
Abstract
Verticillium species are soilborne plant pathogens, responsible for big yield losses worldwide. Here, we report improved procedures to generate DNA from Verticillium species imbedded in farm soils. Using new genomic sequence information, primers for V. dahliae, V. albo-atrum, V. tricorpus, and V. longisporum were designed. In a survey of 429 samples from intensively farmed soil of two Swedish regions, only V. dahliae and V. longisporum were identified. A bias towards V. longisporum (40%) was seen in the south, whereas V. dahliae was more frequent in the western region (19%). Analyses of soil and leaf samples from 20 sugar beet fields, where foliar wilting had been observed, revealed V. dahliae DNA in all leaf and soil samples and V. longisporum in 18 soil samples, illustrating host choice and longevity of the V. longisporum microsclerotia. This study demonstrates the applicability of new molecular diagnostic tools that are important for growers of variable crops.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The angiosperm family Brassicaceae includes many economically important vegetables, condiment and fodder crops as well as edible and industrial oilseed (Rakow 2004). In terms of production, winter oilseed rape (Brassica napus) is the most important oil crop in Europe and globally third behind palm and soybean oil. Annual yields of winter oilseed rape in northern Europe are about one metric ton less compared to Central Europe (http://ec.europa.eu/eurostat), and the cause of this difference is debated. Besides slightly different climate and somewhat unpredictable climate conditions, one important factor for yield losses of Brassica crops in Sweden could be related to soilborne pathogens, such as Verticillium longisporum and Plasmodiophora brassicae. The latter causes the clubroot disease. P. brassicae was detected in more than 50% of analyzed soils from southern Swedish fields 2013 and 2014 and constitute an emergence disease threat to Brassica crop production (Wallenhammar et al. 2016).
Verticillium dahliae causes wilting disease on more than 200 different host plants (Pegg and Brady 2002), whereas V. longisporum is more restricted in its host range, attacking mainly plants in the Brassicaceae family. Although V. longisporum substantially shares the disease cycle characteristics with the more studied V. dahliae, it does not cause wilting of the plants. Therefore, the V. longisporum disease is now suggested to be named Verticillium stem stripe instead of Verticillium wilt (Depotter et al. 2016). A major problem for the disease management of these two plant pathogens is the longevity of their microsclerotia that are released into the soil from infected plant residues at the end of the disease cycle. These resting structures have the capacity to remain dormant in the soil for many years. It is anticipated that microsclerotia are stimulated to germinate by root exudates released from a host plant growing nearby. The hyphae of V. longisporum invade the lateral roots and root hairs (Zhou et al. 2006), colonizing the root tissues before enter the xylem elements. Here conidia are formed and can be spread via the plant transpiration stream. The colonization of the vessel tissue restricts the access to xylem sap of the plant cells and thereby affects the plant growth. Since microsclerotia are not formed and protruded in stems and leaves until the plant is in the senescence phase, a process associated with induction of abscisic acid (Roos et al. 2014), the infection commonly remains unnoticed.
Although not necessarily observed every year, the Verticillium stripe disease is more frequently reported in recent decades (Dixelius et al. 2005). Fluctuation in disease reporting is mostly explained by the late occurrence of disease symptoms in the season and a combination of new more tolerant crop varieties and impact of environmental factors influencing the microsclerotia germination frequency. However, early detection of Verticillium infested soils is crucial to prevent further spread and multiplication of these pathogens. Currently, detection methods used for V. dahliae or V. longisporum are either based on soil sieving and plating on agar plates followed by counting of germinating microsclerotia (e.g. Goud and Termorshuizen 2003; Kabir et al. 2004; Goud et al. 2011) or deployment of quantitative polymerase chain reaction (qPCR). Plate methods are laborious and require expertise when monitoring the colonies on the plates, whereas qPCR is a fast, unbiased and a sensitive method to detect and quantify individual species. However, qPCR relies on the use of proper primers and reasonable pure DNA template and fails to discriminate between DNA from viable and dead fungal structures.
This work had the overall aim to lay the grounds for a reliable, sensitive and high-throughput molecular detection method of natural Verticillium infested soils. This knowledge could give the ability to growers to test the level of soil infestation prior to sowing and to optimize their crop rotation schemes.
Materials and methods
Fungal isolates, cultures and DNA extraction
DNA from single-spore isolates of four Verticillium species (Table 1) was used as reference materials to test the specificity of the primers design. All individual fungal isolates were grown on potato dextrose agar (PDA) for two weeks and DNA was extracted from mycelium using a CTAB method (Moller et al. 1992). The DNA was quantified using a NanoDrop Micro Photometer. Microsclerotia of V. dahliae and V. longisporum were obtained from in vitro grown oilseed rape stems and further cultured as described by Neumann and Dobinson (2003). Finally, water suspensions were made by serially dilutions to achieve 25, 50, 500, 5000 and 10,000 microsclerotia per ml. One millilitre of these suspensions was used to artificially infest 500 g soils (Hasselfors, autoclaved and pre-analyzed for traces of Verticillium) followed by DNA extractions.
DNA extractions from artificially infested soils
Soil samples were air dried at room temperature, milled and sieved through 106 and 20 µm nested sieves prior to any analysis. To avoid overloading, 100 mg soil powder was used when evaluating tested DNA extraction methods. DNA was extracted using (1) a bead-beating technique, (2) a freeze–thaw technique, (3) density flotation or (4) using commercial DNA kits. In the manual DNA extraction methods, soil was homogenized by vortexing in 1 ml extraction buffer (500 mM Tris–HCl pH 8.0, 100 mM sodium EDTA pH 8.0, 1.5 M NaCl). For the bead beating cell disruption (100 mg), samples were homogenized using the FastPrep®-24 (MPbio) with 100 mg of 0.1 mm diameter zirconia beads for 2 × 30 s at maximum speed 6 with intermittent storage on ice for 10 min. Freeze–thaw disruption of cells in the soil samples was achieved by three cycles of freezing in −70 °C overnight and thawing at 55 °C in a water bath for 15 min. Alternatively, a density flotation of the soil samples was carried out prior to the DNA extraction to further enhance yields. Here, 10 g soil was sedimented in 65% sucrose by centrifugation at 3000g for 15 min. The supernatant was passed through 0.2 μM filters (Millipore), the material retained on the filter was washed with sterile water and dissolved in water.
Samples obtained from either approach were incubated with the addition of proteinase K for 30 min at 37 °C, mixed with 20% (w/v) SDS and incubated at 65 °C for 1 h before centrifugation, at 4000g for 10 min. The supernatants were transferred to new tubes containing 0.5 volume of polyethylene glycol (PEG) solution [30% (w/v) PEG, in 1.5 M NaCl] and incubated at room temperature for 2 h prior to centrifugation. Pellets were dissolved in TE buffer (pH 8.0), mixed with 10 M NH4OAc and incubated on ice for 5 min. After centrifugation, the DNA in the supernatant was purified by phenol chloroform–isoamyl alcohol extraction, and isopropanol precipitation. The DNA was re-suspended in TE buffer and treated with RNAse before storage at −20 °C. DNA extraction using the commercial E.Z.N.A. soil DNA kit (Omega Bio-Tek), the ZR soil microbe DNA kit (Zymo Research) and the FastDNA spin kit (MP Biomedicals) for soil was performed with 350 mg soil according to the manufacturer’s instructions. DNA from leaf tissues was extracted using the GeneJET Plant Genomic purification kit (Thermo Fisher).
PCR analysis
Genomic fungal DNA (5–10 ng) was used as template for PCR analysis using Phusion high-fidelity DNA polymerase (Thermo Fischer) in the following conditions: initial denaturation at 95 °C for 2 min followed by 30 cycles of 96 °C for 15 s, melting T +3 °C for 30 s, 72 °C for 35 s, followed by final extension of 72 °C for 5 min before storage.
Real-time PCR
Real-time PCR assays were carried out using 5–10 ng total genomic DNA as template in a SYBR Green setup. All reactions were done at least as triplicates under following conditions: initial denaturation at 95 °C for 10 min followed by 30–40 cycles of 96 °C for 15 s, 60 °C for 30 s, 72 °C for 30 s, using the CFX Connect Real-Time System (Bio-Rad). The same protocol was used for all the primers tested. A melting curve analysis was done at the end of each run to verify the presence or absence of target sequences and to distinguish specific amplifications from nonspecific ones and primer dimers.
Amplification inhibition
Any potential qPCR inhibition was monitored by comparing the internal positive control Ct value obtained with soil extracts to those obtained with nuclease-free water samples (negative control). DNA extracted with the conventional methods described above, required 200-fold dilution to overcome inhibition or reactions in the amplification step. For the three commercial kit extracts, a tenfold dilution was enough to eliminate any traces of inhibition in the qPCR. The soil samples spiked with various amounts of microsclerotia from V. dahliae and V. longisporum used for the DNA extractions were also used to determine the sensitivity of the qPCR analysis.
DNA quantification
Purified DNA, isolated from the Verticillium strains, was quantified using Qubit (Invitrogen). The standard curve technique was applied with a tenfold dilution series of DNA from each Verticillium species in sterile water ranging from 100 to 1 × 106 fg DNA/5 μl. The slope of the standard curve was used to determine the PCR efficiency (E = 10−1/slope − 1). For quantification of Verticillium species in DNA from sugar beet leaves, primers of the endogenous adenylate transporter gene developed by Chaouachi et al. (2013) were used. All calculations and statistical analyses were performed as described in the ABI PRISM 7700 Sequence Detection System User Bulletin #2 (Applied Biosystems, USA) as outlined by Martin et al. (2011).
Naturally infested field samples
Soil samples from southern and western regions in Sweden were collected during springtime of 2013, 2014 and 2015. Each soil sample consisted of 10 subsamples taken to a depth of 20 cm in a W-pattern from each field. The samples were homogenized, air dried at room temperature, milled and sieved (Wallenhammar et al. 2012). Three hundred fifty mg soil powder was used in all analysis of field materials, the quantity used by commercial companies and DNA was extracted using the FastDNA spin kit (MP Biomedicals). In addition, samples of Verticillium infected leaves and soil from 20 sugar beet fields from the southern region were collected and analyzed.
Results and discussion
DNA preparation from artificially infested soil
Soil is a complex substrate for DNA extraction. Factors such as texture, pH and organic matter, including humic substances, impact quality and quantity of DNA and may influence downstream molecular analysis (Schena et al. 2013). To improve this part of the analysis, we first used samples with known amount of microsclerotia mixed in greenhouse soil. Pretreatments such as bead-beating and freeze–thaw methods did not yield in any significant increase in the DNA amounts compared to density floatation. The latter method was a less complex density floatation procedure compared to Debode et al. (2011). Our protocol yielded about 2.6 times higher amount of DNA compared to the other two methods, but the procedure is still more labor intense than the bead-beating and freeze–thaw methods.
Three commercial kits were also included in the comparisons. In our conditions, the FastDNA SPIN kit yielded the highest average amount of fungal DNA per gram of soil compared to all other methods and kits used. The lowest detection level obtained from the artificially infested soils (25 microsclerotia) was 63 fg DNA of V. dahliae and 100 fg DNA for V. longisporum.
Primer design and specificity assays
Earlier published qPCR primers were first tested on DNA of our reference strains. Those primers showed either reduced amplification efficiency (Atallah et al. 2007), formation of primer dimers (Pantou et al. 2006; Atallah et al. 2007; Lievens et al. 2006), or absence of species specificity (Pantou et al. 2006; Eynck et al. 2007; Atallah et al. 2007; Inderbitzin et al. 2011, 2013).
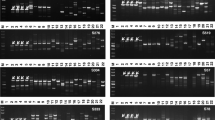
Universal species-specific primers for four Verticillium species (V. dahliae, V. albo-atrum. V. tricorpus, V. longisporum) were designed, by comparing the genome sequences of V. dahliae and V. albo-atrum (Klosterman et al. 2011) and the genome assembly data of two in house-generated Swedish V. longisporum isolates (unpublished data). Sequences were aligned and searched for single nucleotide polymorphisms (SNPs) and species-specific regions. Twenty-four primer pairs were designed in regions potentially unique to each species. Specificity of each primer pairs was tested by PCR and qPCR analysis using DNA extracted from isolates of the four species (Table 1). To test for any cross-reactivity, mixtures of DNA from one Verticillium species, and from another Verticillium strain were tested followed by qPCR analysis with primers for each individual species. All information together with melting curve analysis and the lowest Ct values, resulting in a single fragment, was the criteria set to identify the most optimal primer pair for each species (Fig. 1; Table 2). Specificity for primers enlisted in Table 2 was further checked against different soilborne pathogens such as Rhizoctonia solani, Phytophthora pisi and Aphanomyces euteiches. No amplification was observed when DNA from any of these species was used in the amplification reactions (data not shown).

Analysis of Verticillium primers specificity. Primers listed in Table 2 of a V. longisporum, b V. dahliae, c V. albo-atrum, d V. tricorpus were tested against the other Verticillium species. Amplification was observed only on-target Verticillium species. No amplification on off-target species and on mock samples was observed. Melt curve analysis shows amplification of a single fragment. Two technical replicates were used
Naturally infested soil and leaf samples
The species-specific primers, listed in Table 2, were used to analyze the level of natural infestation in 429 soil samples from two Swedish regions with intensive farming. The soil samples had all high clay content, representing characteristics as in soil mixture number four analyzed by Almquist et al. (2016). A study showing that the limit of DNA detection was lowest in samples with high clay content using similar soil handling and DNA extraction methodology as presented here. Among all 429 samples only V. dahliae and V. longisporum were found, whereas V. albo-atrum, V. tricorpus were not detected. The fungal DNA content detected in the samples ranged from 5 fg DNA/g soil to 121.62 pg DNA/g soil of V. longisporum and from 6 fg DNA/g soil to 137.84 pg DNA/g soil of V. dahliae. In the soil samples from southern Sweden, 40% were infested with V. longisporum and 7% were infested with V. dahliae (Table 3). The distribution was opposite in the western region, where 19% of the soil samples contained V. dahliae and only in 9% of the samples V. longisporum was identified (Table 3). Both Verticillium species were found in two soil samples from southern Sweden, while in the western region each species was only found individually. Most likely this result reflects the random sampling of the soil samples.
The restriction to V. longisporum and V. dahliae in the Swedish agricultural soils is in line with earlier observations (Johansson et al. 2006). Detection of both species varied from zero to remarkable high levels, which is most likely related to the different crop rotation schemes practiced in these two regions. In the south, combinations of winter oilseed rape, winter wheat, sugar beets and barley are common, whereas in the western regions sugar beets are replaced by oat or field bean. Due to the high market prices, particularly winter oilseed rape cultivation in the southern region has expanded from 24,100 ha in 2006 to 44,700 ha in 2016 (http://www.svenskraps.se). This increased oilseed cultivation likely attributed to the more frequent occurrence of V. longisporum. Using the wet sieving method, as high as 164.8 colony-forming units of V. longisporum per gram soil were found when sampling 3 weeks post-harvest from a field in the southern region, where winter oilseed rape had been grown (Andersson 2003). This observation highlights the importance of when soil sampling is done.
In addition, we collected soil and leaf samples from 20 sugar beet fields where foliar wilting had been observed. Here, only V. dahliae DNA was found in the leaf samples. In the contrary, species distribution in the 20 soil samples was: 20 positive for V. dahliae (26.39–103.66 pg DNA/g soil) and 18 positive for V. longisporum (0.72–42.37 pg DNA/g soil). These results correspond with earlier experience in Sweden that sugar beets are only infected by V. dahliae. It also shows that degradation of V. longisporum takes time and it highlights the risk of mechanical spreading long periods after growing host species besides questions concerning crop rotation.
Successful and precise diagnostic analysis of soilborne pathogens depends on many factors, such as the soil sampling procedure and subsequent treatments (drying, milling, sieving, and sample storage), efficient DNA extraction and suitable nucleic acid target sequence to achieve species-specificity. The many sources for non-optimal conditions for this type of molecular diagnosis imply that a careful interpretation of the results is needed. We suggest that soil tests for advisory work on farming practices should be based on different indicative threshold intervals, since the number of cells and thereby DNA in each individual microsclerotium of the two Verticillium species studied here varies substantially. It means that any strict correlation between the amount of DNA per gram soil and number of microsclerotia would be misleading. Cells of V. longisporum contain approximately twice more DNA than V. dahliae (Steventon et al. 2002) which adds to the complexity. A rough estimate based on 50 cells for each individual microsclerotium would equal 1 pg V. dahliae DNA alternatively 2 pg V. longisporum DNA per gram soil, which could be applied as thumb-rules for calculations based on qPCR-based soil analyses. Similarly, quantities below 100 fg DNA per gram soil should be regarded as trace levels. In light of estimations in other countries particularly on V. dahliae, levels above 10 microsclerotia/g soil should lead to serious concerns (Wei et al. 2015). However, economic losses related to different crops cannot be directly compared since there are various degrees of resistance genes introduced in the different crop varieties grown worldwide.
Molecular monitoring of presumed infested soils is becoming an increasingly important management tool and analytic commercial services have been established for example to detect the Brassica clubroot pathogen P. brassicae (Wallenhammar et al. 2016). We foresee similar progress comprising other pathogens. In many cases, the same soil sample could be analyzed for the presence of DNA of multiple plant pathogens. Such multi-test would significantly increase the efficiency and at the same time lower the analytic costs for the growers. Proper interpretation of the analytic results and information on the farm history of the soil, the sample is taken from, are keys for implementation of this type of services by advisory groups.
References
Almquist C, Persson L, Olsson Å, Sundström J, Jonsson A (2016) Disease risk assessment of sugar beet root rot using quantitative real-time PCR analysis of Aphanomyces cochioides in natural infested soil samples. Eur J Plant Pathol 145:731–742
Andersson C (2003) Detection methods of Verticillium longisporum in soil and in oilseed rape. M.Sc. Dissertation 45: ISSN 1651-5196, Swedish University of Agricultural Sciences (SLU)
Atallah ZK, Bae J, Jansky SH, Rouse DI, Stevenson WR (2007) Multiplex real-time quantitative PCR to detect and quantify Verticillium dahliae colonization in potato lines that differ in response to Verticillium wilt. Phytopathology 97:865–872
Chaouachi M, Alaya A, Ali IB, Ben Hafsa A, Nabi N, Berard A, Romaniuk M, Skhiri F, Said K (2013) Development of real-time PCR method for the detection and the quantification of a new endogenous reference gene in sugar beet “Beta vulgaris L.”: GMO application. Plant Cell Rep 32:117–128
Debode J, van Poucke K, Franca SC, Maes M, Hofte M, Heungens K (2011) Detection of multiple Verticillium species in soil using density flotation and Real-Time Polymerase Chain reaction. Plant Dis 95:1571–1580
Depotter JR, Deketelaere S, Inderbitzin P, Tiedemann AV, Hofte M, Subbarao KV, Wood TA, Thomma BP (2016) Verticillium longisporum, the invisible threat to oilseed rape and other brassicaceous plant hosts. Mol Plant Pathol 17:1004–1016
Dixelius C, Happstadius I, Berg G (2005) Verticillium wilt on Brassica oilseed crops: a Swedish perspective. J Swed Seed Assoc 115:36–48
Eynck C, Koopmann B, Grunewaldt-Stoecker G, Karlovsky P, Von TIedemann A (2007) Differential interactions of Verticillium longisporum and V. dahliae with Brassica napus detected with molecular and histological techniques. Eur J Plant Pathol 118:259–274
Fahleson J, Lagercrantz U, Hu Q, Steventon LA, Dixelius C (2003) Estimation of genetic variation among Verticillium isolates using AFLP analysis. Eur J Plant Pathol 109:361–371
Goud JC, Termorshuizen AJ (2003) Quality of methods to quantify microsclerotia of Verticillium dahliae in soil. Eur J Plant Pathol 109:523–534
Goud JKC, Termorshuizen AJ, van Bruggen AHC (2011) Verticillium wilt in nursery trees: damage thresholds, spatial and temporal aspects. Eur J Plant Pathol 131:451–465
Inderbitzin P, Bostock R, Davis R, Usami T, Platt H, Subbarao K (2011) Phylogenetics and taxonomy of the fungal vascular wilt pathogen Verticillium, with the descriptions of five new species. PLoS ONE 6:e28341
Inderbitzin P, Davis RM, Bostock RM, Subbarao KV (2013) Identification and differentiation of Verticillium species and V. longisporum lineages by simplex and multiplex PCR assays. PLoS ONE 8:e65990
Johansson A, Goud JKC, Dixelius C (2006) Plant host range of Verticillium longisporum and microsclerotia density in Swedish soils. Eur J Plant Pathol 114:139–149
Kabir Z, Bhat RG, Subbarao KV (2004) Comparison of media for recovery of Verticillium dahliae from soil. Plant Dis 88:49–55
Klosterman SJ, Subbarao KV, Kang SC, Veronese P et al (2011) Comparative genomics yields insights into niche adaptation of plant vascular wilt Pathogens. PLoS Pathog 7:e1002137
Lievens B, Brouwer M, Vanachter ACRC, Cammue BPA, Thomma BPHJ (2006) Real-time PCR for detection and quantification of fungal and oomycete tomato pathogens in plant and soil samples. Plant Sci 171:155–165
Martin T, Biruma M, Fridborg I, Okori P, Dixelius C (2011) A highly conserved NB-LRR encoding gene cluster effective against Setosphaeria turcica in sorghum. BMC Plant Biol 11:151
Moller EM, Bahnweg G, Sandermann H, Geiger HH (1992) A simple and efficient protocol for isolation of high molecular weight DNA from filamentous fungi, fruit bodies, and infected plant tissues. Nucleic Acids Res 20:6115–6116
Neumann MJ, Dobinson KF (2003) Sequence tag analysis of gene expression during pathogenic growth and microsclerotia development in the vascular wilt pathogen Verticillium dahliae. Fungal Genet Biol 38:54–62
Pantou MP, Kouvelis VN, Typas MA (2006) The complete mitochondrial genome of the vascular wilt fungus Verticillium dahliae: a novel gene order for Verticillium and a diagnostic tool for species identification. Curr Genet 50:125–136
Pegg GF, Brady BL (2002) Verticillium Wilts. CABI Publishing, Wallingford
Rakow G (2004) Species origin and economic importance of Brassica. In: Pua EC, Douglas CJ (eds) Biotech in Agriculture and Forestry. Springer, New York
Roos J, Bejai S, Oide S, Dixelius C (2014) RabGAP22 is required for defense to the vascular pathogen Verticillium longisporum and contributes to stomata immunity. PLoS ONE 9:e88187
Schena L, Nicosia MGLD, Sanzani SM, Faedda R, Ippolito A, Cacciola SO (2013) Development of quantitative PCR: detection methods for phytopathogenic fungi and oomycetes. J Plant Pathol 95:7–24
Steventon LA, Fahleson J, Hu Q, Dixelius C (2002) Identification of the causal agent of Verticillium wilt of winter oilseed rape in Sweden as Verticillium longisporum. Mycol Res 106:70–578
Wallenhammar AC, Almquist C, Söderström M, Jonsson A (2012) In field distribution of Plasmodiophora brassicae measured using quantitative real-time PCR. Plant Pathol 61:16–28
Wallenhammar AC, Gunnarson A, Hansson F, Jonsson A (2016) Quantification of Plasmodiophora brassicae using a DNA-based soil test facilitates sustainable oilseed rape production. Plants (Basel) 5:21
Wei F, Fan R, Dong HT, Shang WJ, Xu XM, Zhu HQ, Yang JR, Hu XP (2015) Threshold microsclerotial inoculum for cotton Verticillium wilt determined through wet-sieving and real-time quantitative PCR. Phytopathol 105:220–229
Zhou L, Hu Q, Johansson A, Dixelius C (2006) Verticillium longisporum and V. dahliae: infection and disease in Brassica napus. Plant Pathol 55:37–144
Acknowledgements
We thank Mattias Myrenås for analyzing the ITS-related qPCR primers. The work was funded by: the BioSoM thematic program, the Swedish Seed and Oilseed Growers Association (SSO), the Research Council Formas, and the Swedish University of Agricultural Sciences (SLU).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Communicated by Erko Stackebrandt.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Tzelepis, G., Bejai, S., Sattar, M.N. et al. Detection of Verticillium species in Swedish soils using real-time PCR. Arch Microbiol 199, 1383–1389 (2017). https://doi.org/10.1007/s00203-017-1412-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00203-017-1412-z