD11B2 mutations were identified that cause mild low renin hypertension (Wilson et al. 1998; Pro227Leu; Leu114del-Glu115del); however, a relevance of HSD11B2 mutations for primary hypertension remains to be established.
Additional inborn errors in glucocorticoid metabolism, including deficiencies in 11β-hydroxylase or 17α-hydroxylase, result in an inappropriate accumulation of deoxycorticosterone and corticosterone, which are potent mineralocorticoids and cause hypertension similar to AME, but are out of the scope of this review (Kagimoto et al. 1988; White et al. 1991).
Mineralocorticoid receptor mutations and hypertension exacerbated in pregnancy
A syndrome similar to AME with autosomal dominant early-onset hypertension arises from activating mutations in the mineralocorticoid receptor itself. An amino acid substitution (Ser810Leu) in the steroid-binding domain has been identified that results in a constitutively active MR which is further activatable by aldosterone (Geller et al. 2000). In addition, the 810Leu mutation in the steroid binding region alters the receptor specificity, with progesterone becoming a potent agonist (Fig. 4). During pregnancy, when progesterone levels rise a hundredfold, high blood pressure crises occur in carriers of the mutation. Whether similar mechanisms contribute to other pregnancy-associated hypertensive states, including preeclampsia, remains to be investigated. Moreover, the activated conformation of the 810Leu mutant MR receptor is further stabilized by spironolactone, switching this MR blocker to a potent agonist. The 810Leu mutation in the MR gene on chromosome 4q in a heterozygous state suffices for severe early-onset hypertension (Geller et al. 2000).

Upper panel: structures of the mineralocorticoid receptor (MR) and the closely related glucocorticoid receptor, both containing a DNA-binding domain with two zinc fingers followed by a steroid binding domain. The activating mutation Ser810Leu in the MR, causing ‘early-onset autosomal hypertension with exacerbation in pregnancy’, is indicated by an arrow. The lower panel depicts steroids that act as agonists on the 810Leu MR mutant. Positions of known loss-of-function mutations (asterisks, frame shift; squares, premature stop codons; triangle, missense) are indicated on top of the MR scheme. These mutations cause the syndrome of autosomal dominant pseudohypoaldosteronism type I that includes hypotension
Liddle’s syndrome
Liddle’s syndrome describes a rare autosomal dominant disorder that comprises early-onset hypertension, frequently accompanied by metabolic alkalosis and hypokalemia, suppressed plasma renin and aldosterone levels (Liddle et al. 1963). In contrast to AME and GRA, Liddle’s syndrome does not respond to treatment with aldosterone antagonists, but is responsive to thiazides, amiloride and triamterene. A case-report demonstrating that a patient with Liddle’s syndrome and renal insufficiency was cured upon renal transplantation indicated that the cause for this disease resides in the kidneys (Botero-Velez et al. 1994). Subsequent linkage analysis based on pedigrees with Liddle’s syndrome detected a locus on chromosome 16 near the position of the β subunit (gene: SCNN1B) of the epithelial Na+ channel (ENaC; Shimkets et al. 1994; Hannson et al. 1995a; Inoue et al. 1998) followed by similar observations in the ENaC γ subunit gene, also on chromosome 16 (gene: SCNN1G; Hannson et al. 1995b), indicating the molecular heterogeneity of the syndrome.
ENaCs consist of three different subunits termed α, β, and γ (Fig. 5). While the α-subunit has been suggested to be the sole unit that supports Na+ transport, β- and γ-subunits do not mediate Na+ channel activity but greatly augment the conductance of the α-subunit upon co-expression. All subunits contain 2 transmembrane spanning domains and share ∼35% amino acid identity. ENaCs are expressed in kidney, pancreas, small intestine, colon, testes, lung and sweat glands. Renal ENaC expression is predominantly confined to the cortical collecting duct, where approximately 2% of the filtered Na+ is reabsorbed in a mineralocortioid-responsive fashion. Crucial for a regulated action of ENaC is the controlled insertion into the plasma membrane and its controlled internalization and degradation. In the absence of mineralocorticoids, ENaC is highly ubiquitinylated by a specific ubiquitin ligase termed Nedd4-2. Ubiquitinylated ENaC is degraded by either the proteasomal or lysosomal pathway. In the presence of aldosterone, SGK1 (sodium and glucocorticoid-induced kinase) expression increases, which results in an enhanced phosphorylation of Nedd4-2 that inhibits ubiquitinylation of ENaC and increases surface ENaC expression. Changes in intracellular Na+ concentrations and variations in cAMP levels also affect ENaC surface expression (Fig. 6; Staub et al. 1996; Kamynina et al. 2001).

Proposed topology of the three ENaC subunits encoded by the genes SCNN1A, SCNN1B and SCNN1G. Indicated is the location of the canonical PPPXY motif, essential for interaction with the WW domain of Nedd4-2. Premature stop codons, frame-shift or missense mutations result in the deletion of the C-terminus, or alter the PPPXY motif. In these cases, the interaction with Nedd4-2 is prevented, giving rise to permanent insertion of the channel into the tubular cell membrane, the molecular mechanism for Liddle’s syndrome. On the contrary, inactivating mutations of ENaC channel subunits diminish sodium reabsorption and cause pseudohypoaldosteronism type I, which is associated with hypotension

Molecular pathomechanism of Liddle’s syndrome. Upper panel: The epithelial sodium channels (ENaC) consists of two α-, one β- and one γ-subunits. Each subunit contains a C terminal PPPXY motif for interaction with the ubiquitin ligase Nedd4-2. Nedd4 proteins consist of a C2 Ca2+-binding, 4 WW domains and a HECT domain, i.e., a canonical domain responsible for ubiquitin ligase activity. Nedd4-2 binds with the third and fourth WW domain to the PPPXY motifs of ENaC subunits, which results in ubiquitinylation and internalization. Lower panel: In Liddle’s syndrome, ENaC subunits are expressed that miss the PPPXY motif, or have an altered motif that prevents Nedd4-2 from binding, resulting in an increased fractional expression of ENaCs. Phosphorylation of Nedd4-2 by the kinase SGK1 results in a decreased affinity of Nedd4-2 for ENaC, which is one mechanism for translation of aldosterone signals to a cellular response
Liddle’s syndrome mutations affect the cytoplasmic tail of ENaC subunits, altering or deleting a crucial PPPXY motif essential for interaction with WW domains of Nedd4-2 (and a homologue Nedd4-1) and degradation as well as for internalization via clathrin-coated pits (Schild et al. 1995). The mutated ENaC channel subunits in Liddle’s syndrome are no longer recycled, and the fraction at the luminal cell surface increases (Fig. 6). The ensuing constant inappropriate Na+ reabsorption causes volume expansion, even under conditions of high salt intake, and ultimately hypertension (Synder et al. 1995; Shimkets et al. 1997). A considerable genetic heterogeneity between different mutations and pedigrees has been observed. Since amiloride and triamteren specifically block ENaC channels, they are key treatment options to ameliorate symptoms in Liddle’s syndrome (Palmer and Alpern 1998).
As with the other forms of monogenic hypertension, it has been postulated that polymorphisms in ENaC genes could contribute to the pathogenesis of primary hypertension. The α-subunit (gene: SCNN1A) is located on the short arm of chromosome 12, a locus that has been linked to hypertension (Nagy et al. 1999; Disse-Nicodeme et al. 2000). Several common polymorphisms were identified in SCNN1A, among them a promoter polymorphism (G2139A) which was in close linkage disequilibrium to a 5′ UTR polymorphism in exon 1 in a large Japanese cohort (Iwai et al. 2002). The 2139G allele occurred in a frequency of ∼60%, and was associated with a higher SCNN1A promoter activity and hypertension, whereas the A allele appeared to protect against hypertension. While of potential interest, these results remain unconfirmed so far. For the SCNN1B gene a–comparably rare–T594M polymorphism located in the C terminus but unrelated to the crucial PPPXY motif has been identified that was more frequent in hypertensive (∼8% heterozygous carriers) than in normotensive (∼2%) blacks (Baker et al. 1998), results that were not reproduced in another black cohort (Nkeh et al. 2003; Hollier et al. 2006). A similar approach for the SCNN1G subunit resulted in the identification of a promoter polymorphism (G-173A) and a silent polymorphism in exon 3 with allele frequencies of ∼25% each (Persu et al. 1999; Iwai et al. 2001). The -173G allele was associated with a higher promoter activity and an increased risk for hypertension in the Japanese but not in the European sample. Interestingly, homozygous carriers of the -173A allele (occurring in only 0.7% of the population) had a higher prevalence of hypotension, and the mean systolic blood pressure was 11 mmHg lower, suggesting a function as a ‘hypotensive or hypertension protection gene’ here.
Independent genome-wide linkage analyses have identified a locus on chromosome 18q in primary hypertension. The Nedd4-2 gene localizes to chromosome 18q21-22. The gene extends over more than 400 kb and consists of at least 40 exons, which complicates genetic studies. So far, no linkage between blood pressure phenotypes and Nedd4-2 has been reported.
Gordon’s syndrome (pseudohypoaldosteronism type II)
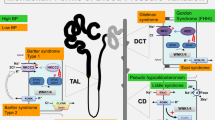
Patients with Gordon’s syndrome, an autosomal dominant disorder, exhibit hypertension associated with hyperkalemia, discrete hyperchloremic metabolic acidosis, normal glomerular filtration rate and low renin activity, which is susceptible to treatment with thiazide diuretics (Gordon et al. 1970) Stroke risk is increased in such families. By linkage analyses in different affected pedigrees loci on chromosomes 1q (PHA2A), 17p (PHA2B) and 12p13 (PHA2C) were identified (Mansfield et al. 1997; Disse-Nicodeme et al. 2000) which explains the marked genetic heterogeneity of the disorder. A 41-kb deletion on chromosome 12 in the linked region in affected family members helped to identify the culprit gene, WNK1 (PRKWNK1 = protein kinase WNK1) and subsequently WNK4 on chromosome 17 (PRKWNK4; Wilson et al. 2001). WNK refers to the acronym ‘with no lysine’ (one-letter abbreviation of lysine is ‘K’) acknowledging the fact that these serine–threonine kinases have no canonical lysine, critical for catalysis, in the beta strand 3 of the kinase domain. Instead, this lysine is present in beta strand 2. This kinase domain is located at the N-terminus, followed by an autoinhibitory domain and coiled-coil domains (Fig. 7). In addition, multiple SH3 domains, proline-rich segments and nuclear localisation signals were identified.

Upper panel: Gene structures of WNK1 and WNK4. An extended deletion in intron 1 of WNK1 causes an increased transcript expression. Lower panel: Conserved domains in WNK1 and WNK4
WNK1 and WNK4 localize specifically to the distal convoluted tubule and cortical collecting duct of the nephron. WNK1 is confined solely to the cytoplasm, whereas WNK4 localizes to tight junctions and to the cytoplasm (Wilson et al. 2001). Both kinases regulate the function of the thiazide-sensitive Na+/Cl− transporter (NCC; NCCT; SLC12A3) and other ion tranport proteins including the ROMK (renal outer medullary) K+ channel (=Kir1.1; KCNJ1) and epithelial Ca2+ channels (TRPV4, 5). Furthermore, WNK4 may also regulate the function of two kinases (SPAK, OSR) that in turn regulate renal cation-Cl− cotransporters (NKCC1, KCC2). WNK1 and WNK4 also phosphorylate synaptotagmin, a protein involved in membrane-trafficking (Bindels 2003; Peng and Bell 2006; Subramanya et al. 2006). WNK4 expressed in tight junctions phosphorylates claudin, a protein that controls paracellular Cl− fluxes. In addition to their specific functions in the kidney, WNKs expressed in other tissues have been implicated in the regulation of cellular growth, organogenesis and insulin signalling (Fig. 8).

a Ion transport regulation by WNK1 and WNK4. There is evidence that WNK4 (including its PHAII mutations) interacts with NCC with its C-terminal part (downstream of the PHAII mutation) and reduces the surface expression of NCC by stimulating lysosomal degradation. WNK4 facilitates the retrieval of ROMK from the plasma membrane by a process that is independent of its kinase activity. Nevertheless, expression of PHAII-WNK4 mutants causes an increased downregulation of ROMK activity. In addition to its intracellular expression, WNK4 has also been detected in the paracellular space, where it stimulates paracellular chloride fluxes by the phosphorylation of claudins. WNK1 acts upstream of WNK4, and WNK1-mediated phosphorylation inhibits WNK4 activity/function. b PHAII-causing mutations in WNK4 are loss-of-function mutations with respect to NCC inhibition. They cause an increased internalization of ROMK and enhanced paracellular Ca2+ fluxes, both per definition gain-of-function mutations, illustrating the complexity of WNK signalling. c The known gain-of-function mutation (deletion of intronic sequences) in the WNK1 gene results in a fivefold increased expression of WNK1, which in turn causes an increased inhibition of WNK4. It should be noted that these are preliminary schemes of an incompletely understood system, especially concerning the effects of WNK1 overexpression on ROMK and paracellular Cl− fluxes in PHA2C. For simplicity, NCC and ROMK are shown in the same cell
Two splice variants of WNK1 have been identified that are controlled by different promoters, a full-length kinase-sufficient WNK1 trancript (L-WNK1), and a shorter isoform that lacks a functional kinase domain (KS-WNK1), which appears to be confined to the aldosterone-sensitive distal nephrone. L-WNK1 is involved in response to osmotic stressors, including hyper- and hypotonicity, while KS-WNK1 appears to regulate processes in aldosterone signalling. Activation of WNK4 is associated with a decreased incorporation of NCC into the membrane. In contrast, activation of WNK1 inhibits the WNK4-mediated suppression of NCC transporter translocalization (Fig. 8; Yang et al. 2003; Wilson et al. 2003) and requires kinase activity of WNK1.
The mutations found in the pedigrees described above cause either a large intronic deletion in the WNK1 gene that is associated with a fivefold increase in the expression of the L-WNK1 protein, or missense mutations in WNK4 (Wilson et al. 2001; Fig. 7). At first glance, gain-of-function by overexpression of L-WNK1 or loss-of-function of WNK4 both result in an increased membranal expression of NCC and an increased Na+ reabsorption in the distal tubule that causes hypertension. Inhibiting the NCC by thiazides is an effective remedy in this situation. The hyperkalemic metabolic acidosis is explained by the fact that less Na+ reaches the distal tubule / collecting duct where the concerted actions of ENaCs and ROMKs constitute a functional Na+ / K+ exchange. If less Na+ is reabsorbed, less K+ and H+ are lost to the urine, resulting in hyperkalemia and metabolic acidosis. A decreased insertion of ROMK into the membrane associated with mutated WNK4 further explains the observed hyperkalemia. On a molecular level, however, we have not yet understood the function of the WNK4 mutations for PHA2. Three of four identified PHA2B-causing WNK4 mutations are charge-changing substitutions (Glu562Lys, Asp564Ala; Gln565Glu) in an amino-acid motif localized directly adjacent to the first coiled-coil domain that is highly conserved in all members of the WNK family. An Arg1185Cys exchange also causes PHA2B. The affected Arg is located just distal of the second coiled-coil domain, and is also highly conserved in all WNK family members (Fig. 7; Wilson et al. 2001). Interestingly, all mutations are remote from the kinase domain. WNK4 kinase activity, however, is required for inhibition of NCC. Hence, the mechanism for the WNK4-mediated decrease in NCC surface expression is not understood. Current evidence suggests that it does not involve trafficking from ER to Golgi or clathrin-dependent internalization. The WNK4-mediated decrease in ROMK expression is mechanistically different since it does not require WNK4 kinase activity, and affects internalization via clathrin-coated pits. PHA2-causing WNK4 mutations cause greater (!) inhibition of ROMK than wild-type WNK4 itself. Available hypotheses view WNK4 (and the isoforms of WNK1) as partners of a multiprotein complex that involves the homo- and heteromultimers of WNKs that exert scaffolding and kinase functions on downstream target proteins. Mice bearing a transgene with the human PHA2-mutant WNK4 develop hypertension, hyperkalemia and hypercalciuria, with dramatic structural changes of the distal convoluted tubule, including hyperplasia and massive expression of NCC (Lalioti et al. 2006). This group suggested that PHA2B-causing mutations in negatively charged domains of WNK4 affect the ability to sense intracellular Ca2+ levels, e.g. in response to AngII, that are required for a functional fine-tuning of WNK4 and ultimately NCC activity. Given the heterogeneity of PHA2, the fact that the culprit gene for PHA2A on chromosome 1 has not been identified, and that PHA2 families are known with most likely mutations in yet other proteins, there is a good chance that additional players in this system must be identified for its full comprehension.
Changing from these molecular considerations to a systemic view of renal WNK-signalling, it appears that WNK1 and WNK4 are important switches for the control of renal sodium and potassium excretion. Adrenal aldosterone release is stimulated either by volume contraction or hyperkalemia, situations which require different adaptations (sodium and water uptake vs sodium uptake coupled to potassium excretion) of the kidneys despite an identical signal. WNK4 activity could be the switch for this adaptation (Lalioti et al. 2006).
In a genome-wide linkage analysis in the Framingham-Heart-Study population, a segment of chromosome 17 was identified that links to hypertension and contains the WNK4 gene (Levy et al. 2000). A G/A polymorphism in intron 10 of the WNK4 gene was identified, and found to occur in 13% vs 7% in white hypertensives and normotensives, a result that was not confirmed in later studies (Erlich et al. 2003; Speirs and Morris 2004; Tobin et al. 2005). In a hypertensive Japanese cohort, three novel missense mutations in the coding sequence of WNK4 were identified, but occurred altogether in only 5 of 956 patients (Kamide et al. 2004). For the WNK1 gene, a British survey with 712 severely hypertensive families detected no association of presumed WNK1 haplotypes with the risk for hypertension, but a significant association with a potential promoter polymorphism (rs1468326; minor allele frequency ∼10%) and the severity of hypertension (Newhouse et al. 2005). A population-based family study with 996 subjects from 250 white European families identified 9 SNPs in WNK1, and reported that five SNPs or their combinations and potential haplotypes were associated with blood-pressure parameters from 24-hr blood pressure recordings. These polymorphisms were confined to intron 1 (rs2369402, rs765250), intron 10 (rs880054), intron 22 (rs953361), intron 23 (rs2301880) and intron 26 (rs2286028), and the minor allele frequencies ranged from 10–40%. It should be noted that the above rs1468326 SNP was not associated with hypertension in this survey (Tobin et al. 2005). In conclusion, the identification of WNK1 and WNK4 5 years ago has sparked a whole new research in the fine regulation of renal Na+ and K+ handling, and has expanded our understanding of kidney physiology. Further studies are required to unravel potential contributions to the genetics of essential hypertension.
Autosomal dominant hypertension with brachydactyly
While the monogenic forms of hypertension presented so far affect renal Na+ and volume control, either by affecting Na+ transporting systems of by interfering with signal transduction systems or the function of the mineralocorticoid receptor, autosomal dominant hypertension with brachydactyly is different. This syndrome was described in a large Turkish pedigree, and includes severe hypertension and strokes in early life as well as brachydactyly (shortened fingers) and short stature (Bilginturan et al. 1973). Luft and co-workers meticulously characterized these patients. Parameters of kidney function (renin, angiotensin II, aldosterone levels), the activity of the autonomous system (catecholamine responses) are in the normal range (Schuster et al. 1996a), and the patients exhibit no characteristic susceptibility towards a specific class of five tested classes of antihypertensives (Schuster et al. 1998).
With respect to an initial finding of a looping vessel in cerebral arteriography in one patient, cranial MRI suggested that 15 affected family members had signs of neurovascular compression, in contrast to 12 non-affected relatives. In the affected subjects, unilateral or bilateral loops of the vertebral artery or the posterior inferior cerebellar arteries were detected (Naraghi et al. 1997). Various neurosurgical, neuroanatomical and neuroradiological studies document that neurovascular abnormalities in the posterior fossa of the skull can result in compression of the ventrolateral medulla near the entry zones of the cranial nerves IX and X. A postulated hyperactivation of medullary autonomus structures may ultimately cause hypertension (Janetta et al. 1985; Naraghi et al. 1994).
Based on a hypothesis of autonomous hyperactivity, in-depth analyses of vegetative functions were conducted in affected patients with brachydactyly and hypertension. Complete ganglionic blockade did not reduce hypertensive blood pressure values in young affected family members, suggesting that autonomic activity is not responsible for increases in basal blood pressure (Jordan et al. 2000). Likewise, muscle sympathetic nerve activity during sympathetic stimulation was not different or even lower at rest and upon stress testing in affected subjects, arguing against an increased sympathetic nerve activity. Sympathetic stimuli including cold pressure, handgrip testing and upright posture, however, caused excessive increases in blood pressure in affected subjects. Similarly, the potency of the non-selective α-adrenoceptor-agonist phenylephrine to increase blood pressure was more than an order-of-magnitude higher in the affected family members. Since the relative sensitivity to phenylephrine was distinctly reduced in these patients during ganglionic blockade—which also blocks the baroreceptor reflex—the authors concluded that an impaired ability of the baroreceptor reflex to compensate increases in blood pressure is a crucial abnormality in this type of monogenic hypertension (Jordan et al. 2000).
Genome-wide linkage analysis in this family, and observations from a chromosomal deletion syndrome in a Japanese child, confined the locus of this syndrome to chromosome 12p (Schuster et al. 1996b; Bähring et al. 1997; Toka et al. 1998). Interestingly, one type of Gordon’s syndrome (Disse-Nicodeme et al. 2000), the gene of the epithelial sodium channel SCNN1A and the G protein β3 subunit—implicated in the genesis of essential hypertension (Siffert et al. 1998)—are also confined to this locus, although fine mapping indicates separate disease entities or loci. For essential hypertension, one family study with dizygotic twins also identified this locus, beside others, to be linked to hypertension (Fig. 9; Nagy et al. 1999). Screens for candidate genes in this region that could affect brachydacytly, vessel looping and hypertension in the Turkish kindred included the parathyroid hormone-related peptide (PTHrP) and the L-SOX5 transcription factor for collagen I synthesis, but these screens have been unsuccessful to date (Fig. 9).

Localization of gene loci implicated in hypertension genetics, including the gene for WNK1, the ENaC channel subunit SCNN1A, the G protein β3 subunit, and the locus implicated in hypertension with brachydactyly (marker D12S1682) and the potential candidate genes phospodiesterase 3A (PDEA3), the ATP-dependent potassium channel Kir6.1 (KCNJ8), and the regulator of the sulfonyl urea receptor SUR2 (ABCC9) at that locus
Another total genome analysis in pedigrees with familial hypertension from a Chinese geographic isolate revealed a significant linkage of the hypertensive trait (without brachydacytly) again to chromosome 12p (Gong et al. 2003), indicating that brachydactyly and hypertension are most likely caused by different genes on chromosome 12p, obviously co-localizing in the Turkish kindred. Subsequent high-resolution cytogenetic analyses of the Turkish kindred revealed a 3.5 Mbp intrachromosomal rearrangement on chromosome 12p that included among uncharacterized genes the genes for the ATP-dependent potassium channel Kir6.1 (KCNJ8), the regulator of the sulfonyl urea receptor SUR2 (ABCC9) and the phosphodiesterase PDE3A as reasonable targets for blood pressure increases. Gene expression studies and pharmacological in vivo characterizations of these candidate genes, however, revealed no consistent differences between affected and non-affected family members (Fig. 9; Bähring et al. 2004).
Monogenic hypotension
What we measure as blood pressure is a highly regulated value, the final integration of blood pressure elevating and lowering effects. To comprehend genetic mechanisms causing hypertension, it is rewarding to face also the ‘hypotensive arm’ of blood pressure control. Having understood the known ‘knowns’ of monogenic hypertension, it is not surprising that mutations with opposite cellular effects in such genes (e.g. loss-of-function instead of gain-of-function) finally cause monogenic hypotension.
Defective mineralocorticoid action and pseudohypoaldosteronism type I
Mineralocorticoid-stimulated Na+ reuptake is critical for volume control, and we have learned that increased mineralocorticoid activity causes volume expansion and hypertension, as shown for GRA or AME. As a mirror image, patients with inborn errors in the biosynthesis of aldosterone present with salt wasting, severe hypotension, and a decreased distal tubular K+ and H+ secretion. Well-characterised syndromes include homozygous loss of aldosterone synthase activity (the actual mirror image of GRA) or 21-hydroxylase activity (Mitsuuchi et al. 1992; Pascoe et al. 1992b; Bongiovanni and Root 1963; Armor et al. 1988).
Similar syndromes arise if the mineralocorticoid receptor is inactive (Fig. 4). These syndromes, termed pseudohypoaldosteronism type I (PHAI), include life-threatening neonatal hypotension, dehydratation and salt wasting, accompanied by hyperkalemia, metabolic acidosis and grossly increased aldosterone levels. Autosomal dominant and recessive forms of PHAI have been identified (Hanukoglu 1991). The autosomal dominant types result from loss-of-function mutations in one MR gene (Geller et al. 1998), which indicates that two functional copies of the MR gene are necessary for regular salt homoeostasis. Salt-rich diets ameliorate the symptoms, and the syndrome itself attenuates with age, nicely illustrating the close interactions of genetic, demographic and environmental factors in blood pressure control.
The autosomal recessive forms of PHAI are phenocopies of the dominant type that do not ameliorate with age. They are caused by a large number of now known inactivating mutations affecting all three subunits of ENaC channels (Figs. 5 and 10; Chang et al. 1996; Strautnieks et al. 1996). ENaCs–as described above–translate the aldosterone signal into an increased Na+ reabsorption, which is prevented by the loss-of-function mutations causing salt wasting and hypotension.

Molecular physiology of sodium absorption in the thick ascending limb of Henle (a) and in the distal tubule (b). Sodium reabsorption in Henle’s loop is mediated by the Na+/K+/2Cl− transporter (SLC12A1), which requires a constant recycling of K+ to the tubule via ROMK K+ channels. At the basolateral membrane, the Na+/K+ ATPase and Cl− channels are critically involved in vectorial transtubular NaCl transport. The epithelial Cl− channel consists of an ion transporting subunit (CLCNKB; CLCNKA) and a subunit required for trafficking (Barttin). Loss-of-function mutations in these proteins cause Bartter’s syndrome. NaCl transport in the distal tubule is mediated by the NaCl− Cotransporter (NCCT; SLC12A3). Loss-of-function mutations cause Gitelman’s syndrome
Bartter’s and Gitelman’s syndromes
While the monogenic forms of hypotension described so far are accompanied by hyperkalemia, hypokalemic forms have also been characterized, including Bartter’s and Gitelman’s syndromes. Bartter’s syndrome is a hereditary hypotensive disorder that causes a severe form of volume depletion with hypercalciuria and hypokalemic alkalosis early in life, mostly in the neonatal period or after premature delivery in association with fetal polyuria and polyhydramnios. The symptoms are accompanied by a strong activation of the renin-angiotensin-aldosterone system. Bartter’s syndromes are genetically heterogenic, and three different forms have been identified. The type 1 of Bartter’s syndrome is caused by missense or frameshift mutations in the Na+/K+/2Cl− transporter (NKCC2; SLC12A1) located in the thick ascending limb of Henle (Simon et al. 1996b; Figs. 10 and 11). The NKCC2 controls up to 30% of renal salt reabsorption and is inhibited by loop diuretics, e.g. furosemide. Hence, Bartter’s syndrome type 1 resembles the picture of a severe intoxication with loop diuretics. Optimal transport conditions of the NKCC2 require a stochiometry of 1 Na+, 1 K+ and 2 Cl− ions, which is limited by the low K+ concentration in the urinary lumen of the thick ascending limb. To maintain NKCC2 transport activity, K+ has to recycle back after cellular uptake into the urinary lumen through ROMK channels (Fig. 10). Genetic analyses of families with hereditary hypotension and hypokalemia not explained by mutations in NKCC2 have led to the identification of loss-of-function mutations in the ROMK gene (Fig. 11; KCNJ1; Simon et al. 1996c). In agreement with the concept depicted in Fig. 10, recycling of K+ to the urinary lumen is blocked in carriers of such mutations which inhibits the activity of NKCC2 and causes severe salt wasting and hypotension.

Topology and localization of mutations in SLC12A1, ROMK1 and CLCNKB that cause Bartter’s syndrome. A gain-of-function mutation in CLCNKB (T481S) that has been associated with primary hypertension is indicated by the legend in the black box. For ROMK1, there exist two major splice variants which are distinguished by a short N-terminal elongation. Since different groups have numbered mutations in ROMK1 in reference to either splice variant, discrepancies occur. To keep the established nomenclature of mutations, we have not adjusted ROMK1 variants detected in splice variant 2 to the depicted variant 1
NaCl, once transported into the cells of Henle’s loop by NKCC2, leaves these cells at the basolateral membrane by a concerted action of the Na+/K+-ATPase and epithelial chloride channels (Fig. 10). A third variant of Bartter’s syndrome is linked to inactivating mutations in the renal chloride channel CLCNKB (Fig. 11; Simon et al. 1997). The epithelial chloride channels CLCNKA and CLCNKB are heterodimers that require a second subunit, termed Barttin, for full activity. While the Barttin protein itself is not involved in chloride transport, it is essential for membrane insertion, ion permeation and gating of chloride channels. Inactivating mutations in Barttin give rise to a fourth type of Bartter’s syndrome, again with familial hypotension, but complicated by sensorineural deafness attributable to the role of chloride channels in epithelia of the inner ear (Fig. 12; Birkenhager et al. 2001).

Topology and localization of mutations in Barttin, that cause Bartter’s syndrome type 4 and in the NaCl-Cotransporter, that cause Gitelman’s syndrome, both monogenic forms of hypotension
Gitelman’s syndrom is the result of a wide variety of loss-of-function mutations in the thiazide-sensitive Na+/Cl+ transporter (Fig. 12; Simon et al. 1996a). NCC in the distal convoluted tube governs the reabsorption of approximately 7% of Na+. Hence, symptoms in Gitelman’s syndrome are less severe than in Bartter’s syndrome, and include hypokalemia, metabolic alkalosis, hypomagnesemia and hypocalciuria (in strict contrast to hypercalciuria in Bartter’s syndrome) in addition to low normal blood pressure or overt hypotension, mirroring the situation of extensive thiazide diuretics intake. Patients typically present with non-specific neuromuscular signs in adolescence or young adulthood (Bettinelli et al. 1992). The tubular loss of salt is counterbalanced by an activation of the renin-angiotensin-aldosterone system that leads to increased Na+ reabsorption via ENaC channels in the cortical collecting duct, at the expense of enhanced K+ and H+ excretion. Heterozygous carriers of these mutations are normotensive but do actually consume more salt, again underscoring the tight interaction of genetic and environmental factors to blood pressure regulation. While homozygous carriers of Gitelman’s or Bartter’s mutations suffer from severe disorders, there is speculation that heterozygotes or carriers of other mutations that cause only mild inhibition of such transporters are protected against hypertension (Cruz et al. 2001). In addition to this considerable number of loss-of-function mutations in these ion transport proteins, a gain-of-function mutation in CLCNKB has been reported that causes a conservative amino acid substitution (T481S; Fig. 11). It occurs in 12% of white Europeans and 22% of black Africans (Jeck et al. 2004). In one study, hypertension frequency increased from 25% in carriers of the wild-type gene to 45% in carriers of the mutation, findings that were not confirmed by other groups (Jeck et al. 2004; Kokubo et al. 2005; Speirs et al. 2005).
So far, we have discussed monogenic forms of arterial hypertension and hypotension. Interestingly, almost all genetic abnormalities relate to renal salt and water handling. It is tempting to speculate that, for polygenic hypertension, genetic abnormalities in renal mechanisms may also exert key effects. Furthermore, we are at the beginning of investigating whether common polymorphisms in these genes have an impact on polygenic hypertension.
Polygenetic forms of hypertension
In contrast to the rare cases of monogenic hypertension, primary hypertension is attributable to polygenetic mechanisms, a mosaic of neural, hormonal and cellular abnormalities with a strong influence of lifestyle and environmental factors. There is evidence that the impact of certain genetic polymorphisms varies between major ethnicities. Within the last decade, numerous genes have been implicated in the genetics of hypertension, in many cases with a narrow basis of evidence only. Here, we focus on several hypertension candidate genes for primary hypertension, for which a considerable amount of knowledge on potential pathomechanisms exists in addition to linkage analyses or association studies.
Components of the renin-angiotensin-aldosteron system
Given the pivotal role of the renin-angiotensin-aldosteron (RAS) system for long-term regulation of blood pressure and volume and as drug targets in the treatment of hypertension, it was obvious to consider RAS components as candidate genes in hypertension. Under conditions of reduced renal perfusion pressure, salt or volume losses or sympathetic activation, renin, an aspartyl protease, is released from juxtaglomerular cells in the kidney. It cleaves the inactive peptide angiotensinogen (AGT), synthesized in the liver, into angiotensin I, which is converted into angiotensin II (Ang II) by the converting enzyme (ACE). AngII binds to its specific G protein-coupled receptor (GPCR) on vascular smooth-muscle cells to cause potent vasoconstriction, and in the adrenal glomerulosa to induce mineralocorticoid secretion. Vasoconstriction and aldosterone-mediated sodium reabsorption counteract the initial fall in renal perfusion pressure.
Polymorphisms in the angiotensin converting enzyme
ACE is a zinc metallopeptidase widely distributed on the surface of endothelial and epithelial cells. The ACE gene is confined to chromosome 17q23, comprises 21 kb and consists of 26 exons. Two promoters give rise to i) a somatic ACE form using exons 1 to 26 except 13 which is widely expressed, and by alternative splicing to ii) a testicular form using exons 13 to 26, which is required for male fertility. Another ACE homologue, ACE2, encoded on the X-chromosome and expressed in heart, kidney and testes, is involved in the inactivation of AngII (for review of the ACE system, see Sayed-Tabatabaei et al. 2006). Numerous reports have linked a common insertion-deletion (I/D) polymorphism in the ACE gene with a wide variety of diseases including hypertension, atherosclerosis, cardiac hypertrophy, stent stenosis, progression of kidney diseases and more. The I/D polymorphism consists of an insertion of a 287-bp portion of DNA into intron 16 of the ACE gene (Fig. 13). Technically, detection of this polymorphism results in approximately 5% genotyping errors for the I allele, since PCR-amplification of the shorter D allele is favoured. Therefore, control genotyping assays are mandatory in presumed DD carriers (Lin et al. 2001). Mechanistically, the D allele is associated with increased levels (approximately twofold in homozygous carriers) of ACE in Caucasian and Asian but not in African populations (Rigat et al. 1990; Nakai et al. 1994; Bloem et al. 1996). Other groups have confirmed that this polymorphism accounts for half of the observed variance in ACE plasma levels. Since the location of the I/D polymorphism in a non-coding region of the ACE gene argues against a functional variant, considerable efforts have been spent to discover the precise location of a functional polymorphism and to unravel predictive haplotypes. Present hypotheses locate functional polymorphisms to a region between intron 18 and the 3’ UTR (Zhu et al. 2000). The possibility remains, however, that a promoter polymorphism in linkage with the I/D polymorphism contributes to the observed variance in ACE levels (Villard et al. 1996; McKenzie et al. 2005). Furthermore, an interaction between the ACE I/D polymorphism and a QTL on chromosome 4 has been discussed as cause for the variances in ACE levels (Kammerer et al. 2004). Despite the comparable shortness of the ACE gene (21 kb only), these conflicting results indicate how strenuous the unravelling of complex genetic mechanisms can be (Fig. 13).

a Genomic organization of the ACE gene consisting of 26 exons. The position of the frequently investigated insertion/deletion polymorphism in intron 16 is depicted. The D allele is associated with enhanced ACE levels. However, causal mutations linked to the I/D polymorphisms have not been identified yet. Current hypothesis locate causal mutations in a stretch from exon 18 to the 3’-UTR. Other authors suggest variants in the promoter region to cause the observed variance in ACE (regions indicated by double arrows). b Genomic organization of the AGT gene on chromosome 1. A signal peptide and the cleavage site for renin are indicated. The most frequently investigated M235T polymorphism and additional associated polymorphisms are depicted. Available evidence suggests that not M235T but sequence variances in the promoter region cause the observed variation in AGT expression (Binding sites at the promoter region: USF, upstream-stimulating factor; ERα, estrogen receptor α response element). c Genomic organization and scheme of the primary structure for the angiotensin II type 1 receptor. The localization of the frequent A1166C polymorphism is indicated. There are important sequence motifs in the extreme end of the 3’ untranslated region that govern mRNA stability. It is not known whether the alleles of the A1166C polymorphism itself or by linkage disequilibrium are associated with these regulatory functions. Coding exons are depicted as black boxes, untranslated sequences in white boxes
While most authors have ruled out linkage between hypertension and the ACE gene (Jeunemaitre et al. 1992a; Schmidt et al. 1993; Schunkert et al. 1994; Iwai et al. 1994; Vasku et al. 1998; Tiret et al. 1998; Castellano et al. 2003), others have reported significant associations between this polymorphism and hypertension in certain subgroups, e.g. males (Fornage et al. 1998; O’Donnel et al. 1998; Kario et al. 1999; Giner et al. 2000). A first meta-analysis based on 28 case-control studies with 6923 individuals revealed a non-significant pooled odds ratio for hypertension in carriers of the DD versus II genotypes, translating into a 10% increased risk for hypertension in carriers of the DD genotype. Subgroup analyses indicated a significantly increased hypertension risk for women and in Asians (Staessen et al. 1997), contrary to studies mentioned above. A more recent meta-analysis in 15,942 Caucasians indicated that the attributable blood pressure increase for carriers of the DD genotype amounted to 0.5 mmHg (95% CI: −0.5–1.8) compared to the II genotype (Agerholm-Larsen et al. 2000). Another recent review counted 12 positive and 14 ‘negative’ publications on this issue (Agarwal et al. 2005). Likewise, available evidence suggest that the D allele causes a modest–and most likely irrelevant–increase in the risk for other cardiovascular and renal diseases (Cambien et al. 1992; Schunkert et al. 1994; Iwai et al. 1994; Pontremoli et al. 1996; Fernandez-Llama et al. 1998). For these entities, meta-analyses revealed odds ratios for carriers of the DD genotype ranging from 1.04–1.43 for myocardial infarction (Agerholm-Larsen et al. 2000; Keavney et al. 2000) and from 1.28–1.56 (Staessen et al. 1997; Ng et al. 2005) for diabetic nephropathy.
Polymorphisms in the angiotensinogen gene
The M235T polymorphism in the AGT gene was the first and the most scrutinized candidate linked to essential hypertension. It is in linkage disequilibrium with a T174M polymorphism, also frequently addressed. Available evidence suggests that the T allele of the M235T variant is associated with higher circulating angiotensinogen levels (∼20% in homozygous carriers) and increased blood pressure and preeclampsia (Jeunemaitre et al. 1992b; Ward et al. 1992; Staessen et al. 1999). Some animal models indicate that increased AGT gene expression results in hypertension (Takahashi and Smithies 1999). AGT belongs to the serpin gene superfamily and is expressed in many tissues, including liver, heart, adipose tissue, vessel wall, brain and kidney. The AGT gene comprises ∼12 kb on chromosome 1 and contains 5 exons (Fig. 13). AGT expression is controlled by a 1.2-kb promoter region, and further augmented by an enhancer immediately downstream of a second polyadenylation site in the 3′ flanking region. The sole exchange of methionine by threonine at position 235 is not a likely mechanism causing hypertension. This exchange is located distantly from the renin cleavage site, and in vitro renin-mediated AngI generation is kinetically not different for the 235T variant. Currently, a promoter polymorphism at position -6 (G-6A) in strong linkage disequilibrium (-6G–235M; -6A–235T) is a more plausible candidate for differing AGT transcriptions (Inoue et al. 1997; Morgan et al. 1997). Many additional polymorphisms, however, have been identified in the AGT gene, including an additional promoter polymorphism at −20. This polymorphism is also interesting, since depending on the genotype different response elements are generated, including those for estrogen receptor α, upstream regulatory factor and members of the COUP-TF family, which correlates with different phenotypes, including pregnancy-dependent variations in AGT levels, non-modulating hypertension and an altered relationship between BMI and blood pressure (Zhao et al. 1999; Morgan et al. 2000; Hilgers et al. 2001; Tiago et al. 2002). Current research efforts address the haplotype structure of the AGT gene, which should be comparatively easy given the short length of the gene of only ∼12 kb. Based on the number of SNPs included in each definition of haplotypes and by the ethnicities analysed, haplotype maps vary markedly. While some reports showed that defined haplotypes contribute to the risk for hypertension (in part modulated by polymorphisms at the ACE locus), others found contrasting results. Available evidence suggests that there are more than five common haplotypes of the AGT gene that include promoter, coding region and 5′ flanking region polymorphisms (Jeunemaitre et al. 1997; Nakajima et al. 2002; Brand et al. 2002; Zhu et al. 2003a,b; Tsai et al. 2003; Gu et al. 2005). While genetic concepts promise that understanding haplotype structures will reduce complexities, at present this is not the case for the AGT locus, and we have to await further clarification of this issue.
For the common AGT M235T polymorphism, more than 500 reports exist that address hypertension and other cardiovascular diseases. Both positive and negative associations were documented for white populations in North America, Australia, Japan, Europe, and China, while no associations were reported from black populations in the Caribbean, USA and Africa (Jeunemaitre et al. 1992b; Caulfield et al. 1994, 1995; Hata et al. 1994; Kunz et al. 1997; Staessen et al. 1999). Meta-analyses confirmed the association between the 235T allele with hypertension in whites, potentially Asians but not in blacks, with significant odds ratios ranging from 1.22–1.60 (Kunz et al. 1997; Staessen et al. 1999; Kato et al. 1999; Sethi et al. 2003). The absolute effect, however, is rather modest, best illustrated by the Italian GENIPER study on 2,461 subjects; this study reported significant odds ratios of 1.35 for hypertension in carriers of the 235T allele, but the absolute blood pressure difference between genotype groups was smaller than 1 mmHg (Castellano et al. 2003).
Genetic variants in the angiotensin II receptor type I
Since AngII-mediated vasoconstriction and aldosterone secretion are mediated by the angiotensin II receptor type 1 (AT1R), several groups searched for common polymorphism in its gene, which comprises more than 55 kb and consists of 5 exons. These efforts resulted in the identification of an A1166C variant that is confined to the 3′ untranslated region of the AT1R cDNA (Bonnardeaux et al. 1994; Wang et al. 1997) and has been implicated in determining mRNA stability and, ultimately, receptor expression (Thekkumkara and Linas 2003; Fig. 13). There is evidence that a 3′-UTR motif of the receptor mRNA involving bases 2179–2195—i.e., far downstream of the A1166C polymorphism—is essential for the binding of regulatory proteins including calreticulin. AngII-stimulated phosphorylation of calreticulin has been demonstrated to increase AGT1R mRNA decay (Nickenig et al. 2001, 2002). It has also been proposed that removal of the entire 3′-UTR from the AT1R changes the coupling of the AT1R from Gαi to Gαq. However, the mechanisms for such an astonishing observation have remained obscure to date (Thekkumkara and Linas 2003). Furthermore, a role for the receptor’s 3′-UTR in tachyphylaxis of AngII-induced inositol trisphosphate and Ca2+ levels has been suggested (Franca et al. 2003). While these novel mechanisms of posttranscriptional mRNA processing are highly interesting, there are no convincing connections of these observations to the A1166C polymorphism.
Several reports have shown an association between the AT1R 1166C allele and hypertension, especially severe hypertension (Bonnardeaux et al. 1994; Hingorani et al. 1995; Wang et al. 1997; Kainulainen et al. 1999; Henskens et al. 2003) and in pregnant women (Nalogowska-Glosnicka et al. 2000; Kobashi et al. 2004). Other groups refuted these results (Schmidt et al. 1997; Zhang et al. 2000; Staessen et al. 2001; Iliadou et al. 2002; Ono et al. 2003). Overall, it remains open whether there is a relevant association between the AT1R A1166C polymorphism and hypertension. Furthermore, the suggested pathoetiology of this silent nucleotide exchange is at best uncompletely understood.
Meanwhile, more than 55 additional SNPs have been identified in the AT1R gene, and there is some evidence for linkage between the A1166C and an A-153G promoter polymorphism (Takahashi et al. 2000; Lajemi et al. 2001). From clinical studies, it has been suggested that homozygous carriers of the 1166C allele exhibit lower decreases in blood pressure (2 mmHg) than homozygous carriers of the 1166A allele (12 mmHg) upon administration of AT1R blockers in a setting of high salt diet (Spiering et al. 2000, 2005). Another group did not observe physiological differences upon AngII infusion between carriers of the various AT1R A1166C genotypes who were on a normal salt diet (Hilgers et al. 1999).
While components of the RAS are obvious candidate genes for hypertension research, we have to conclude after many studies that the contribution of known variations in these genes to the observed blood pressure variation is modest, if present at all. The reason for this sobering notion is most likely that we deal with a highly regulated system. From the many genetically engineered mouse models of the RAS, it appears that just changing the expression levels (the suggested mechanisms of the common RAS polymorphisms in man) of RAS components (as exemplified by 1, 2, 3 or 4 ACE genes) does not change blood pressure (or only modestly), as long as the secretion of renin (and other pressure regulating systems) is adapted to this situation. Only in situations when the regulatory balance is hampered (e.g. in diabetes) do such mice develop a pronounced phenotype (Bernstein et al. 2005), a situation in some agreement with observations in man.
Polymorphisms in adducin genes
A manifold of animal models for various aspects of hypertension have been inbred or generated by direct transgenic manipulations. The identification of the α-adducin gene is the sole example that discoveries in animal studies have directly resulted in the identification of genetic polymorphisms in man. Crucial for the identification of α-adducin polymorphisms was the generation of hypertensive and normotensive rat strains by classical selection and crossing experiments leading to the Milan hypertensive (MHS) and normotensive (MNS) strains. Refined physiologic analyses of these animals revealed that prehypertensive young MHS animals exhibited—among other features—an increased glomerular filtration rate, an enhanced 24-hr Na+ urinary output and a reduced plasma renin activity (Ferrari and Bianchi 1995), a phenotype also observed in some young human individuals prone to develop hypertension (Cusi and Bianchi 1996). The further development of hypertension in MHS is accompanied by an increased renal Na+ reabsorption. Na+ transport abnormalities were also detected in erythrocytes (but not erythrocyte membranes) from MHS, findings that sparked cross-immunization experiments between MHS and MNS. Raised antibodies detected adducin, which is ubiquitously expressed and found in erythrocytes in considerable amounts. Adducin is a heterodimeric cytoskeleton protein that consists of an α-subunit (103 kDa) and either a β-subunit (97 kDa) or a γ-subunit (90 kDa) encoded by three different genes referred to as Add1, Add2 and Add3. They sustain spectrin–actin binding, and control as end-capping actin proteins the rate of actin polymerization and, thus, the organization of the spectrin-actin lattice. This activity of adducin is further regulated by Ca2+/calmodulin and the kinases PKA, PKC and Rho-kinase (Matsuoka et al. 2000).
Systematic sequence analyses of the adducin genes in MHS and MNS led to the identification of polymorphisms in the rat α-adducin (Add1, F316Y), β-adducin (Add2, Q529R) and γ-adducin (Add3, Q572K), that segregated in part with the hypertensive trait (Bianchi et al. 1994; Tripodi et al. 1997). Backcross experiments indicated that the Add1 F316Y polymorphism (but not the variants in Add2 and Add3) accounted for about 50% of the blood pressure variation in the F2 hybrid population (Bianchi et al. 1994). Furthermore, reciprocal gene transfer of the α-adducin chromosomal region modulated the pressure of MHS and MNS strains (Tripodi et al. 2004).
An intensive search for similar polymorphisms in humans resulted in the identification of two common polymorphisms (G460W [rs4961], S586C [rs4963]) in the human α-adducin gene (Add1; Cusi et al. 1997). Additional polymorphisms were identified in all three human adducin genes. At present, most studies were conducted with the G460W polymorphism, which has been shown to be associated with increased blood pressure in case-control and linkage studies (Cusi et al. 1997). It is worthy of note that the human Add1 G460W exchange is not homologous to the F316Y exchange in rats. Hence, it is important to distinguish biochemical and physiological effects observed within different species. Many studies following these initial observations have addressed the potential association of the α-adducin 460W allele with hypertension and other cardiovascular diseases. As with many association studies, results were variable and in part contradictory, attributed partially to confounders of ethnicity, lifestyle and other environmental factors (reviewed in Bianchi et al. 2005). Hence, some family studies showed associations between adducin alleles and blood-pressure parameters or demonstrated linkage with the α-adducin locus, while others failed to do so (Cusi et al. 1997; Niu et al. 1999; Chu et al. 2002; Ju et al. 2003; He et al. 2003; Wang et al. 2004). Similarly, the majority of studies addressing blood pressure in general, predominantly normotensive populations failed to show associations with adducin polymorphisms, reported positive associations for subgroups only (e.g. postmenopausal women), or depended on epistatic interactions with other polymorphisms, especially the ACE I/D polymorphism. Eleven case-control studies demonstrated a positive association between adducin polymorphisms and hypertension, whereas 12 studies reported contradictory results (reviewed in Bianchi et al. 2005). Carriers of the 460W allele reportedly have lower renin levels and a larger decrease in blood pressure upon administration of diuretics (Glorioso et al. 1999). At the cellular level, hypertensive carriers of the 460W allele are characterised by a decreased red cell Na+ content and an increased Na+/K+ transport (Glorioso et al. 2002). For the human Add2 C1967T polymorphism, evidence for an association between the 1967T allele and increased blood pressure in a high salt intake population exists (Tikhonoff et al. 2003).
The Na+/K+ ATPase located at the basolateral membrane in kidney tubules is critical for vectorial transport of many solutes including Na+ (Fig. 14). Catecholamines, especially dopamine, are involved in the regulation of Na+ excretion during salt load or deprivation (Aperia 2000). Dopamine stimulates the phosphorylation of Na+/K+ ATPase α-subunits in kidney tubules (Chibalin et al. 1999) and activates PI3 kinases, the products of which, such as phosphatidyl-inositol-3-phosphate, are essential for the clathrin-dependent endocytosis of the Na+/K+ ATPase by mediating the interaction of the adaptor protein adaptin (AP2) with a tyrosine motif in the Na+/K+ ATPase α-subunit (Ogimoto et al. 2000; Done et al. 2002).

a Vectorial Na+ transport in the renal tubule depends on the regulated action of different Na+ transporters (NaCl Cotransporter, NCC; Na+/K+/Cl2− transporter, NKCC2; Na+/H+ exchanger, NHE3 in the proximal tubule; ENaC channels) in the luminal membrane and the Na+/K+ ATPase in the basolateral membrane. In this scheme, various transporters were depicted in one cell. Physiologically, these are confined to different parts of the nephron. Among others, dopamine is an important regulator of renal Na+ transport. In the presence of dopamine, Na+/K+ ATPase activity is inhibited by phosphorylation and by increased internalization, which ultimately results in decreased salt uptake. Dopamine also regulates numerous other renal transporters, and renal signalling mechanisms are complex depending on dopamine receptor subtypes and renal cell types. b Adducin is involved in the internalization of the Na+/K+ ATPase, a process that involves a multiprotein complex leading to the formation of clathrin-coated vesicles. Adaptin binds Na+/K+ ATPase molecules, and interacts with cargo proteins including clathrin. Stimulation with dopamine results in an increased Na+/K+ ATPase and adaptin phosphorylation and promotes internalization. Adducin is involved in this process, potentially linking it to the actin cytoskeleton. Expression of mutant adducin interferes with this internalization process and is accompanied by an increased basal adaptin phosphorylation which does not further increase upon dopamine stimulation. An altered function of a protein phosphatase has been implicated in the presence of mutant adducin (proposed scheme according to Efendiev et al. 2004 and Bianchi et al. 2005). Many questions remain open, but it appears that in the presence of mutant adducin Na+/K+ ATPase internalization is reduced, leading to an increased renal Na+ reabsorption. c Increased renal Na+ uptake is the final effect of gain-of-function mutations in GRK4, too. As described above, dopamine stimulation results in an inhibition of Na+/K+ ATPase activity that is initiated by the activated dopamine receptors and heterotrimeric G proteins, followed by the activation of different effector systems. Stimulation of G protein-coupled receptors causes the immediate activation of several desensitization processes to limit the extent of a cellular activation. GRK4 is a G protein-coupled receptor kinase that specifically inhibits the renal dopamine receptor type 1. Gain-of-function mutations in GRK4 cause a permanent phosphorylation and down-regulation of dopamine receptors. Under such conditions dopamine-mediated inhibition of Na+/K+ ATPase activity is blunted, which causes an increased Na+ reabsorption
Interestingly, the p85 subunit of PI3 kinase can also directly interact with the Na+/K+ ATPase (Chibalin et al. 1998; Yudowski et al. 2000). Adaptin binds to cargo proteins, recruits clathrin and promotes the formation of clathrin-coated pits (Kirchhausen 2000), triggering an endocytotic process of the Na+/K+ ATPase that reduces sodium pump activity in the kidney and causes a decreased Na+ reabsorption upon dopamine stimulation.
Early transfection studies with the mutant rat α-adducin (316Y) into renal cells revealed a rearrangement of the cytoskeleton (potentially by an accelerated actin polymerization) associated with an increased Na+/K+ ATPase activity (Tripodi et al. 1996). More recently, expression of rat and human α-adducin variants has been implicated in a reduced endocytosis of Na+/K+ ATPase molecules (Efendiev et al. 2004). Stimulation with dopamine resulted in phosphorylation and internalization of Na+/K+ ATPase subunits, an effect that was blunted in the presence of variant α-adducin subunits (Fig. 14). The reduced internalization was accompanied by a decreased phosphorylation of adaptin (or more accurately, of its μ2 subunit) which governs the affinity for binding to cargo proteins (Efendiev et al. 2004). Although these findings have remarkably extended our knowledge concerning the molecular pathology of α-adducin variants, many questions remain open. It is completely unknown where α-adducin, the actin-cytoskeleton and the dopamine-mediated mechanisms of adaptin- and clathrin-dependent Na+/K+ ATPase internalization meet. In non-stimulated renal epithelial cells expressing the rat α-adducin variant, adaptin phosphorylation status was increased despite reduced Na+/K+ ATPase internalization. This puzzling result was interpreted as a defect in the transition of phosphorylated to unphosphorylated adaptin, caused by a protein phosphatase that is part of a complex with adducin. Complexation of such a protein phosphatase with adducin and the cytoskeleton, required for proper phosphorylation / dephosphorylation of adaptin, may be disturbed in adducin mutants (Efendiev et al. 2004). Further studies are required to resolve these questions and to explain why the proposed internalization abnormality is confined to the Na+/K+ ATPase and does not occur in other membrane proteins, e.g. the dopamine or Ang II receptor as shown in this report (Efendiev et al. 2004). It remains open how these mechanisms that affect the dynamics of Na+/K+ membrane expression are in accordance with results from another study suggesting that the kinetic activity of the Na+/K+ ATPase is increased and its ATP affinity is higher in the presence of the rat 316Y and the human 460W variants compared to wildtype α-adducin in cell-free preparations (Ferrandi et al. 1999).
In summary, we have to conclude that numerous reports describe abnormalities in cellular and renal Na+ handling, renin-angiotensin status, efficacy of diuretics and in blood pressure control in carriers of the Add1 460W allele, although many contradictory findings exist. We begin to understand how mutants in α-adducin may affect the internalization of the Na+/K+ ATPase. Nevertheless, many questions remain open related to the tissue-specific expression of the adducin heterodimers, potential splice variants, the haplotype blocks within the adducin genes and the blood pressure control in adducin knock-out and knock-in models.
Polymorphisms in G protein-coupled receptor signalling mechanisms
According to biophysical concepts, primary hypertension is basically characterized by an increased peripheral resistance, which reflects an altered balance between vasoconstriction and vasodilation. Therefore, hormones and signalling mechanisms that regulate vascular tone have gained interest as candidate genes by many workers in the field of hypertension genetics.
G protein-coupled receptors are a huge family of membrane receptors that comprise more than 3% of all genes in our genome. In the cardiovascular system, typical GPCRs are the α- and β-adrenoceptors, receptors for endothelins, angiotensin II, vasopressin and many other agonists.
Genetic variants in β-adrenoceptor genes
The β1-adrenoceptor (β1AR) is encoded by an intronless gene located on chromosome 10. Two common polymorphisms in the β1AR are Gly389Arg and Ser49Gly (Fig. 15; for review see Leineweber et al. 2004). The Gly389Arg amino acid exchange is confined to the C-terminus region of the receptor that couples to G proteins. In line with this localization, in vitro studies suggested 2- to 3-fold higher increases in adenylyl cyclase activity for the 389Arg allele as a gain-of-function mutation (Mason et al. 1999; since phylogenetic comparisons suggest that the 389Arg allele is the true ancestral variant, the 389Gly is actually a loss-of-function mutation, an issue of mere definition than of relevance). Whether these functional differences remain under in vivo conditions is still a matter of debate. The Ser49Gly polymorphism is located in the N-terminal region of the receptor protein, and the 49Gly allele is associated with enhanced agonist-stimulated down-regulation (Rhatz et al. 2002). Mechanistically, this differential behaviour is attributable to an altered N-glycosylation leading to an increased resistance towards receptor degradation upon internalization in the 49Ser allele. Furthermore, the 49Gly variant is associated with an increased basal activity (constitutive activity), a finding, however, that depends on the degree of receptor expression. For genetic studies, it is important to note that both polymorphisms are linked which requires the analysis of haplotypes.

Topology and common missense amino acid substitutions in the human β1- and β2-adrenoceptors
A Scandinavian sib and case-control study documented that homozygous carriers of the 389Arg allele, i.e., those with improved coupling to G proteins, had significantly higher diastolic blood pressure and heart rates. Odds ratios for hypertension in this cohort amounted to 1.9. The Ser49Gly polymorphism, however, did not affect blood pressure (Bengtsson et al. 2001a,b). In Caucasians, but not in other ethnicities, this result has been confirmed once (Humma et al. 2001) and contradicted by other investigators (Büscher et al. 2001; Ranade et al. 2002; O’Shaughnessy et al. 2000; Sofowora et al. 2003).
The β2 adrenoceptor (β2AR) is also encoded by an intronless gene, located on chromosome 5. Upstream of its coding region is the coding region for a 19 amino acid peptide, termed 5′ leader cistron, that has been implicated in receptor expression. Since blunted β-adrenoceptor-mediated vasodilation has been observed in hypertension, the β2AR has become a candidate gene for this disease (Stein et al. 1995). To date, several polymorphisms have been identified in the β2AR, five of which change the primary sequence (Fig. 15). A Val34Met polymorphism (in the first transmembrane domain) occurs rarely, and the Cys(-19)Arg affects the 5′ leader cistron. The Arg16Gly, Gln27Glu and Thr164Ile polymorphisms have been frequently investigated. These polymorphisms display marked linkage disequilibirum, which results in the occurrence of three dominant haplotypes: -19Arg/16Gly/27Glu, -19Cys/16Gly/27Gln and -19Cys/16Arg/27Gln (Wang et al. 2001). Functional analysis revealed that the Arg16Gly and the Gln27Glu polymorphisms, located in the amino-terminus of the protein, neither alter ligand binding nor adenylyl cyclase activity (Green et al. 1994). The 16Arg and the 27Glu forms of the receptor, however, were resistant to agonist-promoted down regulation upon individual expression in in vitro studies (Green et al. 1994). In double-mutation studies, however, the 16Gly mutation confered a dominant phenotype over the 27Glu variant, while the 16Arg/27Glu variant (the frequency of which in the population is <1%) was completely resistant to down-regulation (Green et al. 1994). In vivo the situation becomes more complex, and the 16Gly has also been linked to reduced (!) desensitization. For the 164Ile variant of the Thr164Ile polymorphism located in the fourth transmembrane domain, considerably lower affinities for isoproterenol, epinephrine and norepinephrine associated with decreased basal and agonist-stimulated adenylyl cyclase activity have been documented (Green et al. 1993). Finally, carriers of the -19Cys allele in the 5′ cistron have been reported to express significantly more β2AR (McGraw et al. 1998).
Potential associations between β2AR polymorphisms and hypertension were addressed in the Rochester Heart Study, which included 55 pedigrees with at least one discordant sib pair and 589 different families, to elucidate a region on chromosome 5 (including the β2AR gene) which was formerly linked to hypertension. They found that the 16Gly and 27Glu alleles were more common among hypertensives than the 16Arg and 27Gln alleles. The β2AR polymorphisms accounted for ∼2% of the observed variation in blood pressure (Bray et al. 2000). While results of some groups confirmed the association between the 16Gly allele and blood pressure (Kotanko et al. 1997; Rosmond et al. 2000; Gratze et al. 1999), others found opposite effects, i.e., an association between the 16Arg allele and hypertension (Busjahn et al. 2000; Bengtsson et al. 2001a,b; Timmermann et al. 1998) or no effects at all (Herrmann et al. 2002; Kato et al. 2001; Xie et al. 2000; for a recent meta-analysis see Hahntow et al. 2006). Again, differing inclusion of haplotypes, variations in ethnicities and different study designs may contribute to these sobering results. Although β1ADR und β2ADR genes may affect blood pressure regulation, the actual contribution of genetic polymorphisms in these genes appears to be small, if present.
Genetic variants in the gene for the Gα subunit (GNAS) and the gene for the Gβ3 subunit (Gβ3)
The next step downstream in G protein-coupled receptor signalling—including the β1 and β2 adrenoceptors, dopamine receptors, endothelin receptors, bradykinin receptors and the AT1R—is the activation of heterotrimeric G proteins. G proteins are key signal transducers which are composed of three subunits, termed Gα, Gβ and Gγ according to their decreasing molecular weight (Fig. 16; this topic is excellently reviewed by Wettschureck and Offermanns (2005). While the Gα subunit carries the enzymatic GTPase activity, the Gβ and Gγ subunits form a stable dimer. The inactive heterotrimeric G protein consists of a Gα subunit and a Gβγ dimer. The inactive Gα subunit is liganded with GDP. Upon interaction with an activated GPCR, the Gα subunit changes its conformation and releases GDP. The GDP is replaced by GTP, and in its GTP-bound state the Gα subunit takes an activated conformation with low affinity for the Gβγ dimer. Subsequently, GTP-bound Gα subunits interact with their respective effector systems (e.g. adenylyl cyclases) and modulate their activity. Likewise, free Gβγ dimers are also effective regulators of many effector systems. The GTPase activity of the Gα subunit mediates a hydrolytic cleavage of GTP into GDP, which terminates its active conformation. Upon association of the GDP-bound Gα with a Gβγ dimer the ‘GTPase cycle’ is closed, and the once-again heterotrimeric G protein is ready for another round of activation. More than 20 different Gα genes, 5 Gβ genes and 12 Gγ genes have been identified in the human genome. Together, they give rise to (theoretically) more than a thousand different heterotrimers, which may explain the observed receptor and effector specificity in G protein-mediated cell signalling. Currently, G proteins are further subdivided according to their Gα subunit. GαS, the ‘stimulatory G protein’, was identified based on its ability to activate adenylyl cyclases. Likewise, Gαi, the ‘inhibitory G protein’, diminishes the activity of adenylyl cyclase isoforms. There are actually several isoforms in the Gαi family (Gαi1, Gαi2, Gαi3, Gαo) with sometimes distinct functions. Besides inhibiting adenylyl cyclase activity, activation of Gαi has been linked to many more cellular effects, including activation of potassium channels, activation of kinase pathways or initiation of cell proliferation. Most likely, these latter functions are attributable to the release of Gβγ subunits. Another subfamily of Gα proteins, the Gαq family with the members Gαq, Gα11, Gα14 and Gα15, is predominantly involved in the activation of phospholipase Cβ isoforms, which results in the generation of inositol trisphosphate and ultimately in an increase of intracellular Ca2+. Gα12 and Gα13 mediate the activation of small G proteins of the rho family, which are implicated in the regulation of cell shape and motility. Free Gβγ dimers are also important and versatile regulators of many effector systems, including adenylyl cyclases, phospholipase Cβ isoforms, PI3 kinases, potassium and calcium channel isoforms, and several kinase systems associated with cell proliferation. The crucial advantage of this highly complex system of different G proteins and effector systems is its ability to integrate and fine-tune many different signals into a cellular response. For instance, adenylyl cyclase activity–and thus the generation of cAMP–is stimulated by the GαS pathway, inhibited by Gαi proteins and can further be modulated by released Gβγ subunits and Ca2+ transients elicited by activation of Gαq.

a GTPase cycle. Upon ligand-binding to a G protein-coupled receptor (GPCR; 1) a conformational change occurs that stimulates a GDP/GTP exchange at the Gα subunit (2). The active GTP-bound Gα subunit separates from the receptor and the Gβγ dimer. Activated Gα and free Gβγ subunits regulate cellular effector systems (3). The endogenous GTPase activity of the Gα subunit causes a hydrolysis of GTP to GDP (4). Again in the GDP-bound state, the inactive Gα subunit re-associates with Gβγ dimers. RGS proteins (‘regulators of G protein signalling’) are GTPase-activating proteins for Gα subunits and greatly accelerate GTP hydrolysis (4a). Another system for limiting G protein activation is generated by the action of G protein receptor kinases (GRK). They interact with an activated receptor and phosphorylate receptor residues that allow for binding of other proteins, e.g. arrestins. Ultimately, activation of GRKs results in desensitization and internalization of GPCRs. b Structure of the Gβ3 gene, GNB3 on chromosome 12. It consists of 11 exons and 10 introns. ‘ATG’ and ‘TGA’ indicate start and stop codons. The T allele of a common silent polymorphism (C825T) in exon 10 favours alternative splice variants termed Gβ3s and Gβ3s2. The alternatively spliced parts of exon 9 (for Gβ3s) or exon 10 (Gβ3s2) are indicated as black boxes. Below, the positions of several polymorphisms are depicted, which are in almost complete linkage disequilibrium in Caucasians. This gives rise to two major prevailing haplotypes. c Proposed model for Gβ3 based on crystallization data of Gβ1. Gβ proteins are propeller proteins made up of seven WD domains. The deletions generated by alternative splicing for Gβ3s and Gβ3s2 are indicated. Each is the equivalent of one entire WD domain. The proposed structures of Gβ3s and Gβ3s2 are propeller proteins with only six WD domains. For other WD proteins, it has been documented by crystallization studies, that such a structure is stable
Since both β adrenoceptors couple to GαS, the GNAS gene became an interesting candidate gene in hypertension research. Loss-of-function and gain-of-function mutations in the GNAS gene cause complex phenotypes involving bone Ca2+ homoiostasis and mesenchymal growth abnormalities, leading to rare diseases which include McCune-Albright osteodystrophy or pseudohypoparathyroidism types Ia and Ib. The GNAS locus, located on chromosome 20q, is subject to a highly complex regulation, and it is partially imprinted in different tissues (for review see Weinstein et al. 2006). Jia et al. (1999) detected a silent polymorphism in exon 5 of GNAS (T393C), and documented an association between increased systolic blood pressure and the 393T allele. Carriers of the 393T allele were more frequently classified as poor responders upon treatment with betablockers (Jia et al. 1999). So far, no mechanistic explanation for this polymorphism has been provided, and information on GαS expression or function is completely lacking. The effect of this polymorphism on hypertension was addressed in three Japanese studies. While no or only modest associations were documented, those studies suggested considerable environmental influences, and reported an enhanced hypertension risk for carriers of the 393T allele in non-heavy smokers (Abe et al. 2002) and in heavy consumers of alcohol (Chen et al. 2003; Yamamoto et al. 2004). It is astonishing that further epidemiologic, genetic and biochemic evidence is lacking for this variant, given the well-established methods for assaying G protein expression and function.
Based on results that G protein-mediated signaling is enhanced in a subgroup of hypertensive patients, and that this phenotype is retained under cell culture conditions (Rosskopf et al. 1993), a systematic sequencing strategy resulted in the discovery of a silent polymorphism in GNB3 (C825T), the gene for the Gβ3 subunit (Siffert et al. 1998). This gene is located on the short arm of chromosome 12, which has frequently been implicated as a hypertension locus, although current evidence suggests that the different loci do not completely overlap (Fig. 9). Mechanistically, this polymorphism is associated with the occurrence of two alternative splice variants termed Gβ3s and Gβ3s2 (Fig. 16; Siffert et al. 1998; Rosskopf et al. 2003a). Protein crystallization studies have resolved the structure of the Gβγ dimer. The Gβ protein consists of seven regular protein motifs which are called WD domains. These seven domains, which are highly conserved in all eukaryotes, form a propeller protein that exhibits a very compact and stable structure which allows Gβ proteins, as other WD proteins, to assemble and coordinate multi-protein complexes, which is the way in which Gβγ dimers regulate effector systems. The splicing to Gβ3s or Gβ3s2 causes the loss of the equivalent of one WD domain upon translation of such transcripts (Fig. 16). It is difficult to explain how the loss of one WD domain translates into the gain-of-function of the system. One theory suggests that Gβγ dimers containing Gβ3s or Gβ3s2 are less suited to stabilize–as a heterotrimer–a Gα subunit in its GDP-bound form. This could result in an increased sensitivity of such a heterotrimer towards activation by a receptor. Although these concepts contain some plausibility, a final conclusive proof is missing. Overexpression of Gβ3s or Gβ3s2 in different cell types and model systems mimics some of these issues, while other features are missing, e.g. the activation of certain effector systems by Gβ3s or Gβ3s2 (Rosskopf et al. 2003a,b; Ruiz-Velasco and Ikeda 2003). Overexpressing G protein components into cell lines is on the one hand a routine method for the analysis of this system, but bears some problems because it grossly disturbs a fine-controlled balance of endogenous G protein signaling components. Presently, we have to conclude that–despite considerable efforts–the molecular process by which a polymorphism in GNB3 translates into a complex phenotype is yet unresolved, which contrasts with the considerable amount of data from genetic association studies.
Given that the 825T allele of the mentioned GNB3 C825T polymorphism actually sensitizes signal transduction in all cell types where Gβ3 is expressed, it is easy to speculate that an enhanced signal transduction may affect vessel reactivity, the activity of the vegetative nerve system or the signaling by vasoactive agonists including angiotensin II or endothelins. This could ultimately result in hypertension. Again, a final proof for these concepts is missing.
Several groups have reproduced the original finding that the GNB3 825T allele is associated with hypertension, whereas other groups refuted that finding (Siffert et al. 1998; Schunkert et al. 1998; Benjafield et al. 1998; Brand et al. 1999; Dong et al. 1999, to cite but a few). The fact that there is a considerable variance in genotype distribution within different ethnicities (Siffert et al. 1999) creates a major problem. While the C allele is the predominant allele in Caucasians, in blacks the T allele prevails. Asians have intermediate frequencies. Hence, there is a high risk for disparate population admixture in case-control studies. The GNB3 gene comprises a comparably short stretch of 6 kB DNA on chromosome 12. This explains why in Caucasians two haplotypes explain more than 95% of the observed variability (Fig. 16; Rosskopf et al. 2000, 2002). In other words, genotyping at the C825T locus provides information for six additional tightly linked polymorphisms. In Asians and in blacks the situation is more complicated. Since we do not know which of the identified polymorphisms are ultimately necessary for alternative splicing to Gβ3s or Gβ3s2, different ethnic-dependent haplotype structures could be one solution to some discrepancies observed.
The 825T allele has also been linked to obesity, and speculations were raised whether this polymorphism contributes to the pathogenesis of a metabolic syndrome (Siffert et al. 1999). While considerable controversy on certain issues remains, the different alleles of the GNB3 C825T polymorphism are obviously associated with complex phenotypes. Some results in this field are promising; nevertheless, many questions regarding the mechanisms remain not yet understood.
Genetic variants of the regulator of G protein signalling (RGS2)
Relatively late identified players in signal transduction via heterotrimeric G proteins are a family of RGS proteins, an acronym for ‘regulators of G protein signalling’ (excellently reviewed by Wieland and Mittmann 2003). More than 20 different RGS proteins have been identified in humans, all of which stimulate the hydrolysis of GTP of the GTP-bound (i.e., activated) Gα subunits and thus accelerate G protein deactivation (Figs. 16 subset 4a and 17). Hallmark of RGS proteins is a canonical 120 amino acid motif that confers the ability to serve as GAPs (‘GTPase activating protein’), a function that protects cells from an unregulated and sustained signalling. Most RGS proteins are able to regulate different Gα subunits. There is, however, a complex system of specificity relating to different Gα subunits, RGS proteins and their temporo-spatial expression in various cell types, issues which are still under active investigation. RGS proteins themselves are subject to regulation by phosphorylation, lipid modifications and the interaction with a wide variety of proteins in addition to Gα subunits. Current evidence suggests that alterations in RGS2 and RGS4 produce cardiovascular phenotypes.

Multiple functions of RGS proteins. RGS proteins are GTPase-activating proteins for activated Gα subunits which limit the activation of Gα subunits. RGS proteins, as shown here for RGS2, not only interact with Gα subunits but also bind to and inhibit adenylyl cyclase isoforms or bind to G protein-coupled receptors. Angiotensin II (AngII) stimulation of vascular smooth-muscle cells causes an increased transcript expression of RGS2, which will in turn diminish Ang II signalling. In addition, RGS2 is an important mediator of NO-mediated vasodilation. The cGMP-dependent protein kinase G (PKG1α) phosphorylates and, thereby, activates RGS2. In turn, vasoconstrictor responses mediated by Gαq proteins are markedly attenuated. Knock-out of RGS2 is associated with severe hypertension in mice
RGS2 is expressed among other tissues in heart, vascular smooth-muscle cells, in the kidneys and in the brain. While RGS2 can act as a GAP protein for Gαi and GαS proteins (Ingi et al. 1998; Sinnarajah et al. 2001), it is the most potent RGS for Gαq, the Gα subunit pivotal for the signalling of most vasoconstrictors, including noradrenaline, AngII, endothelin-1, thromboxane, vasopressin and thrombin (Heximer et al. 1997). RGS2 will not only attenuate Gαq-mediated vasoconstrictor signals, vasoconstrictors such as AngII can also stimulate RGS2 transcript expression, which will ultimately result in a further feedback inhibition of Gαq-mediated signals (Grant et al. 2000). In line with these findings, RGS2 knock-out mice exhibit increased and protracted vasoconstriction, severe hypertension and vascular hypertrophy, already in the heterozygous state (Heximer et al. 2003). Unexplained so far remain the facts that these mice did not develop pronounced cardiac hypertrophy, and that pressor responses to common agonists including AngII and noradrenaline were distinctly blunted. Abnormalities in renal sodium handling have not been observed in these animals.
Another fascinating discovery was that RGS2 is an integrator making two different worlds of cell signalling meet: GPCRs and NO-mediated signalling. NO, released from endothelial cells is known to stimulate cGMP production in vascular smooth-muscle cells via a soluble guanylyl cyclase. cGMP in turn stimulates a cGMP-dependent protein kinase, PKGI-α, that is supposed to further relay the vasodilatory signal to target proteins. Tang et al. (2003) identified RGS2 as a substrate of PKGI-α, and showed that PKGI-α-mediated phosphorylation of RGS2 results in an increased GAP activity and ultimately in the inhibition of Gαq-mediated vasoconstrictor signals (Fig. 17). In addition, the N-terminus of RGS2 can bind to and inhibit type V adenylyl cyclase (Salim et al. 2003). GPCRs themselves have become partners for RGS2 interaction, as shown recently for the α1A adrenoceptor, potentially by interaction of the N-terminal domain of RGS2 with the third intracellular loop of GPCRs mediated by the protein spinophilin in a Ca2+ sensitive fashion (Fig. 17; Bernstein et al. 2004; Hague et al. 2005; Wang et al. 2005). Apart from the cardiovascular functions mentioned so far, RGS2 activity has also been implicated in the regulation of immune functions and neuronal plasticity.
Altogether, these fascinating new findings make RGS2 a prime candidate in the search for hypertension candidate genes. The RGS2 gene maps to chromosome 1q31, the locus for one form of monogenic hypertension, pseudohypoaldosteronism II (Mansfield et al. 1997). Yang et al. (2003) identified five missense variations in the RGS2 gene in a Japanese population. All of them, however, occurred as rare mutations (<1% allele frequency) only in their hypertensive cohort. Functional studies are presently missing; however, sequence analyses revealed that one mutation affects the core RGS motif, while the others change phylogenetically highly conserved amino acids, predominantly confined to the adenylyl cyclase interacting N-terminus and to a helix implicated in plasma membrane localization. In addition to these rare mutations, they identified two common RGS2 polymorphisms (T1026A located in intron 1, and 1891-1892del TC, located in intron 2) which were associated with hypertension. These associations, however, were only significant in women, the biological plausibility of which remains to be studied (Yang et al. 2003). Riddle and colleagues (2006) followed a similar approach in white and black Americans, and identified 16 variants in the RGS2 gene. Three variants affected the coding region but occurred at or below 1%. They also identified the 1891-1892TC insertion/deletion polymorphism, which was in complete linkage disequilibrium with the 2138-2139AA insertion/deletion polymorphism. While the 1891-1892TC deletion allele was significantly increased in black hypertensives (here without gender effect), there was no effect in whites (Riddle et al. 2006).
Genetic variants of G protein-coupled kinase 4
In addition to RGS proteins, G protein-coupled kinases (GRKs) are another system for the fine-tuning of GPCR-mediated signals. Phosphorylation of GPCRs by GRKs is an initial step in desensitization following receptor occupation with agonists. Next, the receptor binds to arrestins and other adaptor proteins, which leads to an uncoupling of the activated receptor from G protein complexes and terminates its function. Although different receptor classes vary, phosphorylated arrestin-complexed receptors undergo internalization via clathrin-coated pits, and cycle to an endosome where the receptor is dephosphorylated and recycled to the membrane or in other cases degraded in the lysosome or proteasome. There are several GRKs, including GRK1 and GRK7 that belong to the rhodopsin kinase family, GRK2 and GRK3 creating the β-adrenergic receptor kinase family, and GRK 4, 5 and 6 that belong to the GRK4 family. GRK2 is not only a receptor kinase but also contains an RGS domain, thus integrating two functions in attenuating GPCR signalling.
Dopamine has long been recognized as an important modulator of blood pressure, sodium balance and renal function. Within the kidneys, locally generated dopamine acts on renal tubular cells (in part from the tubular lumen) to decrease sodium transport under conditions of sodium excess (Fig. 14). While all dopamine receptor isoforms have been identified in the kidney, the D1 isoform appears to be the most important for the regulation of sodium balance, and the D1 receptor agonist fenoldopam is suitable for treatment of hypertensive emergencies. From human and animal studies, there is evidence for a renal dopamine receptor—or better signalling defect—in hypertension. Studies in the D1 receptor protein, G proteins, adenylyl cyclases, phospholipase C, sodium transporters or the sodium pump revealed no causal abnormalities, so far (for review of the renal dopaminergic system in hypertension see Zeng et al. 2004). Since the hypertensive phenotype with respect to renal dopamine signalling resembles processes observed in uncontrolled receptor desensitization, Felder et al. (2002) studied D1 receptor desensitization processes and observed an enhanced phosphorylation of the receptor protein at serine residues, which was attributed to an enhanced activity of the G protein-coupled receptor kinase 4 (GRK4). Four different splice variants of GRK4 are known, termed α, β, γ and δ. In contrast to other GRKs, GRK4 exhibits a restricted pattern of expression that includes brain, testes and the renal proximal tubule (Felder et al. 2002). Basal and stimulated GRK4 activity was enhanced in renal tubulus from hypertensive individuals, causing an increased desensitization of D1 receptor signalling. The GRK4 locus on chromosome 4p16 has been linked to hypertension (Allayee et al. 2001) and several amino acid exchanges in GRK4 were identified, including Arg65Leu, Ala142Val and Ala486Val (Felder et al. 2002). In vitro experiments revealed that the 65Leu, the 142Val and the 486Val variant appeared to be constitutively active and caused an increased phosphorylation of the D1 receptor, explaining the phenotype observed in some individuals with human hypertension (Fig. 14). Transgenic mice, overexpressing the GRK4γ isoform with the 142Val variant, exhibited increases in mean blood pressure by ∼30 mmHg compared to mice overexpressing the human wildtype variant (Felder et al. 2002). Mice overexpressing the GRK4 486Val variant were initally normotensive but developed hypertension on a sodium diet (Zeng et al. 2004).
In human hypertension, the common 486Val variant has been reported to be associated with salt-sensitive hypertension in Italians (Bengra et al. 2002). These results were confirmed in white Australians for the 486Val polymorphism, and were extended to several haplotypes that included the Arg65Leu and the Ala142Val variants (Speirs et al. 2004). Results published in abstract form indicate that GRK4 variants also affect the risk for hypertension in black African and Japanese populations (Zeng et al. 2004).
Evidence, mostly from animal studies, suggests that other GRKs may also contribute to the pathogenesis of hypertension. Targeted overexpression of GRK2 to the vasculature is accompanied by increases in resting blood pressure and attenuated β-adrenergic signalling (Eckhart et al. 2002). Similar results were obtained for the GRK5 isoform (Keys et al. 2005). In addition to receptor desensitization, there is evidence that GRK2 is involved in the regulation of endothelial NO synthase (eNOS) activity, at least in hepatic portal endothelial cells (Fig. 18). Here, GPCRs (e.g. the endothelin B receptor), activate the proteinkinase Akt via free Gβγ subunits and the PI3 kinase, which in turn phosphorylates and stimulates eNOS. GRK2 has been shown to bind to and inhibit Akt and thus NO generation, which may constitute the link to hypertension in transgenic GRK2 mice (Liu et al. 2005). It remains to be elucidated whether this mechanism is representative for the whole vasculature. At present, we stand at the beginning of a novel field of research that addresses the function of regulatory mechanisms in G protein-mediated signal transduction on vascular tone. First studies into the genetics of these target proteins have revealed promising results which require corroboration in the future.

Role of GRK2 in endothelial NO generation. Stimulation of G protein-coupled receptors (e.g. endothelin receptor) causes activation of the endothelial NO-synthase (eNOS) and the generation of NO. In addition to increases in cellular Ca2+, Gβγ-mediated activation of PI3 kinase contributes to the stimulation of eNOS. PI3 kinase phosphorylates the serine-threonine kinase AKT, which in turn phosphorylates and, thereby, activates eNOS. GRK2 is involved in the desensitization of activated GPCRs and also interferes with the PI3 kinase-mediated activation of AKT. This leads to an attenuated NO generation and to a decreased vasodilatory response. Targeted overexpression of GRK2 to the vasculature in mice causes severe hypertension
Polymorphisms in rho kinase gene (ROCK2)
Most recently, a polymorphism in the rho kinase gene (ROCK2; Thr431Asn) has been identified. Homozygous carriers of the 431Asn allele had an increased risk for hypertension and this allele accounted for ∼5% of the observed blood pressure variation (Seasholtz et al. 2006). Rho and Rho kinases present an additional pathway in GPCR signalling that is involved in stress-fiber formation, process retraction and cell rounding. RhoA, together with Rac1 and Cdc42, forms a subfamily within the huge family of small GTPases. GPCR-stimulated activation of RhoA involves G proteins of the Gα12/13 family. Subsequently, activated RhoA stimulates Rho kinases which are serine-threonine kinases. Two isoforms of Rho kinases have been identified; ROCK1, also referred to as 160ROCK, and ROCK2, which is predominantly expressed in vascular smooth-muscle cells. ROCK2-mediated phosphorylation of myosin phosphatase in vascular smooth-muscle cells inhibits the phosphatase activity and results in an enhanced accumulation of phosphorylated myosin light chains and, ultimately, vascular smooth muscle contraction (Fig. 19). ROCK inhibitors have been demonstrated to lower blood pressure (for review Riento and Ridley 2003). The identified 431Asn exchange is confined to a potential coiled-coil domain that has been implicated in homodimerization of ROCK2, which could be disturbed in carriers of the 431Asn allele (Seasholtz et al. 2006). Again, additional studies are required to further clarify the mechanistic and genetic roles of this protein for human hypertension.

a Domain structure of Rho kinase 2 (ROCK2), consisting of a kinase domain and a coiled-coil domain that is involved in the binding of activated Rho and, potentially, in the homodimerization. b Contribution of the Rho/ROCK system to the regulation of the vasoconstriction of vascular smooth-muscle cells. Here, GPCRs activate Gα subunits of the Gα12/13 family that stimulate a guanine nucleotide exchange factor for Rho (Rho GEF). GTP-bound Rho is active, and stimulates the ROCK2 which phosphorylates and, thereby, inhibits myosin phosphatase (MPP). Phosphorylation of the regulatory chain of myosin II is mediated by the myosin light chain kinase (MLCK) and results in smooth-muscle contraction. MPP counteracts MLCK. Upon inhibition of MPP by ROCK, MLCK activity prevails which causes an increased vasoconstriction. Of note, other signalling pathways (Ca2+/ calmodulin; cAMP, PKA; NO) also contribute to the fine-tuning of vascular smooth muscle tone
Hypertension and stroke
Randomized controlled studies of pharmacological intervention in hypertension have unequivocally documented that blood pressure reduction lowers morbidity and mortality, with dramatic reductions in stroke mortality and smaller reductions in myocardial infarction. Despite this overwhelming evidence, stroke prevention has remained an area where academic medicine, with its evidence-based guidelines, and daily routine practice appear to belong to different worlds. Target blood pressure values are seldom reached, in part related to behaviours of physicians and patients. Current epidemiological data indicate that only ∼50% of individuals with hypertension are diagnosed. Of these, 41% receive an antihypertensive treatment that results in 60% and 30% response rates at thresholds of 160/95 and 140/90 mmHg respectively. This leads to the most unfavourable situation that only 8% of all hypertensive individuals are controlled at guideline levels of <140/90 mmHg (Wolf-Maier et al. 2004). Under these conditions, genetic abnormalities that markedly impact blood pressure variation should also materialize as stroke genes. For monogenic forms of hypertension, several reports of increased stroke frequency exist, including hypertension with brachydacytly (Bilginturan et al. 1973), glucocorticoid remediable hypertension (Rich et al. 1992; Litchfield et al. 1998), apparent mineralocorticoid excess (New et al. 1977), and Gordon’s syndrome (PHA2).
It is noteworthy that for genes implicated to essential hypertension, most studies failed so far to demonstrate an effect on stroke risk. On one hand, this may indicate that the contributions of each of those genes to blood pressure variation are so small that effects on stroke frequency remain undetectable, particularly if only hypertensive populations and not stroke samples are analysed. On the other hand, this also questions the relevance of such genes for clinical medicine. However, some exceptions exist.
Two studies investigated the contribution of adducin polymorphisms to the genetics of stroke. While one report failed to demonstrate an association between the Add1 460W allele and ischemic stroke (Morrison et al. 2001), Psaty and co-workers reported an increased risk for combined hemorrhagic and ischemic stroke in carriers of the Add1 460W allele, although there was no association to hypertension itself (Psaty et al. 2002). In the same study mentioned just above, Morrison and coworkers (2001) documented an increased frequency for subclinical strokes in carriers of the GNB3 T allele, again independent of an effect on blood pressure. Several groups have addressed the effect of the common ACE polymorphism on stroke. Two meta-analyses that reported significant positive associations between the ACE D allele and ischemic stroke (Maeda et al. 1996; Sharma 1998) prompted a prospective matched case-control study within the Physicians’ Health Study. Here, non-significant odds ratios for carriers of the D allele for ischemic stroke, ranging from 1.10–1.22, were observed (Zee et al. 1999). No association was shown between the AT1R polymorphism and stroke (Tiret et al. 1998; Sierra et al. 2002).
Pharmacogenetics
The key rationale for all efforts in unravelling the genetic basis of hypertension etiology is the search for novel antihypertensive drug targets and the prospect for a tailored individualized drug therapy. A given antihypertensive shows effective blood pressure reduction (>10 mmHg) in 50–60% of the patients. Hence, most hypertensive patients require two or more antihypertensive medications to reach target blood pressures, and the optimization of drug therapy for the individual patient is frequently driven by ‘trial and error’. Genetic mechanisms are supposed to contribute to these inter-individual variations addressed by the field of pharmacogenetics, which can further be divided into a pharmacokinetic part that analyzes genetic variations in drug metabolism and transport with respect to ultimate drug effects, and a pharmacodynamic part that addresses variations in drug targets. It deserves mention that key concepts in pharmagenetics have been derived from antihypertensive drugs. CYP2D6 polymorphisms were identified for their dramatic effects on debrisoquine plasma levels, an antihypertensive no longer in use for these reasons (Eichelbaum and Gross 1990). Frequently used β-blockers including metoprolol, however, are also subject to CYP2D6 metabolism, which explains inter-individual differences in their efficacies and adverse effects (Fux et al. 2005).
Pharmacogenetic aspects of antihypertensive therapy have been covered by several excellent recent reviews (Mellen and Herrington 2005; Koopmans et al. 2003; Turner and Boerwinkle 2003; Turner et al. 2001a,b). Therefore, we will summarize relevant results only pertaining to genes discussed above, which completely neglects the whole field of drug disposition.
Since adducin polymorphisms are supposed to affect renal sodium handling, some groups have addressed potential implications for diuretic therapy, and confirmed a greater blood pressure response to hydrochlorothiazide in carriers of the 460W allele (Cusi et al. 1997; Glorioso et al. 1999, 2002), in part as an interaction with ACE gene polymorphisms (Sciarrone et al. 2003). In a large study with 595 hypertensive white and African Americans, however, no effect of the Add1 460W allele on blood pressure responses to hydrochlorothiazide was observed (Turner et al. 2003). As mentioned above, Psaty et al. (2002) followed a managed-care group of 1038 hypertensive individuals for several years. They observed that treatment with diuretics led to a relative reduction in incidences for stroke and myocardial infarction of almost 50% in carriers of the Add1 460W allele compared to other antihypertensive agents that produced a similar blood pressure reduction. A straightforward interpretation of these results indicates that the α-adducin system affects blood pressure-independent mechanisms in stroke pathogenesis that remain to be elucidated. The same group documented that therapy with ACE inhibitors resulted in differential protection from non-fatal stroke, depending on the AGT M235T genotype status (Bis et al. 2003).
Common polymorphisms in components of the RAS system have also been investigated for potential effects on antihypertensive treatments. One report showed that homozygous carriers of the I allele of the common ACE I/D polymorphism exhibited larger reductions in systolic blood pressure after 2 weeks treatment with hydrochlorothiazide (Sciarrone et al. 2003). Another group, however, were able to confirm the effect of the I allele on thiazide effects only for women, while they observed opposite reponses in males, a result that underscores the complexities of genetic interaction, if confirmed (Schwartz et al. 2002). For homozygous carriers of the wild-type AGT 235M allele or of the AGT promoter polymorphism -6G allele atenolol responses on systolic blood pressure were blunted (Kurland et al. 2004), a finding that may relate to antihypertensive mechanisms of β-blockers via inhibition of renin release. Another group observed a similar trend for blood pressure responses, which did, however, not reach statistical significance (Dudley et al. 1996). Several trials have been conducted for effects of the ACE I/D polymorphism on blood pressure responses to ACE inhibitors, documenting contradictory results, with greater responses in homozygous carriers of the D allele (Mondorf et al. 1998; Ueda et al. 1998; Stavroulakis et al. 2000), no difference (Hingorani et al. 1995; Dudley et al. 1996; Sasaki et al. 1996; Nakano et al. 1997; Harrap et al. 2003) and greater responses in homozygous carriers of the I allele (Ohmichi et al. 1997). For treatment with angiotensin receptor blockers, again one group reported that homozygous carriers of the I allele exhibited increased responses (Kurland et al. 2001). Similarly, for the AGT M235T polymorphism, one trial with ACE inhibitors provided evidence for an association of homozygous carriers of the M allele with ‘poor responder’ status (Hingorani et al. 1995), while two other groups refuted this finding (Dudley et al. 1996; Mondorf et al. 1998).
Since β-blockers modulate the actions of the β1 adrenoceptor, obvious candidates for pharmacogenetic effects are components of the G protein-coupled receptor system. While some groups reported enhanced β-blocker responses in homozygous carriers of the gain-of-function 389R allele in small studies (metoprolol: Liu et al. 2003; Johnson et al. 2003; atenolol: Sofowora et al. 2003), others did not observe effects of the G389R and the S49G polymorphisms in the β1 adrenoceptor gene on β-blocker responses (Filigheddu et al. 2004; Karlsson et al. 2004; O’Shaughnessy et al. 2000). A recent study reported 9% decreases in mean blood pressures for homozygous hypertensives with the 49S389R haplotypes, while blood pressure decreases were virtually absent in homozygous carriers of the 49S389G haplotypes or in 49S389G/49G389G diplotypes (Liu et al. 2006). Mixed results were also obtained for the T393C polymorphism in the GαS gene, the next step in signal transduction of the β1 adrenoceptor. While Jia et al. (1999) documented that the T-allele that was associated with enhanced blood pressure, predicted a poor response to β-blocker, others refuted this association (Filigheddu et al. 2004). Blood pressure-lowering responses of Atenolol in 221 hypertensive individuals were significantly enhanced in homozygote carriers of the GNB3 C allele. One interpretation is that under conditions of increased signalling (in case of the GNB3 T-allele) higher doses of a β-blocker could be necessary to achieve a comparable blood pressure response (Filigheddu et al. 2004). On the other hand, Turner et al. documented significantly greater decreases in systolic and diastolic blood pressure after a 4-week treatment period with hydrochlorothiazide for carriers of the GNB3 T-allele (Turner et al. 2001a,b), an astonishing result, since heterotrimeric G proteins do not contribute to early effects of diuretics. Full antihypertensive effects of diuretics, however, require latencies of several weeks, indicating complex adaptation processes in vascular reactivity for which an involvement of G protein-coupled mechanisms is highly probable. In a follow-up survey, this group, however, was not able to reproduce this observation (Turner et al. 2003). Such adaptation processes could also explain the effect of a common polymorphism in the gene for endothelial nitric oxide synthase on hydrochlorothiazide-induced responses in hypertensives. In this report, the effect of this polymorphism contributed to only 1% to the observed inter-individual variation after adjusting for age, gender, race, and waist-to-hip ratio, a ‘magnitude’ of effect that was similar with other polymorphisms, at best (Turner et al. 2003).
Conclusions and future developments
Considerable financial funding, vast human resources and immeasurable individual efforts have been spent to unravel the enigma ‘genetics of hypertension’. The promises of the clarification of the etiology of primary hypertension are suitable preclinical diagnosis procedures, the delineation of hypertensive pathways, the discovery of novel drug targets which will ultimately result in tailored individualized therapeutic regimes. Actually, lessons from monogenetic forms of hypertension have considerably expanded our molecular, cellular and systemic understanding of salt und volume regulation, of mineralocorticoid action and of the signalling involved in regulation of ion transporter and channel activity. Given the diversity of physiologic systems that affect blood pressure, it is perhaps surprising that almost all gene products in monogenic hypertension affect salt absorption in the kidneys. While monogenic diseases impart large effects to small numbers of patients, we are still at the beginning of addressing the small effects of polygenic ‘complex’ mechanisms on large numbers of patients. At best, analyses of common variants linked to essential hypertension explain only a small fraction of the observed variance in blood pressure in the general population. Lack of consistent reproduction appears to be the destiny of many common polymorphisms implicated in the genesis of complex diseases, which underscores the difficulties of genetic research into multifactorial traits. While we have evidence for modest effects of some polymorphisms on a statistical level, for individuals we are far from predicting the genetic risk for hypertension with sufficient precision (if at all) to allow us to improve diagnosis, prevention or differential therapy. Upon demonstration of an initial association, subsequent studies fail to confirm or may refute earlier findings (Anonymous 1999; Hirschhorn et al. 2002). Low sample sizes, limited statistical power, low genotype-relative risks, heterogeneity of studied populations and genetic admixture, publication bias, imperfect characterization of patients and controls, context dependency and epistasis may contribute to these sobering results. Hence, adequate sample sizes, adequate recognition of the ethnic identity of the enrolled subjects, use of family studies if possible, preference of haplotype-based analyses over single SNPs, and the careful consideration of biological plausibilities from the molecular to the systemic level are urgently needed.
With regard to pharmacogenetics, the available basis of evidence cannot be taken as sound. Currently, patient numbers in trials has never exceeded 200 per ethnicity at best, and most trials have addressed several polymorphisms in parallel, generating considerable statistical problems. For instance, the SILVHIA study—which is the largest trial addressing pharmacogenetic effects of angiotensin receptor blockade or atenolol treatment in hypertension—analyzed 74 SNPs in each 100 patients, providing ample opportunity for spurious associations (Kurland et al. 2004). It will be important for future pharmacogenetic studies that independent researchers get access to industry-sponsored trials, and that the consideration of pharmacogenetic issues is mandatory for regulatory measures. To nurture the growth of pharmacogenetics from basic science to clinical medicine, outcome-oriented research representative of clinical reality is required to provide a useable basis for a genetic assessment of risks and benefits.
Current approaches try to unravel the genetic mysteries of essential hypertension in genome-wide linkage analyses, in part analyzing huge and well-characterized human samples, e.g. from the Framingham Heart Study, the MRC-funded British Genetics of Hypertension (BRIGHT; 1599 families with 2010 sibling pairs) study, and the NIH-funded Family Blood Pressure Program (FBPP). Again, the results published so far are disappointing, with either no linkage or comparably weak linkages, e.g. to chromosomes 1, 2, 5, 6, 9 or 17, which do not overlap between different trials (Hsueh et al. 2000; Levy et al. 2000; Thiel et al. 2003; Caulfield et al. 2003; Wallace et al. 2006). Candidate gene approaches revealed a wealth of interesting and not so interesting associations, but also failed to uncover relevant hypertension candidate genes so far. The future will show whether hypertension research will succeed in the identification of a manageable number of hypertension candidate genes that will be useful for clinical medicine. Otherwise, it is also possible that we will face a situation with a large number of genes and a myriad of polymorphism that may actually affect blood pressure values in individuals, in ways, however, not suitable for prediction in daily routine. In the meantime, we have already learned and we will learn how—at first glance—completely different systems of ion transport, membrane trafficking, and signal transduction interplay to control salt and volume homeostasis and vasoconstriction. Hypertension research has sparked fascinating work into novel protein families including WNK kinases, special RGS proteins or GRKs. Whether this research will ultimately unravel hypertension genetics remains open. However, we should be optimistic that these efforts open the way for a better understanding of the complex renal and vascular regulation, and in the end may provide us with novel drug targets and potentially innovative classes of antihypertensive therapeutics.
Abbreviations
- β1AR:
-
β1-adrenoceptor
- β2AR:
-
β2-adrenoceptor
- ACE:
-
angiotensin converting enzyme
- Add:
-
adducin
- AGT:
-
angiotensinogen
- AKT:
-
protein kinase AKT
- AME:
-
apparent mineralocorticoid excess
- Ang II:
-
angiotensin II
- AT1R:
-
angiotensin II receptor, type 1
- CLCNKA, CLCNKB:
-
renal epithelial chloride channels
- ENaC:
-
epithelial sodium channel
- eNOS:
-
endothelial NO synthase
- GAP:
-
GTPase activating protein
- GNAS:
-
gene of the G protein Gαs subunit
- GNB3:
-
gene of the G protein Gβ3 subunit
- GPCR:
-
G protein-coupled receptor
- GRA:
-
glucocorticoid-remediable aldosteronism
- GRK:
-
G protein-coupled receptor kinase
- HSD11B2:
-
11β-hydroxysteroid dehydrogenase type 2
- MHS, MNS:
-
Milan hypertensive/normotensive strain of rats
- MR:
-
mineralocorticoid receptor
- NCC=NCCT=SLC12A3:
-
thiazide-sensitive Na+/Cl− transporter
- NKCC2=SCL12A1:
-
Na+/K+/2Cl− transporter
- PHA:
-
pseudohypoaldosteronism
- PKA, PKC, PKG:
-
protein kinases A, C, G
- QTL:
-
quantitative trait locus
- RAS:
-
renin-angiotensin-system
- RGS:
-
regulators of G protein signalling
- ROCK:
-
rho kinase
- ROMK:
-
renal outer medulla K+ channel
- SCNN1A,SCNN1B, SCNN1G:
-
genes for the epithelial sodium channel subunits α, β, γ
- SGK1:
-
sodium and glucocorticoid-induced kinase
- SNP:
-
single nucleotide polymorphism
- UTR:
-
untranslated region
- WNK=PRKWNK:
-
protein kinase with no lysine
References
Abe M, Nakura J, Yamamoto M, Jin JJ, Wu Z, Tabra Y, Yamamoto Y, Igase M, Kohara K, Miki T (2002) Association of GNAS1 gene variant with hypertension depending on smoking status. Hypertension 40:261–265
Agarwal A, Williams GH, Fisher ND (2005) Genetics of human hypertension. Trends Endocrinol Metab 16:127–133
Agerholm-Larsen B, Nordestgaard BG, Tybjaerg-Hansen A (2000) ACE gene polymorphisms in cardiovascular disease: meta-analysis of small and large studies in whites. Arterioscler Thromb Vasc Biol 20:484–492
Allayee H, Dominguez KM, Aouizerat BE, Krauss RM, Rotter JI, Lu J, Cantor RM, de Bruin TW, Lusis AJ (2001) Genome scan for blood pressure in Dutch dyslipidemic families reveals linkage to a locus on chromosome 4p. Hypertension 38:773–778
Anonymous (1999) Freely associating. Nat Genet 22:1–2
Aperia AC (2000) Intrarenal dopamine: a key signal in the interactive regulation of sodium metabolism. Annu Rev Physiol 62:621–647
Ariza JL, Weinberger C, Cerelli G, Glaser TM, Handelin BL, Housman DE, Evans RM (1987) Cloning of human mineralocorticoid receptor complementary DNA: structural and functional kinship with the glucocorticoid receptor. Science 237:268–275
Armor M, Parker KL, Globerman H, New MI, White PC (1988) Mutation in the CYP21B gene (ile 172 to asn) causes steroid-21-hydroxylase deficiency. Proc Natl Acad Sci USA 85:1600–1604
Baker EH, Dong YB, Sagnella GA, Rothwell M, Onipinla AK, Markandu ND, Cappuccio FP, Cook DG, Persu A, Corvol P, Jeunemaitre X, Carter ND, MacGregor GA (1998) Association of hypertension with T594M mutation in the beta subunit of epithelial sodium channels in black people resident in London. Lancet 351:1388–1392
Bähring S, Nagai T, Toka HR, Nitz I, Toka O, Aydin A, Muhl A, Wienker TF, Schuster H, Luft FC (1997) Deletion at 12p in a Japanese child with brachydactyly overlaps the assigned locus of brachydactyly with hypertension in a Turkish family. Am J Hum Genet 60:732–735
Bähring S, Rauch A, Toka O, Schroeder C, Hesse C, Siedler H, Fesüs G, Haefeli WE, Busjahn A, Aydin A, Neuenfeld Y, Mühl A, Toka HR, Gollasch M, Jordan J, Luft FC (2004) Autosomal-dominant hypertension type E brachydactyly is caused by rearrangement on the short arm of chromosome 12. Hypertension 43:471–476
Bengra C, Mifflin TE, Khripin Y, Manunta P, Williams SM, Jose PA, Felder RA (2002) Genotyping essential hypertension SNPs using a homogenous PCR method with universal energy transfer primers. Clin Chem 48:2131–2140
Bengtsson K, Melander O, Orho-Melander M, Lindblad U, Ranstam J, Ranstam L, Groop L (2001a) Polymorphisms in the β1-adrenergic receptor gene and hypertension. Circulation 104:187–190
Bengtsson K, Orho-Melander M, Melander U, Lindblad U, Ranstam J, Ranstam L, Groop L (2001b) Beta(2) adrenergic receptor gene variation and hypertension in subjects with type 2 diabetes. Hypertension 37:1303–1308
Benjafield AV, Jeyasingam CL, Nyholt DR, Griffiths LR, Morris BJ (1998) G-protein beta3 subunit gene (GNB3) variant in causation of essential hypertension. Hypertension 32:1094–1097
Bernstein LS, Ramineni S, Hague C, Cladman W, Chidiac P, Levey AI, Hepler JR (2004) RGS2 binds directly and selectively to the M1 muscarinic acetylcholine receptor third intracellular loop to modulate Gq/11alpha signaling. J Biol Chem 279:21248–21256
Bernstein KE, Xiao HD, Frenzel K, Li P, Shen XZ, Adams JW, Fuchs S (2005) Six truisms concerning ACE and the renin-angiotensin-system educed from the genetic analysis of mice. Circ Res 96:1135–1144
Bettinelli A, Bianchetti MG, Giardin E, Caringella A, Cecconi M, Appiani AC, Ravanello L, Gastaldi R, Isimbaldi C, Lama G (1992) Use of calcium excretion values to distinguish two forms of primari renal tubular hypokalemic alkalosis: Bartter and Gitelmans syndromes. J Pediatr 120:38–43
Bianchi G, Tripodi MG, Casari G, Salardi S, Barber BR, Garcia R, Leoni P, Torielli L, Cusi D, Ferrandi M, Pinna LA, Baralle FE, Ferrari P (1994) Two point mutations within the adducin genes are involved in blood pressure variation. Proc Natl Acad Sci USA 91:3999–4003
Bianchi G, Ferrari P, Staessen JA (2005) Adducin polymorphism. Detection and impact on hypertension and related disorders. Hypertension 45:331–340
Bilginturan N, Zileli S, Karacadag S, Pirnar T (1973) Hereditary brachydactyly with hypertension. J Med Genet 10:253–259
Bindels RJM (2003) A molecular switch controlling renal sodium and potassium excretion. Nat Genet 35:302–303
Birkenhager R, Otto E, Schurmann MJ, Vollmer M, Rufe EM, Maier-Lutz I, Beekmann F, Fekete A, Imran H, Feldmann D, Milford DV, Jeck N, Konrad M, Landau D, Knoers NVAM, Antignac C, Sudbrak R, Kispert A, Hildebrandt F (2001) Mutations of BSND causes Bartter syndrome with sensorineural deafness and kidney failure. Nature Genet 29:310–314
Biron P, Mongeau JG, Bertrand D (1976) Familial aggregation of blood pressure in 558 adopted children. Can Med Assoc J 115:773–774
Bis JC, Smith NL, Psaty BM, Heckbert SR, Edwards KL, Lemaitre RN, Lumley T, Rosendaal FR (2003) Angiotensinogen Met235Thr polymorphism, angiotensin-covnerting enzyme inhibition therapy, and the risk for nonfatal stroke or myocardial infarction in hypertension patients. Am J Hypertens 16:1011–1017
Bloem LJ, Manantunga AK, Pratt JH (1996) Racial differences in the relationship of an angiotensin-I-converting enzyme gene polymorphism to serum angiontensin I-converting enzyme activity. Hypertension 27:62–66
Bongiovanni AM, Root AW (1963) The adrenogenital syndrome. N Engl J Med 268:1283–1289; 1342–1351; 1391–1399
Bonnardeaux A, Davies E, Jeunemaitre X, Fery I, Charru A, Clauser E, Tiret L, Cambien F, Corvol P, Soubrier F (1994) Angiotensin II type I receptor gene polymorphisms in human essential hypertension. Hypertension 24:63–69
Botero-Velez M, Curtis JJ, Warnock DG (1994) Liddle’s syndrome revisited–a disorder of sodium reabsorption in the distal tubule. N Engl J Med 330:178–181
Brand E, Herrmann SM, Nicaud V, Ruidavets JB, Evans A, Arveiler D, Luc G, Plouin PF, Tiret L, Cambien F (1999) The 825C/T polymorphism of the G-protein subunit β3 is not related to hypertension. Hypertension 33:1175–1178
Brand E, Chatelain N, Paillard F, Tiret L, Visvikis S, Lathrop M, Soubrier F, Demenais F (2002) Detection of putative functional angiotensinogen (AGT) gene variants controlling plasma AGT levels by combined segregation-linkage analysis. Eur J Hum Genet 10:715–723
Brand E, Wang JG, Herrmann SM, Staessen JA (2003) An epidemiological study of blood pressure and metabolic phenotypes in relation to the Gbeta3 C825T polymorphism. J Hypertens 21:729–737
Bray MS, Krushkal J, Li L, Ferrell R, Kardia S, Sing CF, Turner ST, Boerwinkle E (2000) Positional genomic analysis identifies the beta(2) adrenergic receptor gene as a susceptibility locus for human hypertension. Circulation 101:2877–2882
Büscher R, Belger H, Eilmes KJ, Tellkamp R, Radke J, Dhein S, Hoyer PF, Mitchel MC, Insel PA, Brodde OE (2001) In-vivo studies do not support a major functional role for the Gly389Arg β1-adrenoceptor polymorphism in humans. Pharmacogenetics 11:199–205
Busjahn A, Li GH, Faulhaber HD, Rosenthal M, Becker A, Jeschke E, Schuster H, Timmermann B, Hoehe MR, Luft FC (2000) Beta-2 adrenergic recptor gene variations, blood pressure, and heart size in normal twins. Hypertension 35:555–560
Cambien F, Poirier O, Lecerf L, Evans A, Cambou JP, Arveiler D, Luc G, Bard JM, Bara L, Ricard S (1992) Deletion polymorphism in the gene for angiotensin-converting enzyme is a potent risk factor for myocardial infarction. Nature 359:641–644
Castellano M, Glorioso N, Cusi D, Sarzani R, Fabris B, Opocher G, Zoccali C, Golin R, Veglio F, Volpe M, Mantero F, Fallo F, Rossi GP, Barlassina C, Tizzoni L, Filigheddu F, Giacche M, Rossi F (2003) Genetic polymorphisms of the renin-angiotensin-aldosterone system and arterial hypertension in the Italian population: the GENIPER project. J Hypertens 21:1853–1860
Caulfield M, Lavender P, Farrall M, Munroe P, Lawson M, Turner P, Clark AJ (1994) Linkage of the angiotensinogen gene to essential hypertension. N Engl J Med 330:1629–1633
Caulfield M, Lavender P, Newell-Price J, Farrall M, Kamdar S, Daniel H, Lawson M, De Freitas P, Fogarty P, Clark AJ (1995) Linkage of the angiotensinogen gene locus to human essential hypertension in African Caribbeans. J Clin Invest 96:687–692
Caulfield M, Munroe P, Pembroke J, Samani N, Dominiczak A, Brown M, Benjamin N, Webster J, Ratcliffe P, O’Shea S, Papp J, Taylor E, Dobson R, Knight J, Newhouse S, Hooper J, Lee W, Brain N, Clayton D, Lathrop GM, Farrall M, Connell J; MRC British Genetics of Hypertension Study (2003) Genome-wide mapping of human loci for essential hypertension. Lancet 361:2118–2123
Chang SS, Grunder S, Hanukoglu A, Rosler A, Mathew PM, Hanukoglu I, Schild L, Lu Y, Shimkets RA, Nelson-Williams C, Rossier BC, Lifton RP (1996) Mutations in subunits of epithelial sodium channels cause salt wasting with hyperkalaemic acidosis, pseudohypoaldosteronism type I. Nat Genet 12:248–253
Chen Y, Nakura J, Jin JJ, Wu Z, Yamamoto M, Abe M, Tabara Y, Yamamoto Y, Igase M, Bo X, Kohara K, Miki T (2003) Association of the GNAS1 gene variant with hypertension is dependent on alcohol consumption. Hypertens Res 26:439–444
Chibalin AV, Zierath JR, Katz AI, Berggren P-O, Bertorello AM (1998) Phosphatidylinositol 3-kinase-mediated endocytosis of renal Na+, K+-ATPase α subunit in response of dopamine. Mol Biol Cell 9:1209–1220
Chibalin AV, Ogimoto G, Pedemonte CH, Pressley TA, Katz AI, Feraille E, Berggren PO, Bertorello AM (1999) Dopamine-induced endocytosis of Na ,K -ATPase is initiated by phosphorylation of Ser-18 in the rat α-subunit and is responsible for the decreased activity in epithelial cells. J Biol Chem 274:1920–1927
Chu SL, Zhu DL, Xiong MM, Wang GL, Zhang WZ, Zhou HF, Shen D, Gao PJ, Zhan YM, Jin L (2002) Linkage analysis of twelve candidate gene loci regulating water and sodium metabolism and membrane ion transport in essential hypertension. Hypertens Res 25:635–639
Cruz DN, Simon DB, Nelson-Williams C, Farhi A, Finberg K, Burleson L, Gill JR, Lifton RP (2001) Mutations in the Na-Cl cotransporter reduce blood pressure in humans. Hypertension 37:1458–1464
Cusi D, Bianchi G (1996) Renal mechanisms of genetic hypertension: from the molecular level to the intact organism. Kidney Intern 49:1754–1759
Cusi D, Barlassina C, Azzani T, Casari G, Citterio L, Devoto M, Glorioso N, Lanzani C, Manunta P, Righetti M, Rivera R, Stella P, Troffa C, Zagato L, Bianchi G (1997) Polymorphisms of α-adducin and salt sensitivity in patients with essential hypertension. Lancet 349:1353–1357
Disse-Nicodeme S, Achard JM, Desitter I, Houot AM, Fournier A, Corvol P, Jeunemaitre X (2000) A new locus on chromose 12p13.3 for pseudohypoaldosteronism type II, an autosomal dominant form of hypertension. Am J Hum Genet 67:302–310
Done SC, Leibiger IB, Efendiev R, Katz AI, Leibiger B, Berggren PO, Pedemonte CH, Bertorello AM (2002) Tyrosine 537 within the Na+, K+-ATPaseα subunit is essential for AP-2 binding and clathrin-dependent endocytosis. J Biol Chem 277:17108–17111
Dong Y, Zhu H, Sagnella GA, Carter ND, Cook DG, Cappuccio FP (1999) Association between the C825T polymorphism of the G protein β3-subunit gene and hypertension in blacks. Hypertension 34:1193–1196
Dudley C, Keavney B, Casadei B, Conway J, Bird R, Ratcliffe P (1996) Prediction of patient responses to antihypertensive drugs using genetic polymorphisms: investigation of the renin-angiotensin system genes. J Hypertens 17:9–17
Eckhart AD, Ozaki T, Teaearai H, Rockman HA, Koch WJ (2002) Vascular-targeted overexpression of G protein-coupled receptor kinase-2 in transgenic mice attenuates beta-adrenergic receptor signaling and increases resting blood pressure. Mol Pharmacol 61:749–758
Efendiev R, Krmar RT, Leibiger IB, Ogimoto G, Zwiller J, Tripodi G, Katz AI, Bianchi G, Pedemonte CH, Bertorello AM (2004) Hypertension-linked mutation in the adducin α-subunit affects AP2-μ2 phosphorylation and impairs Na+,K+-ATPase endocytosis. Circ Res 95:1100 –1108
Eichelbaum M, Gross AS (1990) The genetic polymorphisms of debrisoquine/sparteine: metabolism–clinical aspects. Pharmacol Ther 46:377–394
Erlich PM, Cui J, Chazaro I, Farrer LA, Baldwin CT, Gavras H, DeStefano AL (2003) Genetic variants of WNK4 in whites and African Americans with hypertension. Hypertension 41:1191–1195
Feinleib M, Garrison RJ, Fabsitz R, Christian JC, Hrubec Z, Borhani NO, Kannel WB, Rosenman R, Schwartz JT, Wagner JO (1977) The NHLBI twin study of cardiovascular disease risk factors: methodology and summary of results. Am J Epidemiol 106:284–285
Felder RA, Sanada H, Xu J, Yu PY, Wang Z, Watanabe H, Asico LD, Wang W, Zheng S, Yamaguchi I, Williams SM, Gainer J, Brown NJ, Hazen-Martin D, Wong LJ, Robillard JE, Carey RM, Eisner GM, Jose PA (2002) G protein-coupled receptor kinase 4 gene variants in human essential hypertension. Proc Natl Acad Sci USA 99:3872–3877
Fernandez-Llama P, Poch E, Oriola J, Botey A, Coll E, Darnell A, Rivera F, Revert L (1998) Angiotensin converting enzyme gene I/D polymorphism in essential hypertension and nephroangiocsclerosis. Kidney Int 53:1743–1747
Ferrandi M, Salardi S, Tripodi MG, Barassi P, Rivera R, Manunta P, Goldshleger R, Ferrari P, Bianchi G, Karlish S (1999) Evidence for an interaction between adducin and Na,K’ATPase: relation to genetic hypertension. Am J Physiol 277:1338–1349
Ferrari P, Bianchi G (1995) Lessons from experimental genetic hypertension. In: Laragh JH, Brenner BM, eds. Hypertension: Pathophysiology, Diagnosis, and Management. 2nd ed. Raven Press, New York, NY, pp 1261–1279
Filigheddu F, Reid JE, Troffa C, PinnaParpaglia P, Argiolas G, Testa A, Skolnick M, Glorioso N (2004) Genetic polymorphisms of the beta-adrenergic system: association with essential hypertension and response to beta-blockade. Pharmacogenomics J 4:154–160
Fornage M, Amos CI, Kardia S, Sing CF, Turner ST, Boerwinkle E (1998) Variations in the region of the angiotensin-converting enzyme gene influences interindividual differences in blood pressure levels in young white males. Circulation 97:1773–1779
Franca LP, Pacheco NA, Correa SA, Han SW, Nakaie CR, Paiva AC, Shimuta SI (2003) Angiotensin II-mediated cellular responses: a role for the 3′-untranslated region of the angiotensin AT1 receptor. Eur J Pharmacol 476:25–30
Funder JW, Pearce PT, Smith R, Smith AI (1987) Mineralocorticoid action: target tissue specificity is enzyme, not receptor, mediated. Science 242:583–585
Fux R, Morike K, Prohmer AM, Delabar U, Schwab M, Schaeffeler E, Lorenz G, Gleiter CH, Eichelbaum M, Kivisto KT (2005) Impact of CYP2D6 genotype on adverse effects during treatment with metoprolol: a prospective clinical study. Clin Pharmacol Ther 78:378–387
Geller DS, Rodriguez-Soriano J, Boado AV, Schifter S, Bayer M, Chang SS, Lifton RP (1998) Mutations in the mineralocorticoid receptor gene cause autosomal-dominant pseudohypoaldosteronism type I. Nat Genet 19:279–281
Geller DS, Farhi A, Pinkerton N, Fradley M, Moritz M, Spitzer A, Meinke G, Tsai FT, Sigler PB, Lifton RP (2000) Activating mineralocorticoid mutations in hypertension exacerbated by pregnancy. Science 289:119–123
Giner V, Poch E, Bragulat E, Oriola J, Gonzalez D, Coca A, de la Sierra A (2000) Renin-angiotensin system genetic polymorphisms and salt sensitivity in essential hypertension. Hypertension 35:512–517
Glorioso N, Manunta P, Filigheddu F, Troffa C, Stella P, Barlassina C, Lombardi C, Soro A, Dettori F, Pinna Parpaglia P, Sciarrone Alibrandi MT, Cusi D, Bianchi G (1999) The role of α-adducin polymorphism in blood pressure and sodium handling regulation may not be excluded by a negative association study. Hypertension 34:649–654
Glorioso N, Filigheddu F, Cusi D, Troffa C, Conti M, Natalizio M, Argiolas G, Barlassina C, Bianchi G (2002) Alpha-Adducin 460Trp allele is associated with erythrocyte Na transport rate in North Sardinian primary hypertensives. Hypertension 39:357–362
Gong M, Zhang H, Schulz H, Lee YA, Sun K, Bähring S, Luft FC, Nürnberg P, Reis A, Rohde K, Ganten D, Hui R, Hübner N (2003) Genome-wide linkage reveals a locus for human essential (primary) hypertension on chromosome 12p. Hum Mol Genet 12:1273–1277
Gordon D, Geddes RA, Pawsey CG, O’Halloran MW (1970) Hypertension and severe hyperkalaemia associated with suppression of renin and aldosterone and completely reversed by dietary sodium restriction. Australas Ann Med 4:287–294
Grant SL, Lassegue B, Griendling KK, Ushio-Fukai M, Lyons PR, Alexander RW (2000) Specific regulation of RGS2 messenger RNA by angiotensin II in cultured vascular smooth muscle cells. Mol Pharmacol 57:460–467
Gratze G, Fortin J, Labugger R, Binder A, Kotanko P, Timmermann B, Luft FC, Hoehe MR, Skrabal F (1999) Beta-2 adrenergic receptor variants affect resting blood pressure and agonist-induced vasodilation in young adult Caucasians. Hypertension 33:1425–1430
Green SA, Cole G, Jacinto M, Inniw M, Liggett SB (1993) A polymorphism of the human beta 2-adrenergic receptor within the fourth transmembrane domain alters ligand binding and functional properties of the receptor. J Biol Chem 268:23116–23121
Green SA, Turki J, Innis M, Liggett SB (1994) Amino-terminal polymorphisms of the human beta 2-adrenergic receptor impart distinct agonist-promoted regulatory properties. Biochemistry 33:9414–9419
Gu CC, Chang YP, Hunt SC, Schwander K, Arnett D, Djousse L, Heiss G, Oberman A, Lalouel JM, Province M, Chakravarti A, Rao DC (2005) Haplotype association analysis of AGT variants with hypertension-related traits: the HyperGEN study. Hum Hered 60:164–176
Hague C, Bernstein LS, Ramineni S, Chen Z, Minneman KP, Hepler JR (2005) Selective inhibition of alpha1A-adrenergic receptor signaling by RGS2 association with the receptor third intracellular loop. J Biol Chem 280:27289 –27295
Hahntow IN, Koopmans RP, Michel MC (2006) The beta2-adrenoceptor gene and hypertension: is it the promoter or the coding region or neither? J Hypertens 24:1003–1007
Hannson JH, Schild L, Lu Y, Wilson TA, Gautschi I, Shimkets R, Nelson-Williams C, Rossier BC, Lifton RP (1995a) A de novo missense mutation of the beta subunit of the epithelial sodium channel causes hypertension and Liddle syndrome, identifying a proline-rich segment critical for regulation of channel activity. Proc Natl Acad Sci USA 92:11495–11499
Hannson JH, Nelson-Williams C, Suzuki H, Schild L, Shimkets R, Canessa CM, Iwasaki T, Rossier B, Lifton RB (1995b) Hypertension caused by a truncated epithelial sodium channel γ subunit: genetic heterogeneity of Liddle syndrome. Nat Genet 11:76–82
Hanukoglu (1991) Type 1 pseudohypoaldosteronism includes two clinically and genetically distinct entities with either renal or multiple target organ defects. J Clin Endocrinol Metab 73:936–944
Harrap SB, Tzourio C, Cambien F, Poirier O, Raoux S, Chalmers J, Chapman N, Colman S, Leguennec S, MacMahon S, Neal B, Ohkubo T, Woodward M; PROGRESS Collaborative Group (2003) The ACE gene I/D polymorphism is not associated with the blood pressure and cardiovascular benefits of ACE inhibition. Hypertension 42:297–303
Hata A, Namikawa C, Sasaki M, Sato K, Nakamura T, Tamura K, Lalouel JM (1994) Angiotensinogen as a risk factor for essential hypertension in Japan. J Clin Invest 93:1285–1287
He X, Wang G, Huang W, Ding-Liang Z (2003) Linkage analysis of five candidate genes and essential hypertension in 106 Chinese nuclear families. J Hum Hypertens 17:69–72
Henskens LH, Spiering W, Stoffers HE, Soomers FL, Vlietinck RF, de Leeuw PW, Kroon AA (2003) Effects of the ACE I/D and AT1R-A1166C polymorphisms on blood pressure in healthy normotensive primary care population: first results of the Hippocrates study. J Hypertens 21:81–86
Herrmann SM, Nicaud V, Tiret L, Evans A, Kee F, Ruidavest JB, Arveiler D, Luc G, Morrison C, Hoehe MR, Paul M, Cambien F (2002) Polymorphisms of the beta2 adrenoceptor (ADRB2) gene and essential hypertension: the ECTIM and PEGASE studies. J Hypertens 20:229–235
Heximer SP, Watson N, Linder ME, Blumer KJ, Hepler JR (1997) RGS2/G0S8 is a selective inhibitor of Gqalpha function. Proc Natl Acad Sci USA 94:14389–14393
Heximer SP, Knutsen RH, Sun X, Kaltenbronn KM, Rhee MH, Peng N, Oliveira-dos-Santos A, Penninger JM, Muslin AJ, Steinberg TH, Wyss JM, Mecham RP, Blumer KJ (2003) Hypertension and prolonged vasoconstrictor signaling in RGS2-deficient mice. J Clin Invest 111:445–452
Hilgers KF, Langenfeld MR, Schlaich M, Veelken R, Schmieder RE (1999) 1166 A/C polymorphism of the angiotensin II type 1 receptor gene and the respone to short-term infusion of angiotensin II. Circulation 100:1394–1399
Hilgers KF, Delles C, Veelken R, Schmieder RE (2001) Angiotensinogen gene core promoter variants and non-modulating hypertension. Hypertension 38:1250–1254
Hingorani AD, Jia H, Stevens PA, Hopper R, Dickerson JE, Brown MJ (1995) Renin-angiotensin system gene polymorphisms influence blood pressure and the response to angiotensin converting enzyme inhibition. J Hypertens 13:1602–1609
Hirschhorn JN, Lohmueller K, Byrne E, Hirschhorn KA (2002) A comprehensive review of genetic association studies. Genet Med 4:45–61
Hollier JM, Martin DF, Bell DM, Li JL, Chirachanchai MG, Menon DV, Leonard D, Wu X, Cooper RS, McKenzie C, Victor RG, Auchus RJ (2006) Epithelial sodium channel allele T594M is not associated with blood pressure or blood pressure response to amiloride. Hypertension 474:428–433
Hsueh WC, Mitchell BD, Schneider JL, Wagner MJ, Bell CJ, Nanthakumar E, Shuldiner AR (2000) QTL influencing blood pressure maps to the region of PPH1 on chromosome 2q31-34 in old order Amish. Circulation 101:2810–2816
Humma LM, Puckett BJ, Richardson HE, Terra SG, Andrisin TE, Lejeune BL, Wallace MR, Lewis JF, McNamara M, Picoult-Newberg L, Pepine CJ, Johnson JA (2001) Effects of β-1 AR genetic polymorphisms on resting haemodynamics in patients undergoing diagnostig testing for ischemia. Am J Cardiol 88:1034–1037
Iliadou A, Lichtenstein P, Morgenstern R, Forsberg L, Svensson R, de Faire U, Martin NG, Pedersen NL (2002) Repeated blood pressure measurements in a sample of Swedish twins: heritabilities and associations with polymorphisms in the renin-angiotensin-aldosterone system. J Hypertens 20:1543–1550
Ingi T, Krumins AM, Chidiac P, Brothers GM, Chung S, Snow BE, Barnes CA, Lanahan AA, Siderovski DP, Ross EM, Gilman AG, Worley PF (1998) Dynamic regulation of RGS2 suggests a novel mechanism in G-protein signaling and neuronal plasticity. J Neurosci 18:7178–7188
Inoue I, Nakajima T, Williams CS, Quackenbush J, Puryear R, Powers M, Cheng T, Ludwig EH, Sharma AM, Hata A, Jeunemaitre X, Lalouel JM (1997) A nucleotide substitution in the promoter of human angiotensinogen is associated with essential hypertension and affects basal transcription in vitro. J Clin Invest 99:1786–1797
Inoue I, Iwaoka T, Tokunaga H, Takamune K, Naomi S, Araki M, Takahama K, Yamaguchi K, Tomita K (1998) A family with Liddle’s syndrome caused by a new missense mutation in the beta subunit of the epithelial sodium channel. J Clin Endocrinol Metab 83:2210–2213
Iwai N, Ohmichi N, Nakamura Y, Kinoshita M (1994) DD genotype of the angiotensin-converting enzyme gene is a risk factor for left ventricular hypertrophy. Circulation 90:2622–2628
Iwai N, Baba S, Mannami T, Katsuya T, Higaki J, Ogihara T, Ogata J (2001) Association of sodium channel gamma-subunit promoter variant with blood pressure. Hypertension 38:86–89
Iwai N, Baba S, Mannami T, Ogihara T, Ogata J (2002) Association of a sodium channel α promoter variant with blood pressure. J Am Soc Neprol 13:80–85
Janetta PJ, Segal R, Wolfson SK (1985) Neurogenic hypertension: etiology and surgical treatment. I. Observations in 53 patients. Ann Surg 201:391–398
Jeck N, Waldegger S, Lampert A, Boehmer C, Waldegger P, Lang PA, Wissinger B, Friedrich B, Risler T, Moehle R, Lang UE, Zill P, Bondy B, Schaefeler E, Asante-Poku S, Seyberth J, Schwab M, Lang F (2004) Activating mutation of the renal epithelial chloride channel ClC-Kb presdisposing to hypertension. Hypertension 43:1175–1181
Jeunemaitre X, Lifton RP, Hunt SC, Williams RR, Lalouel JM (1992a) Absence of linkage between the angiotensin converting enzyme locus and human essential hypertension. Nat Genet 1:72–75
Jeunemaitre X, Soubrier F, Kotelevtsev YV, Lifton RP, Williams SC, Charru A, Hunt SC, Hopkins PN, Williams RR, Lalouel JM (1992b) Molecular basis for human hypertension: role of angiotensinogen. Cell 71:169–180
Jeunemaitre X, Inoue I, Williams C, Charru A, Tichet J, Powers M, Sharma AM, Gimenez-Roqueplo AP, Hata A, Corvol P, Lalouel JM (1997) Haplotypes of angiotensinogen in essential hypertension. Am J Hum Genet 60:1448–1460
Jia H, Hingorani AD, Sharma P, Hopper R, Dickerson C, Trutwein D, Lloyd DD, Brown MJ (1999) Association of the G(s)alpha gene with essential hypertension and response to beta blockade. Hypertension 34:8–14
Johnson JA, Zineh I, Puckett BJ, McGorray SP, Yarandi HN, Pauly DF (2003) Beta1-adrenergic receptor polymorphisms and antihypertensive response to metoprolol. Clin Pharmacol Ther 74:372–379
Jordan J, Toka HR, Heusser K, Toka O, Shannon JR, Tank J, Diedrich A, Stabroth C, Stoffels M, Naraghi R, Oelkers W, Schuster H, Schobel HP, Haller H, Luft FC (2000) Severely impaired baroreflex buffering in patients with monogenic hypertension and neurovascular contact. Circulation 102:2611–2618
Ju Z, Zhang H, Sun K, Song Y, Lu H, Hui R, Huang X (2003) Alpha-adducin gene polymorphism is associated with essential hypertension in Chinese: a case-control and family-based study. J Hypertens 21:1861–1868
Kagimoto M, Winter JSD, Kagimoto K, Simpson ER, Waterman MR (1988) Structural characterization of normal and mutant human steroid 17-α-hydrocylase genes: molecular basis of one example of combined 17-α-hydroxylase/ 17, 20 lyase deficiency. Mol Endocr 2:564–570
Kainulainen K, Perola M, Terwilliger J, Kaprio J, Koskenvuo M, Syvanen AC, Vartiainen E, Peltonen L, Kontula K (1999) Evidence for involvement of the type 1 angiotensin II receptor locus in essential hypertension. Hypertension 33:844–849
Kamide K, Takiuchi S, Tanaka C, Miwa Y, Yoshii M, Horio T, Mannami T, Kokubo Y, Tomoike H, Kawano Y, Miyata T (2004) Three novel missense mutations of WNK4, a kinase mutated in inherited hypertension, in Japanese hypertensives: implication of clinical phenotypes. Am J Hypertens 17:446–449
Kammerer CM, Gouin N, Samollow PB, Vandeberg JF, Hixson JE, Cole SA, MacCluer JW, Atwood LD (2004) Two quantiative trait loci affect ACE activities in Mexican-Americans. Hypertension 25:1266–1269
Kamynina E, Debonneville C, Bens M, Candewalle A, Staub O (2001) A novel mouse Nedd4 protein suppresses the activity of the epithelial Na+ channel. FASEB J 15:204–214
Kario K, Hoshide S, Umeda Y, Sato Y, Ikeda U, Nishiuma S, Matsuo M, Shimada K (1999) Angiotensinogen and angiotensin-converting enzyme genotypes, and day and night blood pressures in elderly Japanese hypertensives. Hypertens Res 22:95–103
Karlsson J, Lind L, Hallberg P, Michaelsson K, Kurland L, Kahan T, Malmqvist K, Ohman KP, Nystrom F, Melhus H (2004) Beta1-adrenergic receptor gene polymorphisms and response to beta 1-adrenergic receptor blockade in patients with essential hypertension. Clin Cardiol 27:347–350
Kato N, Sugiyama T, Morita H, Kurihara H, Yamori Y, Yazaki Y (1999) Angiotensinogen gene and essential hypertension in the Japanese: extensive association study and meta-analysis on six reported studies. J Hypertens 17:757–763
Kato N, Sugiyama T, Morita H, Kurihara H, Sato T, Yamori Y, Yazaki Y (2001) Association analysis of beta(2) adrenergic receptor polymorphisms with hypertension in Japanese. Hypertension 37:286–292
Keavney B, McKenzie C, Parish S, Palmer A, Clark S, Youngman L, Delephine M, Lathrop M, Peto R, Collins R (2000) Large-scale test of hypothessised associations between the angiotensin-converting enzyme insertion / deletion polymorphism and myocardial infarction in about 5000 cases and 6000 controls. International Studies of Infarct Survival (ISIS) Collaborators. Lancet 355:434–442
Keys JR, Zhou RH, Harris DM, Druckman CA, Eckhart AD (2005) Vascular smooth muscle overexpression of G protein-coupled receptor kinase 5 elevated blood pressure, which segregates with sex and is dependent on Gi-mediated signaling. Circulation 112:1145–1153
Kirchhausen T (2000) Three ways to make a vesicle. Nat Rev Mol Cell Biol 3:187–198
Kobashi G, Hata A, Ohta K, Yamada H, Kato EH, Minakami H, Fujimoto S, Kondo K (2004) A1166C variant of angiotensin II type 1 receptor gene is associated with severe hypertension in pregnancy independently of T235 variant of angiotensinogen gene. J Human Genet 49:182–186
Kokubo Y, Iwai N, Tago N, Inamoto N, Okayama A, Yamawaki H, Naraba H, Tomoike H (2005) Association analysis between hypertension and CYBA, CLCNKB, and KCNMB1 functional polymorphisms in the Japanese population–the Suita Study. Circ J 69:138–142
Koopmans RP, Insel PA, Michel MC (2003) Pharmacogenetics of hypertension treatment: a structured review. Pharmacogenetics 13:705–713
Kotanko P, Binder A, Tasker J, DeFreitas P, Kamdar S, Clark AJ, Skrabal F, Caulfield M (1997) Essential hypertension in African Caribbeans associates with a variant of the beta2-adrenoceptor. Hypertension 30:773–776
Kunz R, Kreutz R, Beige J, Distler A, Sharma AM (1997) Association between the angiotensinogen 235T variant and essential hypertension in whites: a systematic review and methodological appraisal. Hypertension 30:1331–1337
Kurland L, Melhus H, Karlsson J, Kahan T, Malmqvist K, Ohman KP, Nystrom F, Hagg A, Lind L; Swedish Irbesartan Left Ventricular Hypertrophy Investigation versus Atenolol (SILVHIA) Trial (2001) Angiotensin converting enzyme gene polymorphism predicts blood pressure responses to angiotensin II receptor type I antagonist treatment in hypertensive patients. J Hypertens 19:1783–1787
Kurland L, Liljedahl U, Karlsson J, Kahan T, Malmqvist K, Melhus H, Syvanen AC, Lind LL (2004) Angiotensinogen gene polymorphisms: relationship to blood pressure response to antihypertensive treatment. Results from the Swedish Irbesartan Left Ventricular Hypertrophy Investigation vs Atenolol (SILVHIA) trial. Am J Hypertens 17:8–13
Lajemi M, Labat C, Gautier S, Lacolley P, Safar M, Asmar R, Cambien F, Benetos A (2001) Angiotensin II type 1 receptor-153A/G and 1166A/C gene polymorphisms and increase in aortic stiffness with age in hypertensive subjects.. J Hypertens 19:407–413
Lalioti MD, Zhang J, Volkman HM, Kahle KT, Hoffmann KE, Toka HR, Nelson-Williams C, Ellison D, Flavell R, Booth CJ, Lu Y, Geller DS, Lifton RP (2006) Wnk4 controls blood pressure and potassium homeostasis via regulation of mass and activity of the distal convoluted tubule. Nat Genet 38:1124–1132
Leineweber K, Büscher R, Bruck H, Brodde OE (2004) β-adrenoceptor polymorphisms. Naunyn-Schmiedebergs Arch Pharmacol 369:1–22
Levy D, DeStefano AL, Larson MG, O’Donnell CJ, Lifton RP, Gavras H, Cupples LA, Myers RH (2000) Evidence for a gene influencing blood pressure on chromosome 17. Genome scan linkage results for longitudinal blood pressure phenotypes in subjects from the framingham heart study. Hypertension 36:477–483
Li A, Li KX, Marui S, Krozowski ZS, Batista MC, Whorwood CB, Arnhold IJ, Shackleton CH, Mendoca BB, Stewart PM (1997) Apparent mineralocorticoid excess in a Brazilian kindred. Hypertension in the heterozygous state. J Hypertens 15:1397–1402
Liddle GW, Bledsoe T, Coppage WS (1963) A familial renal disorder simulating primary aldosteronism but with negligible aldosterone secretion. Trans Am Assoc Physicians 76:199–213
Lifton RP, Dluhy RG, Powers M, Rich GM, Cook S, Ulick S, Lalouel JM (1992a) A chimaeric 11 β-hydroxylase/aldosterone synthase gene causes glucocorticoid-remediable aldosteronism and human hypertension. Nature 355:262–265
Lifton RP, Dluhy RG, Powers M, Rich GM, Gutkin M, Fallo F, Gill JR Jr, Feld L, Ganguly A, Laidlaw JC (1992b) Hereditary hypertension caused by chimaeric gene duplications and ectopic expression of aldosterone synthase. Nat Genet 2:66–74
Lin MH, Tseng CH, Tseng CC, Huang CH, Chong CK, Tseng CP (2001) Real-time PCR for rapid genotyping of angiotensin-converting enzyme insertion/deletion polymorphism. Clin Biochem 34:661–666
Litchfield WR, Anderson BF, Weiss RJ, Lifton RP, Dluhy RG (1998) Intracranial aneurysms and hemorrhagic stroke in glucocorticoid-remediable aldosteronism. Hypertension 31:445–450
Liu J, Liu ZQ, Tan ZR, Chen XP, Wang LS, Zhou G, Zhou HH (2003) Gly389Arg polymorphism of β1-adrenergic receptor is associated with the cardiovascular response to metoprolol. Clin Pharmacol Ther 74:372–379
Liu S, Premont RT, Kontos CD, Zho S, Rockey DC (2005) A crucial role for GRK2 in regulatin of endothelial cell nitric oxide synthase function in portal hypertension. Nat Med 11:952–958
Liu J, Liu ZQ, Yu BN, Xu FH, Mo W, Zhou G, Liu YZ, Li Q, Zhou HH (2006) Beta1-Adrenergic receptor polymorphisms influence the response to metoprolol monotherapy in patients with essential hypertension. Clin Pharm Ther 80:23–32
Longini IM, Higgins MW, Hinton PC, Moll PC, Keller JB (1984) Environmental and genetic sources of familial aggregation of blood pressure in Tecumseh, Michigan. Am J Epidemiol 120:131–144
Maeda Y, Ikeda U, Ebata H, Hojo Y, Seino Y, Hayashi Y, Kuroki S, Shimada K (1996) Angiotensin-converting enzyme gene polymorphisms in hypertensive individuals with parental history of stroke. Stroke 27:1521–1523
Mansfield TA, Simon DB, Farfel Z, Bia M, Tucci JR, Lebel M, Gutkin M, Vialettes B, Christofilis MA, Kauppinen-Makelin R, Mayan H, Risch N, Lifton RP (1997) Multilocus linkage of familial hyperkalemia and hypertension, pseudohypoaldosteronism type II, to chromosomes 1q31-42 and 17p11-q21. Nat Genet 16:202–205
Mason DA, Moore JD, Green SA, Liggett SB (1999) A gain-of-function polymorphism in a G-protein coupling domain of the human beta 1-adrenergic receptor. J Biol Chem 274:12670–12674
Matsuoka Y, Li X, Bennett V (2000) Adducin: structure, function and regulation. Cell Mol Life Sci 57:884–895
McGraw DW, Forbes SL, Kramer LA, Ligett SB (1998) Polymorphisms of the 5′ leader cistron of the human beta2-adrenergic receptor regulate receptor expression. J Clin Invest 102:1927–1932
McKenzie CA, Sinsheimer JS, Adeyemo AA, Cox RD, Southam L, Hugill A, Bouzekri N, Lathrop M, Forrester TE, Cooper RS, Ward R (2005) SNP haplotypes in the angiotensin I-converting enzyme (ACE) gene analysis of Nigerian family data using gamete competition models. Ann Hum Genet 69:227–232
Mellen PB, Herrington DM (2005) Pharmacogenomics of blood pressure response to antihypertensive treatment. J Hypertens 25:1311–1325
Mitchell A, Buhrmann S, Seifert A, Nüurnberger J, Wenzel RR, Siffert W, Philipp T, Schäfers RF (2003) Venous response to nitroglycerin is enhanced in young, healthy carriers of the 825T allele of the G protein beta3 subunit gene (GNB3). Clin Pharmacol Ther 74:499–504
Mitchell A, Luckebergfeld B, Buhrmann S, Rushentsova U, Nürnberger J, Siffert W, Schäfers RF, Philipp T, Wenzel RR (2004) Effects of systemic endothelin A receptor antagonism in various vascular beds in men: in vivo interactions of the major blood pressure-regulating systems and associations with the GNB3 C825T polymorphism. Clin Pharmacol Ther 76:396–408
Mitchell A, Pace M, Nürnberger J, Wenzel RR, Siffert W, Philipp T, Schäfers RF (2005) Insulin-mediated venodilation is impaired in young, healthy carriers of the 825T allele of the G-protein beta3 subunit gene (GNB3). Clin Pharmacol Ther 77:495–502
Mitsuuchi Y, Kawamoto T, Rosier A, Naiki Y, Miyahara K, Toda K, Kuribayashi I, Orii T, Yasuda K, Miura K, Nakao K (1992) Congenitally defective aldosterone biosynthesis in humans: the involvement of point mutations of the P-450(C18) gene (CYP11B2) in CMO II deficient patients. Biochem Biophys Res Commun 182:974–979
Mondorf UF, Russ A, Wiesemann A, Herrero M, Oremek G, Lenz T (1998) Contribution of angiotensin I converting enzyme gene polymorphisms and angiotensinogen polymorphisms to blood pressure regulation in essential hypertension. Am J Hypertens 11:174–183
Morgan T, Craven C, Nelson L, Lalouel JM, Ward K (1997) Angiotensinogen T235 expression is elevated in decidual spiral arteries. J Clin Invest 100:1406–1415
Morgan L, Crawshaw S, Baker PN, Pipkin FB, Kalsheker N (2000) Polymorphism in oestrogen response element associated with variation in plasma angiotensinogen concentrations in healthy pregnant women. J Hypertens 18:553–557
Morrison AC, Doris PA, Folsom AR, Nieto FJ, Boerwinkle E; Atherosclerosis Risk in Communities Study (2001) G-protein β3 subunit and α-adducin polymorphisms and risk of subclinical and clinical stroke. Stroke 32:822–829
Mune T, Roberson FM, Nikkiä H, Agarwal AK, White PC (1995) Human hypertension caused by mutations in the kidney isozyme of 11β-hydroxysteroid dehydrogenase. Nat Genet 10:394–399
Nagy Z, Busjahn A, Bähring S, Faulhaber HD, Gohlke HR, Knoblauch H, Rosenthal M, Müller-Myhsok B, Schuster H, Luft FC (1999) Quantitative trait loci for blood pressure exist near the IGF-1, the Liddle syndrome, the angiotensin II receptor gene and the renin loci in man. J Am Soc Nephrol 10:1709–1716
Nakai K, Itoh C, Miura Y, Hotta K, Musha T, Itoh T, Miyakawa T, Iwasaki R, Hiramori K (1994) Deletion polymorphism of the angiotensin-I-converting enzyme gene is associated with serum ACE concentrations and increased risk for CAD in the Japanese. Circulation 90:2199–2202
Nakajima T, Jorde LB, Ishigama T, Umemura S, Emi M, Lalouel JM, Inoue I (2002) Nucleotide diversity and haplotype structure of the human angiotensinogen gene in two populations. Am J Hum Genet 70:108–123
Nakano Y, Oshima T, Watanabe M, Matsuura H, Kajiyama G, Kambe M (1997) Angiotensin-I converting enzyme gene polymorphism and acute responses to captopril in essential hypertension. Am J Hypertens 10:1064–1068
Nalogowska-Glosnicka K, Lacka BI, Zychma MJ, Grzeszczak W, Zukowska-Szczechowska E, Poreba R, Michalski B, Kniazewski B, Rzempoluch J; PIH Study Group (2000) Angiotensin II type 1 receptor gene A1166C polymorphism associated with the increased risk of pregnancy-induced hypertension. Med Sci Monitor 6:523–529
Naraghi R, Geiger H, Crnac J, Huk W, Fahlbusch R, Engels G, Luft FC (1994) Posterior fossa neurovascular abnormalities in essential hypertension. Lancet 344:1466–1470
Naraghi R, Schuster H, Toka HR, Bähring S, Toka O, Oztekin O, Bilginturan N, Knoblauch H, Wienker TF, Busjahn A, Haller H, Fahlbusch R, Luft FC (1997) Neurovascular compression at the ventrolateral medulla in autosomal dominant hypertension and brachydactyly. Stroke 28:1749–1754
New MI, Levine LS, Biglieri EG, Pareira J, Ulick S (1977) Evidence for an unidentified steroid in a child with apparent mineralocorticoid hypertension. J Clin Endocrinol Metab 44:924–933
Newhouse SJ, Wallace C, Dobson R, Mein C, Pembroke J, Farrall M, Clayton D, Brown M, Samani N, Dominiczak A, Connell JM, Webster J, Lathrop GM, Caulfield M, Munroe PB (2005) Haplotypes of the WNK1 gene associate with blood pressure variation in a severely hypertensive population from the British Genetics of Hypertension study. Hum Mol Genet 14:1805–1814
Ng DP, Tai BC, Koh D, Tan KW, Chia KS (2005) Angiotensin-I-converting enzyme insertion/deletion polymorphism and its association with diabetic nephropathy: a meta analysis of studies reported between 1994 and 2004 and comprising 14,727 subjects. Diabetologia 54:1238–1244
Nickenig G, Michaelsen F, Müller C, Vogel T, Strehlow K, Böhm M (2001) Post-transcriptional regulation of the AT1 receptor mRNA. Identification of the mRNA binding motif and functional characterization. FASEB J 15:1490–1492
Nickenig G, Michaelsen F, Muller C, Berger A, Vogel T, Sachinidis A, Vetter H, Böhm M (2002) Destabilization of AT1 receptor mRNA by calreticulin. Circ Res 90:53–58
Niu T, Xu X, Cordell HJ, Rogus J, Zhou Y, Fang Z, Lindpaintner K (1999) Linkage analysis of candidate genes and gene-gene interactions in Chinese hypertensive sib pairs. Hypertension 33:1332–1337
Nkeh B, Samani NJ, Badenhorst D, Libhaber E, Sareli P, Norton GR, Woodiwiss AJ (2003) T594M variant of the epithelial sodium channel beta-subunit gene and hypertension in individuals of African ancestry in South Africa. Am J Hypertens 16:847–852
Nürnberger J, Dammer S, Mitchell A, Siffert W, Wenzel RR, Gossl M, Philipp T, Michel MC, Schäfers RF (2003) Effect of the C825T polymorphism of the G protein beta 3 subunit on the systolic blood pressure-lowering effect of clonidine in young, healthy male subjects. Clin Pharmacol Ther 74:53–60
Nürnberger J, Opazo Saez A, Mitchell A, Buhrmann S, Wenzel RR, Siffert W, Philipp T, Schäfers RF (2004) The T-allele of the C825T polymorphism is associated with higher arterial stiffness in young healthy males. J Hum Hypertens 18:267–271
O’Donnel CJ, Lindpaintner K, Larson MG, Rao VS, Ordovas JM, Schaefer EJ, Myers RH, Levy D (1998) Evidence for association and genetic linkage of the angiotensinogen-converting enzyme locus with hypertension and blood pressure in men but not in women of the Framingham Heart Study. Circulation 97:1766–1772
Ogimoto G, Yudowski GA, Barker CJ, Kohler M, Katz AI, Feraille E, Pedemonte CH, Berggren PO, Bertorello AM (2000) G-protein-coupled receptors regulate Na+, K+-ATPase activity and endocytosis by modulating the recruitment of adaptor protein 2 and clathrin. Proc Natl Acad Sci USA 97:3242–3247
Ohmichi N, Iwai N, Uchida Y, Shichiri G, Nakamura Y, Kinoshita M (1997) Relationship between the response to angiotensin-converting enzyme inhibitor imidapril and the angiotensin converting enzyme genotype. Am J Hypertens 10:951–955
Ono K, Mannami T, Baba S, Yasui N, Ogihara T, Iwai N (2003) Lack of association between angiotensin II typ I receptor gene polymorphism and essential hypertension in Japanese. Hypertens Res 26:131–134
O’Shaughnessy KM, Fu B, Dickerson C, Thurston D, Brown MJ (2000) The gain-of-function G389R variant of the beta 1-adrenoceptor does not influence blood pressure or heart rate response to beta-blockade in hypertensive subjects. Clin Sci 99:233–238
Palmer BF, Alpern RJ (1998) Liddle’s syndrome. Am J Med 104:301–309
Pascoe L, Curnow KM, Slutsker L, Connell JM, Speiser PW, New MI, White PC (1992a) Glucocorticoid-suppressible hyperaldosteronism results from hybrid genes created by unequal cross-overs between CYP11B1 and CYP11B2. Proc Natl Acad Sci USA 89:8327–8331
Pascoe L, Curnow KM, Slutsker L, Rosier A, White PC (1992b) Mutations in the human CYP11B2 (aldosterone synthase) gene causing corticosterone methyloxidase II deficiency. Proc Natl Acad Sci USA 89:4996–5000
Peng JB, Bell PD (2006) Cellular mechanisms of WNK4-mediated regulation of ion transport proteins in the distal tubule. Kidney Int 69:2116–2118
Persu A, Coscoy S, Houot AM, Corvol P, Barbry P, Jeunemaitre X (1999) Polymorphisms of the gamma subunit of the epithelial Na+ channel in essential hypertension. J Hypertens 17:639–645
Poch E, Gonzalez D, Gomez-Angelats E, Enjuto M, Pare JC, Rivera F, de La Sierra A (2000) G-Protein β3 subunit gene variant and left ventricular hypertrophy in essential hypertension. Hypertension 35:214–218
Pontremoli R, Sofia A, Tirotta A, Ravera M, Nicolella C, Viazzi F, Bezante GP, Borgia L, Bobola N, Ravazzolo R, Sacchi G, Deferrari G (1996) The deletion polymorphism of the angiotensin I-converting enzyme gene is associated with target organ damage in essential hypertension. J Am Soc Nephrol 7:2550–2558
Psaty BM, Smith NL, Heckbert SR, Vos HL, Lemaitre RN, Reiner AP, Siscovick DS, Bis J, Lumley T, Longstreth WT Jr, Rosendaal FR (2002) Diuretic therapy, the alpha-adducin gene variant, and the risk of myocardial infarction or stroke in persons with treated hypertension. J Am Med Assoc 287:1680–1689
Ranade K, Jorgensohn EH, Sheu W, Pei D, Hsiung CA, Chiang F, Chen YI, Pratt R, Olshen RA, Curb D, Cox DR, Botstein D, Risch N (2002) A polymorphism in the β1 adrenergic receptor is associated with resting heart rate. Am J Hum Genet 70:935–942
Rhatz DA, Brown KM, Kramer LA, Liggett SB (2002) Amino acid 49 polymorphism of the human beta1-adrenergic receptor affect agonist-promoted trafficking. J Cardiovasc Pharmacol 39:155–160
Rice T, Vogler GP, Perusse L, Bouchard C, Rao DC (1989) Cardiovascular risk factors in a French Canadian population: resolution of genetic and familial environmental effects on blood pressure using twin, adoptees, and extensive information on environmental correlates. Genet Epidemiol 6:571–588
Rich GM, Ulick S, Cook S, Wang JZ, Lifton RP, Dluhy RG (1992) Glucocorticoid-remediable aldosteronism in a large kindred: clinical spectrum and diagnosis using a characteristic biochemical phenotype. Ann Intern Med 15:813–820
Riddle EL, Rana BK, Murthy KK, Rao F, Eskin E, O’Connor DT, Insel PA (2006) Polymorphisms and haplotypes of the regulator of G protein signaling-2 gene in normotensives and hypertensives. Hypertension 47:415–420
Riento K, Ridley AJ (2003) Rocks: multifunctional kinases in cell behaviour. Nat Rev Mol Cell Biol 4:446–456
Rigat B, Hubert C, Alhenc-Gelas F, Cambien F, Corvol P, Soubrier F (1990) An insertion/deletion polymorphism in the angiotensin I-converting enzym gene accounting for half the variance of serum enzyme levels. J Clin Invest 86:1343–1346
Rosmond R, Ukkola O, Changnon M, Bouchard C, Bjorntorp P (2000) Polymorphisms of the beta2-adrenergic receptor gene (ADRB2) in relation to cardiovascular risk factors in men. J Intern Med 248:239–244
Rosskopf D, Frömter E, Siffert W (1993) Hypertensive sodium-proton exchanger phenotype persists in immortalized lymphoblasts from essential hypertensive patients. A cell culture model for human hypertension. J Clin Invest 92:2553–2559
Rosskopf D, Busch S, Manthey I, Siffert W (2000) G protein beta 3 gene: structure, promoter, and additional polymorphisms. Hypertension 36:33–41
Rosskopf D, Manthey I, Siffert W (2002) Identification and ethnic distribution of major haplotypes in the gene GNB3 encoding the G-protein beta3 subunit. Pharmacogenetics 12:209–220
Rosskopf D, Manthey I, Habich C, Kielbik M, Eisenhardt A, Nikula C, Urban M, Kohnen S, Graf E, Ravens U, Siffert W (2003a) Identification and characterization of Gβ3s2, a novel splice variant of the G-protein β3 subunit. Biochem J 371:223–232
Rosskopf D, Koch K, Habich C, Geerdes J, Ludwig A, Wilhelms S, Jakobs KH, Siffert W (2003b) Interaction of Gβ3s, a splice variant of the G-protein Gβ3, with Gγ- and Gα-proteins. Cell Signal 15:479–488
Ruiz-Velasco V, Ikeda SR (2003) A splice variant of the G protein beta 3-subunit implicated in disease states does not modulate ion channels. Physiol Genomics 13:85–95
Salim S, Sinnarajah S, Kehrl JH, Dessauer CW (2003) Identification of RGS2 and type V adenylyl cyclase interaction sites. J Biol Chem 278:15842–15849
Sasaki M, Oki T, Iuchi A, Tabata T, Yamada H, Manabe K, Fukuda K, Abe M, Ito S (1996) Relationship between the angiotensin converting enzyme gene polymorphism and the effects of enalapril on left ventricular hypertrophy and impaired diastolic filling in essential hypertension: M-mode and pulsed Doppler echocardiographic studies. J Hypertens 14:1403–1408
Sayed-Tabatabaei FA, Oostra BA, Isaacs A, van Duijn CM, Witteman JCM (2006) ACE polymorphisms. Circ Res 98:1123–1133
Schild L, Lu Y, Gautschi I, Schneeberger E, Lifton RP, Rossier BC (1995) Identification of a PY motif in the epithelial Na channel subunits as target sequences for mutations causing channel activation found in Liddle syndrome. EMBO J 15:2381–2387
Schmidt S, van Hooft IM, Grobbee DE, Ganten D, Ritz E (1993) Polymorphism of the angiotensin I converting enzyme gene is apparently not related to high blood pressure: Dutch Hypertension and Offspring Study. J Hypertens 11:345–348
Schmidt S, Beige S, Walla-Friedel M, Michel MC, Sharma AM, Ritz E (1997) A polymorphism in the gene for the angiotensin II type 1 receptor is not associated with hypertension. J Hypertens 15:1385–1388
Schunkert H, Hense HW, Holmer SR, Stender M, Perz S, Keil U, Lorell BH, Riegger GA (1994) Association between a deletion polymorphism of the angiotensin-converting-enzyme gene and left ventricular hypertrophy. N Engl J Med 330:1634–1638
Schunkert H, Hense HW, Döring A, Riegger GA, Siffert W (1998) Association between a polymorphism in the G protein beta3 subunit gene and lower renin and elevated diastolic blood pressure levels. Hypertension 32:510–513
Schuster H, Wienker TF, Toka HR, Bähring S, Jeschke E, Toka O, Busjahn A, Hempel A, Talhammer C, Oelkers W, Kunze J, Bilginturan N, Haller H, Luft FC (1996a) Autosomal dominant hypertension and brachydactyly in a Turkish kindred resembles essential hypertension. Hypertension 28:1085–1092
Schuster H, Wienker TE, Bähring S, Bilginturan N, Toka HR, Neitzel H, Jeschke E, Toka O, Gilbert D, Lowe A, Ott J, Haller H, Luft FC (1996b) Severe autosomal dominant hypertension and brachydactyly in a unique Turkish kindred maps to human chromosome 12. Nat Genet 13:98–100
Schuster H, Toka O, Toka HR, Busjahn A, Öztekin Ö, Wienker TF, Bilginturan N, Bähring S, Skrabal F, Haller H, Luft FC (1998) A cross-over medication trial for autosomal-dominant hypertension with brachydactytly. Kidney Int 53:167–172
Schwartz GL, Turner ST, Chapman AB, Boerwinkle E (2002) Interacting effects of gender and genotype on blood pressure response to hydrochlorothiazide. Kidney Int 62:1718–1723
Sciarrone MT, Stella P, Barlassina C, Manunta P, Lanzani C, Bianchi G, Cusi D (2003) ACE and alpha-adducin polymorphism as markers of individual response to diuretic therapy. Hypertension 41:398–403
Seasholtz TM, Wessel J, Rao F, Rana BK, Khandrika S, Kennedy BP, Lillie EO, Ziegler MG, Smith DW, Schork NJ, Brown JH, O’Connor DT (2006) Rho kinase polymorphism influences blood pressure and systemic vascular resistance in human twins: role of heredity. Hypertension 47:937–947
Sethi AA, Nordestgaard BG, Tybjaerg-Hansen A (2003) Angiotensinogen gene polymorphism, plasma angiotensinogen, and risk of hypertension and ischemic heart diseases. A meta-analysis. Arterioscler Thromb Vasc Biol 23:1269–1275
Sharma P (1998) Meta-analysis of the ACE gene in ischemic stroke. J Neurol Neurosurg Psychiatry 64:227–230
Shimkets RA, Warnock DG, Bositis CM, Nelson-Williams C, Hanson JH, Schambelan M, Gill JR, Ulick S, Milora RV, Findling JW, Canessa CM, Rossier BC, Lifton RP (1994) Liddle’s syndrome. Heritable human hypertension caused by mutations in the β subunit of the epithelial sodium channel. Cell 79:407–414
Shimkets RA, Lifton RP, Cannessa CM (1997) The activity of the epithelial sodium channel is regulated by clathrin-mediated endocytosis. J Biol Chem 272:25537–25541
Sierra C, Coca A, Gomez-Angelats E, Poch E, Sobrino J, de la Sierra A (2002) Renin-angiotensin system genetic polymorphisms and white cerebral white matter lesions in essential hypertension. Hypertension 39:343–347
Siffert W, Rosskopf D, Siffert G, Busch S, Moritz A, Erbel R, Sharma AM, Ritz E, Wichmann HE, Jakobs KH, Horsthemke B (1998) Asociation of a human G-protein beta 3 subunit variant with hypertension. Nat Genet 18:45–48
Siffert W, Forster P, Jockel KH, Mvere DA, Brinkmann B, Naber C, Crookes R, Du P, Heyns A, Epplen JT, Fridey J, Freedman BI, Muller N, Stolke D, Sharma AM, Al Moutaery K, Grosse-Wilde H, Buerbaum B, Ehrlich T, Ahmad HR, Horsthemke B, Du Toit ED, Tiilikainen A, Ge J, Wang Y, Rosskopf D (1999) Worldwide ethnic distribution of the G protein β3 subunit 825T allele and its association with obesity in Caucasian, Chinese, and Black African individuals. J Am Soc Nephrol 10:1921–1930
Simon DB, Nelson-Williams C, Johnson Bia M, Ellison D, Karet FE, Molina AM, Vaara I, Iwaa F, Cushner HM, Koolen M, Gainza FJ, Gitelman HJ, Lifton RP (1996a) Gitelman’s variant of Bartter’s syndrome, inherited hypokalaemic alkalosis, is caused by mutations in the thiazide-sensitive NaCl cotransporter. Nat Genet 12:24–30
Simon DB, Karet FE, Hamdan JM, diPietro A, Sanjad SA, Lifton RP (1996b) Bartter’s syndrome, hypokalaemic alkalosis with hyperclaciuria, is caused by mutations in the Na-K-2Cl cotransporter NKCC2. Nat Genet 13:183–188
Simon DB, Karet FE, Rodriguez-Soriano J, Hamdan JH, diPietro A, Trachtman H, Sanjad SA, Lifton RP (1996c) Genetic heterogeneity of Bartter’s syndrome revealed by mutations in the K+ channel ROMK. Nat Genet 14:152–156
Simon DB, Bindra RS, Mansfield TA, Nelson-Williams C, Mendonca E, Stone R, Schurman S, Nayir A, Alpay H, Bakkaloglu A, Rodriguez-Soriano J, Morales JM, Sanjad SA, Taylor CM, Pilz D, Brem A, Trachtman H, Griswold W, Richard GA, John E, Lifton RP (1997) Mutations in the chloride channel gene, CLCNKB, cause Bartter’s syndrome type III. Nat Genet 17:171–178
Sinnarajah S, Dessauer CW, Srikumar D, Chen J, Yuen J, Yilma S, Dennis JC, Morrison EE, Vodyanoy V, Kehrl JH (2001) RGS2 regulates signal transduction in olfactory neurons by attenuating activation of adenylyl cyclase III. Nature 409:1051–1055
Sofowora GG, Dishy V, Muszkat M, Xie HG, Kim RB, Harris PA, Prasad HC, Byrne DW, Nair UB, Wood AJ, Stein CM (2003) A common β1-adrenergic receptor polymorphism (Arg389Gly) affects blood pressure response to β-blockade. Clin Pharmacol Ther 73:366–371
Speirs HJ, Morris BJ (2004) WNK4 intron 10 polymorphism is not associated with hypertension. Hypertension 43:766–768
Speirs HJL, Katyk K, Kumar NN, Benjafield AV, Wang WYS, Morris BJ (2004) Association of G-protein-coupled receptor kinase 4 haplotypes, but not HSD3B1 or PTP1B polymorphisms, with essential hypertension. J Hypertens 22:931–936
Speirs HJ, Wang WYS, Benjafield AV, Morris BJ (2005) No association with hypertension of CLCNKB and TNFRS1B polymorphism at a hypertension locus on chromosome 1p36. J Hypertens 23:1491–1496
Spiering W, Kroon AA, Fuss-Lejeune MM, Daemen MJ, de Leeuw PW (2000) Angiotensin II sensitivity is associated with the angiotensin II type 1 receptor A(1166)C polymorphism in essential hypertensives on a high sodium diet. Hypertension 36:411–416
Spiering W, Kroon AA, Fuss-Lejeune MM, de Leeuw PP (2005) Genetic contribution to the acute effects of angiotensin II type 1 receptor blockade. J Hypertens 23:753–758
Staessen JA, Wang JG, Ginocchio G, Petrov V, Saavedra AP, Soubrier F, Vlietinck R, Fagard R (1997) The deletion / insertion polymorphism of the angiotensin converting enzyme gene and cardiovascular-renal risk. J Hypertens 15:1579–1592
Staessen JA, Kuznetsova T, Wang JG, Emelianov D, Vlietnick R, Fagard R (1999) M235T angiotensinogen gene polymorphism and cardiovascular risk. J Hypertens 17:9–17
Staessen JA, Wang JG, Brand E, Barlassina C, Birkenhager WH, Herrmann SM, Fagard R, Tizzoni L, Bianchi G (2001) Effects of three candidate genes on prevalence and incidence of hypertension in a Caucasian population. J Hypertens 19:1349–1358
Staub O, Dho S, Henry P, Correa J, Ishikawa T, McGlade J, Rotin D (1996) WW domains of Nedd4 bind to the praline-rich PY motifs in the epithelial sodium channel deleted in Liddle’s syndrome. EMBO J 15:2371–2380
Stavroulakis GA, Makris TK, Krespi PG, Hatzizacharias AN, Gialeraki AE, Anastasiadis G, Triposkiadis P, Kyriakidis M (2000) Predicting response to chronic antihypertensive treatment with fosinopril: the role of angiotensin-converting enzyme gene polymorphisms. Cardiovasc Drugs Ther 14:427–432
Stein CM, Nelson R, Deegan H, He H, Wood M, Wood AJ (1995) Forearm beta adrenergic receptor-mediated vasodilation is impaired, without alteration of forearm norepinephrine spillover, in borderline hypertension. J Clin Invest 96:579–585
Stewart PM, Wallace AM, Valentino R, Burt D, Shackleton CH, Edwards CR (1987) Mineralocorticoid activity of liquorice: 11-beta-hydroxysteroid dehydrogenase deficiency comes of age. Lancet 2:821–824
Strautnieks SS, Thompson RJ, Gardiner RM, Chung E (1996) A novel splice-site mutation in the γ subunit of the epithelial sodium channel gene in three pseudohypoaldosteronism type 1 families. Nat Genet 13:248–250
Subramanya AR, Yang CL, McCormick JA, Ellison DH (2006) WNK kinases regualte sodium chloride and potassium transport by the aldosterone-sensitive distal nephron. Kidney Int 70:630–634
Sutherland DJ, Ruse JL, Laidlaw JC (1966) Hypertension, increased aldosterone secretion and low plasma renin activity relieved by dexamethasone. Can Med Assoc J 95:1109–1119
Synder PM, Price MP, McDonald FJ, Adams CM, Volk KA, Zeiher BG, Stokes JB, Welsh MJ (1995) Mechanisms by which Liddle’s syndrome mutations increase activity of a human epithelial Na+ channel. Cell 83:969–978
Takahashi N, Smithies O (1999) Gene targeting approaches to analyzing hypertension. J Am Soc Nephrol 17:1598–1605
Takahashi N, Murakami H, Kodama K, Kasagi F, Yamada M, Nishishita T, Inagami T (2000) Association of a polymorphism at the 5′-region of the angiotensin II type 1 receptor with hypertension. Ann Hum Genet 64:197–205
Tang KM, Wang GR, Lu P, Karas RH, Aronovitz M, Heximer SP, Kaltenbronn KM, Blumer KJ, Siderovski DP, Zhu Y, Mendelsohn ME (2003) Regulator of G-protein signaling-2 mediates vascular smooth muscle relaxation and blood pressure. Nat Med 9:1506–1512
Thekkumkara TJ, Linas SL (2003) Evidence for involvement of 3′-untranslated region on determining angiotensin II receptor coupling specificity with G proteins. Biochem J 370:631–639
Thiel BA, Chakravarti A, Cooper RS, Luke A, Lewis S, Lynn A, Tiwari H, Schork NJ, Weder AB (2003) A genome-wide linkage analysis investigating the determination of blood pressure in whites and African Americans. Am J Hypertens 16:151–153
Tiago AD, Samani NJ, Candy GP, Brooksbank R, Libhaber EN, Sareli P, Woodiwiss AJ, Norton GR (2002) Angiotensinogen gene promoter region variant modifies body size-ambulatory blood pressure relations in hypertension. Circulation 106:1483–1487
Tikhonoff V, Kuznetsova T, Stolarz K, Bianchi G, Casiglia E, Kawecka-Jaszcz K, Nikitin Y, Tizzone L, Wang JG, Staessen JA (2003) β-Adducin polymorphisms, blood pressure, and sodium excretion in three European populations. Am J Hypertens 16:840–846
Timmermann B, Mo R, Luft FC, Gerdts E, Busjahn A, Omvik P, Li GH, Schuster H, Wienker TF, Hoehe MR, Lund-Johansen P (1998) Beta-2 adrenoceptor genetic variation is associated with genetic predisposition to essential hypertension. The Bergen Blood Pressure Study. Kidney Int 53:1455–1460
Tiret L, Blanc H, Ruidavets JB, Arveiler D, Luc G, Jeunemaitre X, Tichet J, Mallet C, Poirier O, Plouin PF, Cambien F (1998) Gene polymorphisms of the renin-angiotensin system in relation to hypertension and parental history of myocardial infarction and stroke: the PEGASE study. J Hypertens 16:37–44
Tobin MD, Raleigh SM, Newhouse S, Braund P, Bodycote C, Ogleby J, Cross D, Gracey J, Hayes S, Smith T, Ridge C, Caulfield M, Sheehan NA, Munroe PB, Burton PR, Samani NJ (2005) Association of WNK1 gene polymorphisms and haplotypes with ambulatory blood pressure in the general population. Circulation 112:3423–3429
Toka HR, Bähring S, Chitayat D, Melby JC, Whitehead R, Jeschke R, Wienker TF, Toka O, Schuster H, Luft FC (1998) Families with autosomal-dominant brachydactyly type E, short stature, and severe hypertension. Ann Intern Med 129:204–208
Tripodi MG, Valtorta F, Torielli L, Chieregatti E, Salardi S, Trusolino L, Menegon A, Ferrari P, Marchisio PC, Bianchi G (1996) Hypertension associated point mutations in the adducin α and β subunits affect actin cytoskeleton and ion transport. J Clin Invest 97:2815–2822
Tripodi MG, Szpirer C, Reina C, Szpirer J, Bianchi G (1997) Polymorphism of γ-adducin gene in genetic hypertension and mapping of the gene to rat chromosome 1q55. Biochem Biophys Res Comm 237:685–689
Tripodi MG, Florio M, Ferrandi M, Modica R, Zimdahl H, Hubner N, Ferrari P, Bianchi G (2004) Effect of Add1 gene transfer on blood pressure in reciprocal congenic strains of Milan rats. Biochem Biophys Res Comm 324:562–568
Tsai CT, Fallin D, Chiang FT, Hwang JJ, Lai LP, Hsu KL, Tseng CD, Liau CS, Tseng YZ (2003) Angiotensionogen gene haplotype and hypertension:interaction with ACE gene I allele. Hypertension 41:9–15
Turner ST, Boerwinkle E (2003) Genetics of blood pressure, hypertensive complications, and antihypertensive drug responses. Pharmacogenomics 4:53–65
Turner ST, Schwartz GL, Chapman AB, Boerwinkle E (2001a) C825T polymorphism of the G protein β3 subunit and antihypertensive response to a thiazide diuretic. Hypertension 37:739–743
Turner ST, Schwartz GL, Chapman AB, Hall WD, Boerwinkle E (2001b) Antihypertensive pharmacogenetics: getting the right drug into the right patient. J Hypertens 19:1–11
Turner ST, Chapman AB, Schwartz GL, Boerwinkle E (2003) Effects of endothelial nitric oxide synthase, α-adducin, and other candidate gene polymorphisms on blood pressure response to hydrochlorothiazide. Am J Hypertens 16:834–839
Ueda S, Meredith PA, Morton JJ, Connell JMC, Elliot HL (1998) ACE (I/D) genotype is a predictor of the magnitude and duration of the response to an ACE inhibitor drug (enalaprilat) in humans. Circulation 98:2148–2153
Vasku A, Soucek M, Znojil V, Rihacek I, Tschoplova S, Strelcova L, Chidl K, Blakova M, Hajek D, Holla L, Vacha J (1998) Angiotensin I-converting enzyme and angiotensinogen gene interaction and prediction of essential hypertension. Kidney Int 53:1479–1482
Villard E, Tiret L, Visvikis S, Rakotovao R, Cambien F, Soubrier F (1996) Identification of new polymorphisms of the angiotensin I-converting enzyme (ACE) gene, and study of their relationship to plasma ACE levels by two QTL segregation-linkage anlysis. Am J Hum Genet 58:1268–1278
Wallace C, Xue MZ, Newhouse SJ, Marcano AC, Onipinla AK, Burke B, Gungadoo J, Dobson RJ, Brown M, Connell JM, Dominiczak A, Lathrop GM, Webster J, Farrall M, Mein C, Samani NJ, Caulfield MJ, Clayton DG, Munroe PB (2006) Linkage analysis using co-phenotypes in the BRIGHT study reveals novel potential susceptibility loci for hypertension. Am J Hum Genet 79:323–331
Wang WYS, Zee RYL, Moris BJ (1997) Association of angiotensin II type I receptor gene polymorphism with essential hypertension. Clin Genet 51:31–34
Wang J, Mougey EB, David CJ, Humma LM, Johnson JA, Lima JJ, Sylvester JE (2001) Determination of human beta(2)adrenoceptor haplotypes by denaturation selective amplification and subtractive genotyping. Am J Pharmacogenomics 1:315–322
Wang JG, Liu L, Zagato L, Xie J, Fagard R, Jin K, Wang J, Li Y, Bianchi G, Staessen JA, Liu L (2004) Blood pressure in relation to three candidate genes in a Chinese population. J Hypertens 22:937–944
Wang X, Zeng W, Soyombo AA, Tang W, Ross EM, Barnes AP, Milgram SL, Penninger JM, Allen PB, Greengard P, Muallem S (2005) Spinophilin regulates Ca2+ signalling by binding the N-terminal domain of RGS2 and the third intracellular loop of G-protein-coupled receptors. Nature Cell Biol 7:405–411
Ward K, Hata A, Jeunemaitre X, Helin C, Nelson L, Namikawa C, Farrington PF, Ogasawara M, Suzumori K, Tomoda S, Berrebi S, Sasaki M, Corvol P, Lifton RP, Lalouel J-M (1992) A molecular variant of angiotensiogen associated with preeclampsia. Nat Genet 4:59–61
Weinstein LS, Chen M, Xie T, Liu J (2006) Genetic diseases associated with heterotrimeric G proteins. Trends Pharmacol Sci 27:260–266
Wenzel RR, Siffert W, Bruck H, Philipp T, Schäfers RF (2002) Enhanced vasoconstriction to endothelin-1, angiotensin II and noradrenaline in carriers of the GNB3 825T allele in the skin microcirculation. Pharmacogenetics 12:489–495
Wettschureck N, Offermanns S (2005) Mammalian G proteins and their cell type specific functions. Physiol Rev 85:1159–1204
White PC, Dupont J, New MI, Leiberman E, Hochberg Z, Rosier A (1991) A mutation in CYP11B1 (arg488 to his) associated with 11-beta-hydroxylase deficiency in Jews of Moroccan origin. J Clin Invest 87:1664–1667
Wieland T, Mittmann C (2003) Regulators of G-protein signalling: multifunctional proteins with impact on signalling in the cardiovascular system. Pharmacol Ther 97:95–115
Wilson RC, Dave-Sharma S, Wei JQ, Obeyesekere VR, Li K, Ferrari P, Krozowski ZS, Shackleton CHL, Bradlow L, Wiens T, New MI (1998) A genetic defect resulting in mild low-renin hypertension. Proc Natl Acad Sci USA 95:10200–10205
Wilson FH, Disse-Nicodeme S, Choate KA, Ishikawa K, Nelson-Williams C, Disitter I, Gunel M, Milford DV, Lipkin GW, Achard JM, Freely MP, Dussol B, Berland Y, Unwin RJ, Mayan H, Simon DB, Farfel Z, Jeunemaitre X, Lifton RP (2001) Human hypertension caused by mutations in WNK kinases. Science 293:1107–1112
Wilson FH, Kahle KT, Sabath E, Lalioti MD, Rapson AK, Hoover RS, Hebert SC, Gamba G, Lifton RP (2003) Molecular pathogenesis of inherited hypertension with hyperkalemia: the Na-Cl cotransporter is inhibited by wild-type but not mutated WNK4. Proc Natl Acad Sci USA 100:680–684
Wolf-Maier K, Cooper RS, Kramer H, Banegas JR, Giampaoli S, Joffres MR, Poulter N, Primatesta P, Stegmayr B, Thamm M (2004) Hypertension Treatment and Control in Five European Countries, Canada, and the United States. Hypertension 43:10–17
Wyckoff JA, Seely EW, Hurwitz S, Anderson BF, Lifton RP, Dluhy RG (2000) Glucocorticoid-remediable aldosteronism and pregnancy. Hypertension 31:445–450
Xie HG, Stein CM, Kim RB, Gainer JV, Sofowora G, Dishy V, Brown NJ, Goree RE, Haines JL, Wood AJ (2000) Human beta2-adrenergic receptor polymorphisms: no association with essential hypertension in black or white Americans. Clin Pharmacol Ther 67:670–675
Yamamoto M, Abe M, Jin JJ, Wu Z, Tabara Y, Mogi M, Kohara K, Miki T, Nakura J (2004) Association of a GNAS1 gene variant with hypertension and diabetes mellitus. Hypertes Res 27:919–924
Yang CL, Angell J, Mitchell R, Ellison DH (2003) WNK kinases regulate thiazide-sensitive Na-Cl cotransport. J Clin Invest 111:1039–1045
Yudowski GA, Efendiev R, Pedemonte CH, Katz AI, Berggren PO, Bertorello AM (2000) Phosphoinositide-3kinase binds to a proline-rich motif in the Na+, K+-ATPase α subunit and regulates its trafficking. Proc Natl Acad Sci USA 97:6556–6561
Zee RY, Ridker PM, Stampfer MJ, Hennekens CH, Lindpaintner K (1999) Prospective evaluation of the angiotensin-converting enzyme insertion/deletion polymorphism and the risk of stroke. Circulation 99:340–343
Zeltner R, Delles C, Schneider M, Siffert W, Schmieder RE (2001) G-protein beta(3) subunit gene (GNB3) 825T allele is associated with enhanced renal perfusion in early hypertension. Hypertension 37:882–886
Zeng C, Sanada H, Watanabe H, Eisner GM, Felder RA, Jose PA (2004) Functional genomics of the dopaminergic system in hypertension. Physiol Genomics 199:233–246
Zhang X, Erdmann J, Regitz-Zagrosek V, Kürzinger S, Hense HW, Schunkert H (2000) Evaluation of three polymorphisms in the promoter region of the angiotensin II type I receptor gene. J Hypertens 18:267–272
Zhao YY, Zhou J, Narayanan CS, Cui Y, Kumar A (1999) Role of C/A polymorphism at -20 on the expression of human angiotensinogen gene. Hypertension 33:108–115
Zhu X, McKenzie CA, Forrester T, Nickerson DA, Broeckel U, Schunkert H, Doering A, Jacob HJ, Cooper RS, Rieder MJ (2000) Localization of a small genomic region associated with elevated ACE. Am J Hum Genet 67:1144–1153
Zhu X, Chang YP, Yan D, Weder A, Cooper R, Luke A, Kan D, Chakravarti A (2003a) Associations between hypertension and genes in the renin-angiotensin system. Hypertension 41:1027–1034
Zhu X, Yan D, Cooper RS, Luke A, Ikeda MA, Chang YP, Weder A, Chakravarti A (2003b) Linkage disequilibrium and haplotype diversity in the genes of the renin-angiotensin system: findings from the family blood pressure program. Genome Res 13:173–181
Author information
Authors and Affiliations
Corresponding author
Additional information
The authors wish to dedicate this paper to Prof. Dr. Karl H. Jakobs, an outstanding scientist, teacher, advisor and fellow, on the occasion of his 65th birthday.
Rights and permissions
About this article
Cite this article
Rosskopf, D., Schürks, M., Rimmbach, C. et al. Genetics of arterial hypertension and hypotension. Naunyn-Schmied Arch Pharmacol 374, 429–469 (2007). https://doi.org/10.1007/s00210-007-0133-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00210-007-0133-2