
Abstract
Summary
In this study, we integrated large-scale GWAS summary data and used the predicted transcriptome-wide association study method to discover novel genes associated with osteoporosis. We identified 204 candidate genes, which provide novel clues for understanding the genetic mechanism of osteoporosis and indicate potential therapeutic targets.
Introduction
Osteoporosis is a highly polygenetic disease characterized by low bone mass and deterioration of the bone microarchitecture. Our objective was to discover novel candidate genes associated with osteoporosis.
Methods
To identify potential causal genes of the associated loci, we investigated trait-gene expression associations using the transcriptome-wide association study (TWAS) method. This method directly imputes gene expression effects from genome-wide association study (GWAS) data using a statistical prediction model trained on GTEx reference transcriptome data. We then performed a colocalization analysis to evaluate the posterior probability of biological patterns: associations characterized by a single causal variant or multiple distinct causal variants. Finally, a functional enrichment analysis of gene sets was performed using the VarElect and CluePedia tools, which assess the causal relationships between genes and a disease and search for potential gene’s functional pathways. The osteoporosis-associated genes were further confirmed based on the differentially expressed genes profiled from mRNA expression data of bone tissue.
Results
Our analysis identified 204 candidate genes, including 154 genes that have been previously associated with osteoporosis, 50 genes that have not been previously discovered. A biological function analysis found that 20 of the candidate genes were directly associated with osteoporosis. Further analysis of multiple gene expression profiles showed that 15 genes were differentially expressed in patients with osteoporosis. Among these, SLC11A2, MAP2K5, NFATC4, and HSP90B1 were enriched in four pathways, namely, mineral absorption pathway, MAPK signaling pathway, Wnt signaling pathway, and PI3K-Akt signaling pathway, which indicates a causal relationship with the occurrence of osteoporosis.
Conclusions
We demonstrated that transcriptome fine-mapping identifies more osteoporosis-related genes and provides key insight into the development of novel targeted therapeutics for the treatment of osteoporosis.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Osteoporosis is a highly polygenetic disease characterized by low bone mass and deterioration in bone microarchitecture, leading to increased skeletal fragility and fracture risk [1,2,3]. A low bone mineral density (BMD), independently of bone quality or structure, measures the mineral component of bone and is a strong clinically relevant risk factor for osteoporosis and a key indicator of its diagnosis and treatment [4, 5]. Although BMD is most often measured by dual-energy X-ray absorptiometry (DXA) scanning in clinical settings, an alternative method of estimating the BMD is derived from ultrasound, typically at the heel (referred to here as estimated BMD (eBMD)). A previous genome-wide association study (GWAS) of eBMD that used heel ultrasound parameters identified 84% of all currently known genome-wide significant loci for DXA-derived BMD [6, 7], and the effect sizes were concordant between the two traits (Pearson’s r = 0.69 for the lumbar spine and 0.64 for the femoral neck) [7]. The ultrasound-derived eBMD values are highly heritable (on the order of 50% to 80%) [8], and a recent GWAS of eBMD with 426,824 individuals identified 1103 independent genome-wide significant associations at 518 loci, which is approximately 6.4-fold greater than the number of discoveries from DXA GWASs [9, 10]. However, the majority of GWAS hits are in noncoding regions, and their biological mechanisms are difficult to interpret [11, 12].
The effect of genetic variation on phenotype is complex and might involve altering the abundance of one or more proteins by regulating gene expression and then affecting the trait (SNP-Expression-Phenotype) [13, 14]. Gene expression is arguably the most impactful and well-studied effect of regulatory genetic variation. GWAS-derived loci are enriched for expression quantitative trait loci (eQTLs), which render these a potential link between the genetic variant and the biology of the disease [15,16,17]. Although most GWAS do not concomitantly measure gene expression, the influence of genetic variation on gene expression allows the use of reference datasets (e.g., GTEx [18]) to predict gene expression given a set of genotypes and to subsequently identify new disease-associated genes [11, 13, 19]. Thus, the transcriptome-wide association study (TWAS) approach has been established to identify genes whose expression is associated with complex traits by integrating genetic and transcriptional variation [20, 21]. Instead of testing millions of SNPs in GWAS, TWAS evaluates the association among the predicted expression of thousands of genes, which greatly reduces the burden of multiple comparisons due to statistical inference. This approach has been shown to have the potential to identify the genes responsible for GWAS-identified associations for complex traits and provide mechanistic insight regarding genes that are regulated via disease-associated genetic variants [22,23,24].
In this study, we conducted a transcriptome-wide association study to identify genes associated with osteoporosis by integrating gene expression data from the Genotype-Tissue Expression (GTEx) and GWAS summary data from the Genetic Factors for Osteoporosis (GEFOS) Consortium and then evaluated the biological patterns of expression-trait associations using the COLOC method. We then used VarElect to understand the biological function of the associations between the significant genes identified by the TWAS and osteoporosis. In comparison to the results from the differential analysis of multiple gene expression profiles of osteoporosis, we further verified the causal associations between the estimated bone mineral density and the significant genes identified by the TWAS.
Methods
GWAS summary datasets of osteoporosis
The GWAS summary statistics for osteoporosis were derived from the GEFOS Consortium website in December 2018. The phenotypic features of osteoporosis were measured based on the bone mineral density estimated from quantitative heel ultrasounds. The large-scale GWAS analysis of eBMD values was performed with a cohort of 426,824 participants (55% female) from the UK Biobank [9]. Briefly, a GWAS was performed based on the HRC imputation panel (hg19), which includes approximately 14,000,000 SNPs with MAF ≥ 0.05% and acceptable imputation quality (info score > 0.3). A detailed description of the sample characteristics, experimental design, and statistical analysis was previously published [25].
Integration of GWAS and gene expression
To integrate the GWAS results and gene expression, we used the TWAS method. TWAS integrates information from expression reference panels (SNP−gene expression correlation), GWAS summary statistics (SNP−phenotype correlation), and linkage disequilibrium reference panels (SNP−SNP correlation) to assess the association between the cis-genetic component of an expression and a trait (expression−osteoporosis correlation) [20, 22]. In practice, the effect sizes of cis-SNP expression in the 500-kb loci region were estimated using a sparse mixed linear model [26]. The TWAS used pre-computed gene expression weights combined with GWAS summary statistics to calculate the association effect for each gene with the disease.
To select trait-related tissues, we used TSED_DB [27], a reference database for trait-associated tissue specificity based on GWAS results. The UK Biobank heel eBMD-related genes were significantly enriched in muscle-skeletal tissue. The muscle-bone is a unit of functional interaction, and the muscle mass is positively correlated with the BMD [28]. Several studies have shown that low muscle mass is significantly associated with osteoporosis in both men and women of all age groups [29], and age-related low muscle mass might increase the risk of osteoporotic hip fractures [30]. In this study, the gene expression weights were pretrained on GTEx v7 muscle-skeletal tissue dataset, and we derived the weights from the FUSION website. The genes with significant association signals were identified based on a p value < 3.7E-06 after strict Bonferroni correction.
Evaluation of trait−gene expression associations
To evaluate the reliability of the TWAS results and understand the biological mechanisms of trait−gene expression associations, we used the COLOC method [31], which utilizes asymptotic Bayes factors with summary statistics and a regional linkage disequilibrium structure to estimate five posterior probabilities: no association with either a GWAS signal or eQTL (PP0), association with a GWAS signal only (PP1), association with a signal eQTL only (PP2), association with a GWAS signal and eQTL with two independent SNPs (PP3), and association with a GWAS signal and eQTL having one shared SNP (PP4). For each of the GWAS hits, we defined a 500-kb region at either side of the index variant and tested for colocalization within the entire cis−region of any overlapping eQTLs (transcription start and end position of an eQTL gene plus and minus 500 kb, as defined by GTEx) in muscle–skeletal tissue from GTEx v7. In this study, we used default priors in which a random variant in the region is associated with either a GWAS or an eQTL individually (prior probabilities = 1E−04) and set the prior probability that the random variant is causal to both GWAS and eQTLs (prior probability = 1E−06). Several studies have shown that PP3+PP4 > 0.8 is a cut-off threshold that provides evidence of colocalization [32, 33]. We used strict thresholds: PP3 > 0.9 for evidence of trait−gene expression associations caused by multiple distinct causal variants from a GWAS and an eQTL and PP4 > 0.8 for evidence of trait−gene expression associations caused by a joint signal from a GWAS and an eQTL [34].
Assessment of gene−disease associations
To investigate the likelihood that functional genes are more likely to be causal, the associations of biological function between the candidate genes and osteoporosis were assessed using VarElect [35, 36], a cutting-edge Variant Election application for disease/phenotype-dependent gene variant prioritization. VarElect provides a robust algorithm for ranking genes within a shortlist, noting their likelihood to be associated with the disease of interest, and producing a list of prioritized, scored, and contextually annotated genes and direct links to supporting evidence and additional information. VarElect utilizes the deep LifeMap Knowledgebase to infer the “direct” or “indirect” association of biological function between genes and phenotypes. A “direct” association between genes and disease has been supported by many studies showing that genes can directly affect disease development, and an “indirect” association between genes and disease is based on shared pathways, protein-protein interaction networks, paralogy relationships, domain-sharing, and mutual publications.
Protein−protein interaction (PPI) network and pathway enrichment analysis
The functional networks of genes that were found to be significantly associated with osteoporosis by the TWAS were further validated using the STRING and CluePedia tools. STRING (Search Tool for the Retrieval of Interacting Genes) is an online tool designed to evaluate PPI networks [37, 38], and CluePedia is a plugin of Cytoscape software that searches for potential genes associated with certain signaling pathways by calculating linear and nonlinear statistical dependencies from experimental data [39, 40]. The PPI networks of the significant genes identified by the TWAS were constructed using STRING. The functional pathways were detected and visualized using CluePedia.
Differential analysis of gene expression
To further validate the functional causality of candidate genes, the Gene Expression Omnibus (GEO) database and European Molecular Biology Laboratory (EMBL-EBI) database were searched to identify gene expression profiling studies of subjects with osteoporosis. The following key search terms were used: “osteoporosis,” “gene expression,” and “microarray.” We obtained gene expression profiles from four different sources and included original microarray studies that analyzed the differential gene expression profiles between patients with osteoporosis and normal controls, as shown in Table 2. The existence of heterogeneity among multiple microarray studies arising from different microarray platforms, gene nomenclature, and clinical samples makes it infeasible to compare the gene expression data directly. Therefore, normalization is necessary to minimize heterogeneity. Consequently, we performed a robust multiarray average approach [41] for background correction and normalization. The original GEO data were then converted into expression measures. The Limma package [42] was used to identify the differentially expressed probe sets between patients with osteoporosis and normal controls. Gene-specific t tests were performed, and p values were calculated. Multiple testing adjustment was performed, and the genes with adjusted p values < 0.05 were selected as differentially expressed genes (DEGs).
Results
TWAS-based identification of candidate genes for the treatment of osteoporosis
We first used the TWAS method with GWAS summary data from the GEFOS consortium to identify candidate genes associated with osteoporosis. In this study, we used the eBMD GWAS summary dataset rather than fragility fractures because fragility fractures were not found to be enriched by the TWAS, as shown in Supplementary Table 7. A gene expression reference panel for muscle-skeletal, which has a total of 13,416 expressed genes, was used. The TWAS identified 204 significantly associated genes with a p value < 3.7E-06, as shown in Fig. 1.

Manhattan plot of the results from the TWAS (upper panel) and GWAS (lower panel) of osteoporosis. The transcriptome-wide significance threshold was p value = 3.7E-06; the genome-wide significance threshold was p value = 6.6E-09. A total of 1103 conditionally independent SNPs at 515 loci among n = 426,824 UK Biobank participants passed the criteria for genome-wide significance
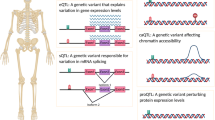
The TWAS method can detect causal genes by effectively predicting genetic variants based on gene expression. The following four biological patterns were identified by the TWAS (Fig. 2). First, for SNPs in coding regions (introns and exons) significantly associated with osteoporosis, the causal genes identified by the GWAS and the TWAS were likely to be consistent, as shown in Fig. 2a. The effect size of rs10411210 (PGWAS = 1.6E-119) on osteoporosis obtained from the GWAS corresponds with that of rs10411210 on RHPN2 (PTWAS = 4.4E-73) gene expression identified from the TWAS. Second, for SNPs in noncoding regions, the candidate genes might be close to the significant eQTLs but different from the GWAS hits, as shown in Fig. 2b. The variant rs2785197 (PGWAS = 6.5E-44) in 11p13 mapped to PDHX in GWAS, but the causal gene for rs2785197 in our TWAS results was more likely to be CD44 (PTWAS = 1.1E-32). The colocalization analysis showed that CD44 (PP4 = 0.99 in Supplementary Table 2) gene expression was regulated by the single variant rs2785197, which might be regarded as its expression regulation element. Third, the candidate genes might be regulated by relatively distant significant SNPs in noncoding regions, as shown in Fig. 2c. Our TWAS results indicated that rs4792909 (PGWAS = 1.5E-74) in 17q21.31 might be associated with G6PC3 (PTWAS = 4.2E-26). The distance between rs4792909 and G6PC3 is 387 kb, but we did not find the gene identified by the GWAS near rs4792909. Fourth, candidate genes were discovered based on SNPs that were not significantly associated with osteoporosis. The nonsignificant region identified from the GWAS was a novel discovery: rs1003260 (PGWAS = 3.6E-08) in 6q13 was associated with RIMS1 (PTWAS = 2.1E-08), as shown in Fig. 2d. RIMS1, as a novel locus, was first reported to be associated with BMD, and further investigation was performed.

Biological patterns identified by the TWAS. a): For significant SNPs in coding regions, rs10411210 (PGWAS = 1.6E-119) in 19q13.11 is associated with RHPN2 (PTWAS = 4.4E-73). b): For SNPs in the noncoding regions, rs2785197 (PGWAS = 6.5E-44) in 11p13 was associated with PDHX, which was marked in green, as determined by the GWAS, but the causal gene for rs2785197 was more likely to be CD44, which is marked in red (PTWAS = 1.1E-32), as determined by our TWAS. c): rs4792909 (PGWAS = 1.5E-74) in 17q21.31 might be associated with G6PC3 (PTWAS = 4.2E-26). The distance between rs4792909 and G6PC3 is 387 kb, but no gene has been identified by a GWAS near rs4792909. d): rs1003260 (PGWAS = 3.6E-08) in 6q13 was associated with RIMS1 (PTWAS = 2.1E-08)
Gene expression differences identified by TWAS might be causally associated with the phenotype of interest but can also be due to variant linkage disequilibrium or gene product co-expression [43, 44]. To pinpoint the causal relationship between the target gene of an eQTL and a complex trait, we performed a colocalization analysis using the COLOC method; see the Methods section. We used a strict threshold for single variant colocalization with PP4 > 0.8 and a stricter threshold for multiple variants colocalization with PP3 > 0.9 as this category seems slightly inflated than PP4; see QQ plot in Supplementary Figure 4. The results showed that 103 TWAS associations provided strong evidence of joint causal variants with PP3 > 0.9, as shown in Supplementary Table 1, and 101 showed evidence of a single causal variant with PP4 > 0.8, as shown in Supplementary Table 2.
Compared with previous GWAS studies, we found that 51 of the identified genes were previously implicated in osteoporosis risk by GWASs, as demonstrated in the literature, and 153 genes have not been reported to be associated with osteoporosis risk in previous GWASs, as shown in Figs. 3a–b.

Significant genes and candidate genes in muscle-skeletal tissue identified by TWAS. (a) Comparison of significant genes found using the TWAS and GWAS methods. (b) The top 20 candidate genes were not reported in previous GWASs, the red bars indicate upregulated gene expression, and the blue bars indicate downregulated gene expression (full lists can be found in Supplementary Figure 1, Supplementary Table 1 and Supplementary Table 2).
Assessment of the candidate gene−osteoporosis associations
For 153 candidate genes, we evaluated the associations between the candidate genes and osteoporosis through an analysis using VarElect. The analytical results showed that 20 genes (Supplementary Table 3) were “directly” associated, 83 genes were “indirectly” associated (Supplementary Table 4), and the remaining genes have not yet been classified. The direct associations indicated that the target genes were supported by rich evidence (the relevant literature, gene function annotation, etc.). The score shown in Supplementary Table 3 indicated the strength of the association between the gene and osteoporosis: a higher score indicates stronger evidence. Indirectly associated genes might interact with intermediaries to influence the development of osteoporosis through a PPI network and pathways (Supplementary Table 5). We considered the remaining unidentified genes as novel candidate genes, which were mainly lncRNAs, pseudogenes, and antisense genes. These novel candidate markers are potential disease factors for which there is no available evidence and thus need further investigation.
Functional pathways of the candidate genes
To further verify the associations between the significant genes identified by the TWAS and osteoporosis, we explored the biological function pathways of these genes using the STRING and CluePedia tools. Four pathways that may promote the understanding of the mechanism of osteoporosis were enriched (adjusted p value < 0.05), as shown in Table 1. However, eBMD-related genes have mostly not yet been well studied, and few overlaps were found with the KEGG database. Other nonsignificant KEGG pathways may also have important roles on osteoporosis and provide additional clues, as shown in Supplementary Table 5. Among them, some of the pathways which interacted with each other were shown in Supplementary Figure 5 (e.g., PI3K-Akt signaling, focal adhesion, and ECM-receptor interaction ). These results showed that the significant genes identified by the TWAS are involved in many biological mechanisms in the development of osteoporosis.
Functional validation for the candidate genes
Previous research based on expression profiling with gene signatures of cellular models to characterize the gene’s involvement in bone metabolism and disease processes revealed that impaired osteoblastic differentiation reduces bone formation and causes severe osteoporosis in animals [45]. We analyzed four gene expression profile datasets from bone, bone marrow, monocyte cells, and B cells of patients with osteoporosis and normal controls and high- and low-BMD control groups. Based on the cut-off criterion for the identification of DEGs (adjusted p value < 0.05), a total of 15 significant genes identified by the TWAS were duplicated, and 11 of these genes were not found by the GWAS, as shown in Table 2. The results of the functional pathway analysis also supported our findings. As shown in Fig. 4, we discovered that four differentially expressed genes were enriched in four KEGG pathways that were significantly and strongly associated with osteoporosis. SLC11A2, which is enriched in the mineral absorption pathway (Ppathway = 0.019), regulates the fine-tuned balance between bone resorption and bone formation and thus affects bone density [46]; MAP2K5 is enriched in the MAPK signalling pathway (Ppathway = 0.388), which is involved in the regulation of many cellular physiological functions, such as proliferation, differentiation, inflammation, and apoptosis, and affects bone formation [47, 48]; NFATC4 is enriched in the Wnt signalling pathway (Ppathway = 0.104) and is a candidate for therapeutic intervention aimed at increasing bone mass and strength in treated patients [49, 50]; HSP90B1 is enriched in the PI3K-AKT signalling pathway (Ppathway= 0.020), which is involved in the inhibition of osteoporosis through the promotion of osteoblast proliferation, differentiation and bone formation [51, 52]. Therefore, we inferred that these genes are very likely to be the causal pathogenic genes of osteoporosis. Due to the small sample size of the mRNA expression datasets, more experiments and other types of RNA datasets are needed in the future.

Biological function verification of the significant genes identified by the TWAS. SLC11A2, NFATC4 and HSP90B1 showed significantly differential expression in bone tissue and bone marrow cells between patients with osteoporosis and normal controls. MAP2K5 gene expression is significantly downregulated in B cells of low-BMD samples
OP osteoporosis, NOR normal, BMD bone mineral density
Discussion
Multiple GWASs have been performed with considerable sample sizes to detect osteoporosis heredity, but the progress toward understanding the mechanism of the disease is limited. Most GWAS hits are in noncoding regions, and it is difficult to understand downstream biological inferences. In most cases, the nearest genes are usually reported [53, 54]. SNPs in noncoding regions do not have to regulate genes based on the distance between SNPs and genes. The integration of GWAS and transcriptome data will empower novel discovery and, most importantly, pinpoint causality. The TWAS method calculates local SNP−gene expression correlations and further calculates the likelihood of gene causality. Therefore, for a significant SNP in a coding region, the causal genes identified by using the GWAS and TWAS methods should be and indeed are consistent, as shown in Fig. 2a. For SNPs in noncoding regions, the causal genes might be close to the significant eQTLs, which might differ from the GWAS hits shown in Fig. 2b. The most relevant GWAS variants and their nearest genes were not enriched as causal variants/genes by the TWAS. Accordingly, we compared the GWAS-reported genes and the TWAS-enriched genes in all significant GWAS regions, as shown in Supplementary Table 8. The TWAS method can even discover causal genes in regions with no significant GWAS hits, as shown in Fig. 2d, and relatively distant significant SNPs, as shown in Fig. 2c. More valuable region plots can be found in Supplementary Figure 2.
We found 204 significant candidate genes through our TWAS. Among these genes, 103 genes were regulated by two distinct causal variants, and 101 genes were regulated by a single causal variant. In comparison with the GWAS, 51 genes were duplicated. For the remaining 153 genes, an analysis of their biological functions revealed that 20 genes directly affected pathways closely related to the development of osteoporosis: IBSP, EIF2B2, CD44, FEN1, UBA7, MARCO, ATF1, CBFB, G6PC3, SLC11A2, MST1R, PLEKHM1, ATRIP, CCDC36, AKAP7, EPRS, CTSB, CRHR1, FADS1, and MAP1LC3A. For example, IBSP (score = 13.74) is the gene most associated with osteoporosis. The COLOC analysis showed that the SNPs rs1471403 and rs1054627 might co-regulate IBSP gene expression. In addition, previous studies have shown that IBSP is expressed in all major bone cells, including osteoblasts, osteocytes, and osteoclasts [55], and encodes a major noncollagenous bone matrix protein that binds to calcium and hydroxyapatite via acidic amino acid clusters in the PI3K-AKT signaling pathway [51]. In contrast, we found that 83 genes appear to exert their biological functions to affect the development of osteoporosis through a PPI network. As shown in Supplementary Figure 3, RAC3 and NFATC4 were enriched in the MAPK signaling pathway (Ppathway = 0.388) through interactions with genes (ESR1, FOS, IGF1, TGFB1, JUN, NFATC1, IGF1, LRP5, TNF, and PRKACA) that are known to be associated with osteoporosis. More information on gene interactions can be found in Supplementary Table 4 and Table 5. In addition, 50 significant markers as novel candidate genes are not associated with osteoporosis based on existing knowledge, and these include 19 genes, 13 lincRNAs, 9 pseudogenes, and 9 antisense genes. Some of these candidate genes, such as AF131215.2 (PTWAS = 1.92E-66) and RP11-73 M18.6 (PTWAS = 2.72E-51), were very significant. Simultaneously, we found that RIMS1 was located in new locus, and its causal SNPs were non-significantly associated with osteoporosis in the GWAS. RIMS1 is an RAS gene superfamily member and plays a role in the regulation of voltage-gated calcium channels during neurotransmitter and insulin release. Although previous studies have not provided evidence supporting a causal association with osteoporosis, these genes might be potential causal biomolecules for osteoporosis, and more experiments are needed to verify their biological function.
Furthermore, we obtained additional evidence by comparing differentially expressed genes through an analysis of four types of gene expression profiles. Our results identified 15 significantly differentially expressed genes, as shown in Table 2. Among them, 11 genes were not discovered by the GWAS, and these included SLC11A2, G6PC3, and MAP1LC3A, which have been proven to be directly associated with osteoporosis, and NFATC4, HSP90B1, TP53I13, MTMR9, PCGF2, MAPT-AS1, and MAP2K5, which are considered to be indirectly associated with osteoporosis based on PPI networks and literature. It is worth mentioning that SLC11A2, NFATC4, HSP90B1, and MAP1LC3A were enriched in four very important pathways. In addition, GPATCH1, SPTBN1, DPP8, and ISYNA1 were also found in the GWAS, and our results once again confirmed their potential as candidate disease markers. The biological function information of these 15 genes can be found in Supplementary Table 6.
This investigation constitutes the largest study integrating the GWAS and TWAS methods to identify osteoporosis susceptibility genes. We used data from the 426,824 individuals with osteoporosis in the GWAS and 860 samples from GTEx in our analyses. Many findings were discovered, although this research still has some limitations. First, the current TWAS method cannot explain the variants influencing disease that are independent of cis expression because it was only trained on cis-eQTL analysis. Second, some bias might exist due to the use of normal muscle-skeletal tissues from GTEx to make predictions. Third, tissue sensitivity and tissue specificity are important issues to consider when performing a TWAS. Prediction models built on gene expression data from osteoblast cells of osteoporosis patients will help identify additional candidate genes associated with osteoporosis [56].
In summary, we integrated data from GWAS and transcriptome expression to identify 204 significant genes associated with osteoporosis. One hundred fifty-four genes have been previously associated with osteoporosis (literature, protein-protein interaction networks, pathway, etc.), and 50 genes have not been previously discovered. Therefore, we analyzed the biological patterns of those loci and explained their pathway interactions. We hope that our findings will provide novel insights for future pathogenetic studies of osteoporosis.
Abbreviations
- BMD:
-
bone mineral density
- DEGs:
-
differentially expressed genes
- eQTLs:
-
expression quantitative trait loci
- GEO:
-
Gene Expression Omnibus
- GEFOS:
-
Genetic Factors for Osteoporosis
- GWAS:
-
genome-wide association study
- GTEx:
-
Genotype-Tissue Expression
- OP:
-
osteoporosis
- TWAS:
-
transcriptome-wide association study
- SNP:
-
single nucleotide polymorphism.
References
Peacock M, Econs MJ, Turner CH, Foroud T (2002) Genetics of Osteoporosis. Endocr Rev 23(3):303–326
Clark GR, Duncan EL (2015) The genetics of osteoporosis. Br Med Bull 113(1):73–81
Consensus development conference: Prophylaxis and treatment of osteoporosis (1991) Osteoporos Int 1(2):114–117
Cauley JA, Hochberg MC, Lui L-Y, Palermo L, Ensrud KE, Hillier TA, Nevitt MC, Cummings SR (2007) Long-term risk of incident vertebral fractures. JAMA 298(23):2761–2767
Jindal M, Lakhwani O, Omkar K, Agarwal S, GJO K (2018) Journals ROA: Bone density versus bone quality as a predictor of bone strength 12(1):21–31
Moayyeri A, Hsu YH, Karasik D, Estrada K, Xiao SM, Nielson C, Srikanth P, Giroux S, Wilson SG, Zheng HF, Smith AV, Pye SR, Leo PJ, Teumer A, Hwang JY, Ohlsson C, McGuigan F, Minster RL, Hayward C, Olmos JM, Lyytikäinen LP, Lewis JR, Swart KMA, Masi L, Oldmeadow C, Holliday EG, Cheng S, van Schoor NM, Harvey NC, Kruk M, del Greco M F, Igl W, Trummer O, Grigoriou E, Luben R, Liu CT, Zhou Y, Oei L, Medina-Gomez C, Zmuda J, Tranah G, Brown SJ, Williams FM, Soranzo N, Jakobsdottir J, Siggeirsdottir K, Holliday KL, Hannemann A, Go MJ, Garcia M, Polasek O, Laaksonen M, Zhu K, Enneman AW, McEvoy M, Peel R, Sham PC, Jaworski M, Johansson Å, Hicks AA, Pludowski P, Scott R, Dhonukshe-Rutten RAM, van der Velde N, Kähönen M, Viikari JS, Sievänen H, Raitakari OT, González-Macías J, Hernández JL, Mellström D, Ljunggren Ö, Cho YS, Völker U, Nauck M, Homuth G, Völzke H, Haring R, Brown MA, McCloskey E, Nicholson GC, Eastell R, Eisman JA, Jones G, Reid IR, Dennison EM, Wark J, Boonen S, Vanderschueren D, Wu FCW, Aspelund T, Richards JB, Bauer D, Hofman A, Khaw KT, Dedoussis G, Obermayer-Pietsch B, Gyllensten U, Pramstaller PP, Lorenc RS, Cooper C, Kung AWC, Lips P, Alen M, Attia J, Brandi ML, de Groot LCPGM, Lehtimäki T, Riancho JA, Campbell H, Liu Y, Harris TB, Akesson K, Karlsson M, Lee JY, Wallaschofski H, Duncan EL, O'Neill TW, Gudnason V, Spector TD, Rousseau F, Orwoll E, Cummings SR, Wareham NJ, Rivadeneira F, Uitterlinden AG, Prince RL, Kiel DP, Reeve J, Kaptoge SK (2014) Genetic determinants of heel bone properties: genome-wide association meta-analysis and replication in the GEFOS/GENOMOS consortium. Hum Mol Genet 23(11):3054–3068
Kemp JP, Morris JA, Medina-Gomez C, Forgetta V, Warrington NM, Youlten SE, Zheng J, Gregson CL, Grundberg E, Trajanoska K, Logan JG, Pollard AS, Sparkes PC, Ghirardello EJ, Allen R, Leitch VD, Butterfield NC, Komla-Ebri D, Adoum AT, Curry KF, White JK, Kussy F, Greenlaw KM, Xu C, Harvey NC, Cooper C, Adams DJ, Greenwood CMT, Maurano MT, Kaptoge S, Rivadeneira F, Tobias JH, Croucher PI, Ackert-Bicknell CL, Bassett JHD, Williams GR, Richards JB, Evans DM (2017) Identification of 153 new loci associated with heel bone mineral density and functional involvement of GPC6 in osteoporosis. Nat Genet 49(10):1468–1475
Lee M, Czerwinski SA, Choh AC, Demerath EW, Sun SS, Chumlea WC, Towne B, Siervogel RM (2006) Unique and common genetic effects between bone mineral density and calcaneal quantitative ultrasound measures: the Fels Longitudinal Study. Osteoporosis international : a journal established as result of cooperation between the European Foundation for Osteoporosis and the National Osteoporosis Foundation of the USA 17(6):865–871
Morris JA, Kemp JP, Youlten SE, Laurent L, Logan JG, Chai RC, Vulpescu NA, Forgetta V, Kleinman A, Mohanty ST et al (2019) An atlas of genetic influences on osteoporosis in humans and mice. Nat Genet 51(2):258–266
Medina-Gomez C, Kemp JP, Trajanoska K, Ja L, Chesi A, Ahluwalia TS, Mook-Kanamori DO, Ham A, Hartwig FP, Evans DS et al (2018) Life-course genome-wide association study meta-analysis of total body BMD and assessment of age-specific effects. Am J Hum Genet 102(1):88–102
Nicolae DL, Gamazon E, Zhang W, Duan SW, Dolan ME, Cox NJ (2010) Trait-associated SNPs are more likely to be eQTLs: annotation to enhance discovery from GWAS. Plos Genetics, 6(4).
Moonesinghe R, Khoury MJ, Liu T, Ioannidis JP (2008) Required sample size and nonreplicability thresholds for heterogeneous genetic associations. Proc Natl Acad Sci U S A 105(2):617–622
Albert FW, Kruglyak L (2015) The role of regulatory variation in complex traits and disease. Nat Rev Genet 16(4):197–212
Zhang X, Joehanes R, Chen BH, Huan T, Ying S, Munson PJ, Johnson AD, Levy D, O'Donnell CJ (2015) Identification of common genetic variants controlling transcript isoform variation in human whole blood. Nat Genet 47(4):345–352
Stranger BE, Nica AC, Forrest MS, Dimas A, Bird CP, Beazley C, Ingle CE, Dunning M, Flicek P, Koller D, Montgomery S, Tavaré S, Deloukas P, Dermitzakis ET (2007) Population genomics of human gene expression. Nat Genet 39(10):1217–1224
Lee D, Gorkin DU, Baker M, Strober BJ, Asoni AL, McCallion AS, Beer MA (2015) A method to predict the impact of regulatory variants from DNA sequence. Nat Genet 47(8):955–961
Gusev A, Lee SH, Trynka G, Finucane H, Vilhjalmsson BJ, Xu H, Zang C, Ripke S, Bulik-Sullivan B, Stahl E et al (2014) Partitioning heritability of regulatory and cell-type-specific variants across 11 common diseases. Am J Hum Genet 95(5):535–552
Carithers LJ, Moore HM (2015) The Genotype-Tissue Expression (GTEx) Project. Biopreserv Biobank 13(5):307–308
Nica AC, Montgomery SB, Dimas AS, Stranger BE, Beazley C, Barroso I, Dermitzakis ET (2010) Candidate causal regulatory effects by integration of expression QTLs with complex trait genetic associations. PLoS Genet 6(4):e1000895
Gusev A, Ko A, Shi H, Bhatia G, Chung W, Penninx BWJH, Jansen R, de Geus EJC, Boomsma DI, Wright FA, Sullivan PF, Nikkola E, Alvarez M, Civelek M, Lusis AJ, Lehtimäki T, Raitoharju E, Kähönen M, Seppälä I, Raitakari OT, Kuusisto J, Laakso M, Price AL, Pajukanta P, Pasaniuc B (2016) Integrative approaches for large-scale transcriptome-wide association studies. Nat Genet 48:245–252
Barbeira AN, Dickinson SP, Bonazzola R, Zheng J, Wheeler HE, Torres JM, Torstenson ES, Shah KP, Garcia T, Edwards TL et al (2018) Exploring the phenotypic consequences of tissue specific gene expression variation inferred from GWAS summary statistics. Nat Commun 9(1):1825
Gusev A, Mancuso N, Won H, Kousi M, Finucane HK, Reshef Y, Song L, Safi A, Schizophrenia Working Group of the Psychiatric, Genomics C, McCarrollm S et al (2018) Transcriptome-wide association study of schizophrenia and chromatin activity yields mechanistic disease insights. Nat Genet 50(4):538–548
Atkins I, Kinnersley B, Ostrom QT, Labreche K, Il'yasova D, Armstrong GN, Eckel-Passow JE, Schoemaker MJ, Nothen MM, Barnholtz-Sloan JS et al (2019) Transcriptome-wide association study identifies new candidate susceptibility genes for glioma. Cancer Res 79:2065–2071
Lu Y, Beeghly-Fadiel A, Wu L, Guo X, Li B, Schildkraut JM, Im HK, Chen YA, Permuth JB, Reid BM, Teer JK, Moysich KB, Andrulis IL, Anton-Culver H, Arun BK, Bandera EV, Barkardottir RB, Barnes DR, Benitez J, Bjorge L, Brenton J, Butzow R, Caldes T, Caligo MA, Campbell I, Chang-Claude J, Claes KBM, Couch FJ, Cramer DW, Daly MB, deFazio A, Dennis J, Diez O, Domchek SM, Dörk T, Easton DF, Eccles DM, Fasching PA, Fortner RT, Fountzilas G, Friedman E, Ganz PA, Garber J, Giles GG, Godwin AK, Goldgar DE, Goodman MT, Greene MH, Gronwald J, Hamann U, Heitz F, Hildebrandt MAT, Høgdall CK, Hollestelle A, Hulick PJ, Huntsman DG, Imyanitov EN, Isaacs C, Jakubowska A, James P, Karlan BY, Kelemen LE, Kiemeney LA, Kjaer SK, Kwong A, le ND, Leslie G, Lesueur F, Levine DA, Mattiello A, May T, McGuffog L, McNeish IA, Merritt MA, Modugno F, Montagna M, Neuhausen SL, Nevanlinna H, Nielsen FC, Nikitina-Zake L, Nussbaum RL, Offit K, Olah E, Olopade OI, Olson SH, Olsson H, Osorio A, Park SK, Parsons MT, Peeters PHM, Pejovic T, Peterlongo P, Phelan CM, Pujana MA, Ramus SJ, Rennert G, Risch H, Rodriguez GC, Rodríguez-Antona C, Romieu I, Rookus MA, Rossing MA, Rzepecka IK, Sandler DP, Schmutzler RK, Setiawan VW, Sharma P, Sieh W, Simard J, Singer CF, Song H, Southey MC, Spurdle AB, Sutphen R, Swerdlow AJ, Teixeira MR, Teo SH, Thomassen M, Tischkowitz M, Toland AE, Trichopoulou A, Tung N, Tworoger SS, van Rensburg EJ, Vanderstichele A, Vega A, Edwards DV, Webb PM, Weitzel JN, Wentzensen N, White E, Wolk A, Wu AH, Yannoukakos D, Zorn KK, Gayther SA, Antoniou AC, Berchuck A, Goode EL, Chenevix-Trench G, Sellers TA, Pharoah PDP, Zheng W, Long J (2018) A transcriptome-wide association study among 97,898 women to identify candidate susceptibility genes for epithelial ovarian cancer risk. Cancer Res 78(18):5419–5430
Sudlow C, Gallacher J, Allen N, Beral V, Burton P, Danesh J, Downey P, Elliott P, Green J, Landray M, Liu B, Matthews P, Ong G, Pell J, Silman A, Young A, Sprosen T, Peakman T, Collins R (2015) UK biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med 12(3):e1001779
Zhou X, Carbonetto P, Stephens M (2013) Polygenic modeling with bayesian sparse linear mixed models. PLoS Genet 9(2):e1003264
Jia P, Dai Y, Hu R, Pei G, Manuel AM, Zhao Z (2020) TSEA-DB: a trait–tissue association map for human complex traits and diseases. Nucleic Acids Res 48(D1):D1022–D1030
Ma HT, Griffith JF, Xu L, Leung PC (2014) The functional muscle–bone unit in subjects of varying BMD. Osteoporos Int 25(3):999–1004
Jang S-Y, Park J, Ryu S-Y, Choi S-W (2020) Low muscle mass is associated with osteoporosis: A nationwide population-based study. Maturitas 133:54–59
Kim BJ, Ahn SH, Kim HM, Lee SH, Koh JM (2015) Low skeletal muscle mass associates with low femoral neck strength, especially in older Korean women: the Fourth Korea National Health and Nutrition Examination Survey (KNHANES IV). Osteoporos Int 26(2):737–747
Giambartolomei C, Vukcevic D, Schadt EE, Franke L, Hingorani AD, Wallace C, Plagnol V (2014) Bayesian Test for Colocalisation between Pairs of Genetic Association Studies Using Summary Statistics. Plos Genetics, 10(5).
Guo H, Fortune MD, Burren OS, Schofield E, Todd JA, Wallace C (2015) Integration of disease association and eQTL data using a Bayesian colocalisation approach highlights six candidate causal genes in immune-mediated diseases. Hum Mol Genet 24(12):3305–3313
Alasoo K, Rodrigues J, Mukhopadhyay S, Knights AJ, Mann AL, Kundu K, Hale C, Dougan G, Gaffney DJ, Consortium H (2018) Shared genetic effects on chromatin and gene expression indicate a role for enhancer priming in immune response. Nat Genet 50(3):424–431
Wu Y, Broadaway KA, Raulerson CK, Scott LJ, Pan C, Ko A, He A, Tilford C, Fuchsberger C, Locke AE, Stringham HM, Jackson AU, Narisu N, Kuusisto J, Pajukanta P, Collins FS, Boehnke M, Laakso M, Lusis AJ, Civelek M, Mohlke KL (2019) Colocalization of GWAS and eQTL signals at loci with multiple signals identifies additional candidate genes for body fat distribution. Hum Mol Genet 28(24):4161–4172
Stelzer G, Plaschkes I, Oz-Levi D, Alkelai A, Olender T, Zimmerman S, Twik M, Belinky F, Fishilevich S, Nudel R et al (2016) VarElect: the phenotype-based variation prioritizer of the GeneCards Suite. BMC Genomics 17(Suppl 2):444
Stelzer G, Rosen N, Plaschkes I, Zimmerman S, Twik M, Fishilevich S, Stein TI, Nudel R, Lieder I, Mazor Y et al (2016) The GeneCards Suite: From Gene Data Mining to Disease Genome Sequence Analyses. Curr Protoc Bioinformatics 54(1):1.30.31–31.30.33
Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J, Simonovic M, Roth A, Santos A, Tsafou KP, Kuhn M, Bork P, Jensen LJ, von Mering C (2015) STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res 43(Database issue):D447–D452
Szklarczyk D, Morris JH, Cook H, Kuhn M, Wyder S, Simonovic M, Santos A, Doncheva NT, Roth A, Bork P, Jensen LJ, von Mering C (2017) The STRING database in 2017: quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res 45(D1):D362–D368
Bindea G, Galon J, Mlecnik B (2013) CluePedia Cytoscape plugin: pathway insights using integrated experimental and in silico data. Bioinformatics 29(5):661–663
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T (2003) Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13(11):2498–2504
Hochreiter S, Clevert D-A, Obermayer K (2006) A new summarization method for affymetrix probe level data. Bioinformatics 22(8):943–949
Smyth GK (2005) In: Gentleman R, Carey VJ, Huber W, Irizarry RA, Dudoit S (eds) limma: Linear Models for Microarray Data. In: Bioinformatics and Computational Biology Solutions Using R and Bioconductor. Springer New York, New York, NY, pp 397–420
Hormozdiari F, van de Bunt M, Segre AV, Li X, Joo JWJ, Bilow M, Sul JH, Sankararaman S, Pasaniuc B, Eskin E (2016) Colocalization of GWAS and eQTL Signals Detects Target Genes. Am J Hum Genet 99(6):1245–1260
Huang YT, Liang LM, Moffatt MF, Cookson WOCM, Lin XH (2015) iGWAS: Integrative genome-wide association studies of genetic and genomic data for disease susceptibility using mediation analysis. Genet Epidemiol 39(5):347–356
Misof BM, Gamsjaeger S, Cohen A, Hofstetter B, Roschger P, Stein E, Nickolas TL, Rogers HF, Dempster D, Zhou H, Recker R, Lappe J, McMahon D, Paschalis EP, Fratzl P, Shane E, Klaushofer K (2012) Bone material properties in premenopausal women with idiopathic osteoporosis. J Bone Miner Res 27(12):2551–2561
Xu X, Jia X, Mo L, Liu C, Zheng L, Yuan Q, Zhou X (2017) Intestinal microbiota: a potential target for the treatment of postmenopausal osteoporosis. Bone Research 5(1):17046
Peng S, Zhou G, Luk KDK, Cheung KMC, Li Z, Lam WM, Zhou Z, Lu WW (2009) Strontium promotes osteogenic differentiation of mesenchymal stem cells through the Ras/MAPK signaling pathway. Cell Physiol Biochem 23(1-3):165–174
Wanachewin O, Boonmaleerat K, Pothacharoen P, Reutrakul V, Kongtawelert P (2012) Sesamin stimulates osteoblast differentiation through p38 and ERK1/2 MAPK signaling pathways. BMC Complement Altern Med 12:71–71
Baron R, Gori F (2018) Targeting WNT signaling in the treatment of osteoporosis. Curr Opin Pharmacol 40:134–141
Amjadi-Moheb F, Akhavan-Niaki H (2019) Wnt signaling pathway in osteoporosis: Epigenetic regulation, interaction with other signaling pathways, and therapeutic promises. J Cell Physiol 234:14641–14650
Xi JC, Zang HY, Guo LX, Xue HB, Liu XD, Bai YB, Ma YZ (2015) The PI3K/AKT cell signaling pathway is involved in regulation of osteoporosis. J Recept Sig Transd 35(6):640–645
Pan JM, Wu LG, Cai JW, Wu LT, Liang M (2019) Dexamethasone suppresses osteogenesis of osteoblast via the PI3K/Akt signaling pathway in vitro and in vivo. J Recept Sig Transd 39(1):80–86
Claussnitzer M, Dankel SN, Kim K-H, Quon G, Meuleman W, Haugen C, Glunk V, Sousa IS, Beaudry JL, Puviindran V, Abdennur NA, Liu J, Svensson PA, Hsu YH, Drucker DJ, Mellgren G, Hui CC, Hauner H, Kellis M (2015) FTO Obesity Variant Circuitry and Adipocyte Browning in Humans. N Engl J Med 373(10):895–907
Spain SL, Barrett JC (2015) Strategies for fine-mapping complex traits. Hum Mol Genet 24(R1):R111–R119
Trost Z, Trebse R, Prezelj J, Komadina R, Logar DB, Marc J (2010) A microarray based identification of osteoporosis-related genes in primary culture of human osteoblasts. Bone 46(1):72–80
Orlic I, Borovecki F, Simic P, Vukicevic S (2007) Gene expression profiling in bone tissue of osteoporotic mice. Arh Hig Rada Toksikol 58(1):3–11
Acknowledgements
We are thankful to our institutes for providing their expertise, which greatly aided this research work. We are also thankful to Diana Czuchry for revising the manuscript.
Funding
This research was supported by the National Natural Science Foundation of China (grant number 11801542), the Strategic Priority Research Program of the Chinese Academy of Sciences (grant number XDB38040200) and the Shenzhen Science and Technology Projects (grant numbers JCYJ20170818164014753, JCYJ20170818163445670, and JCYJ20180703145002040). The funding body had no role in the design of the study, the collection, analysis, and interpretation of the data or the writing of the manuscript.
Author information
Authors and Affiliations
Contributions
MZ and PY conceived the project and designed the experiments. MZ, HF, JJ, LY, WS, and YL analyzed the data. MZ and PY wrote the manuscript. All the authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable
Consent for publication
Not applicable
Availability of data and materials
The GWAS summary data are available from the Genetic Factors for Osteoporosis (GEFOS) Consortium; the gene expression weights of muscle were derived from the FUSION website; the gene expression profiles are available from NCBI GEO and EMBL-EBI.
Websites
GEFOS, http://www.gefos.org/; FUSION, https://gusevlab.org/projects/fusion/; GEO, https://www.ncbi.nlm.nih.gov/geo/; EMBL-EBI, http://wwwdev.ebi.ac.uk/; and STRING, https://www.string-db.org/cgi/
Code availability
Not applicable
Competing interests
Muchun Zhu, Peng Yin, Fan Hu, Jiaxin Jiang, Li Yin, Yingxiang Li, and Shuqiang Wang declare that they have no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Zhu, M., Yin, P., Hu, F. et al. Integrating genome-wide association and transcriptome prediction model identifies novel target genes for osteoporosis. Osteoporos Int 32, 2493–2503 (2021). https://doi.org/10.1007/s00198-021-06024-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00198-021-06024-z