
Abstract
Purpose
The anesthetic-conserving device AnaConDa®, a miniature vaporizer, allows volatile sedation in the intensive care unit (ICU). We investigated the effects of isoflurane sedation on cerebral and systemic physiology parameters in neuromonitored ICU stroke patients.
Methods
Included in the study were 19 consecutive ventilated patients with intracerebral hemorrhage (12), subarachnoid hemorrhage (4), and ischemic stroke (3) who were switched from intravenous propofol or midazolam to inhalative isoflurane sedation for an average of 3.5 days. During the sedation transition, the following parameters were assessed: mean arterial pressure (MAP), intracranial pressure (ICP), cerebral perfusion pressure (CPP), middle cerebral artery mean flow velocity (MFV) and cerebral fractional tissue oxygen extraction (FTOE), as well as systemic cardiopulmonary parameters and administered drugs.
Results
After the first hour, mean ICP showed an increase of 2.1 mmHg that was not clinically relevant. Likewise, MFV did not change. MAP and CPP, however, decreased by 6.5 and 6.3 mmHg, respectively. FTOE was reduced slightly from 0.24 to 0.21 (p = 0.03). Over an observation period of 12 h, ICP remained stable, while MAP and thus CPP showed distinct decreases (CPP: −10 mmHg at 6 h, p < 0.001; −7.5 mmHg at 12 h, p = 0.005, when compared to preswitch levels) despite a 1.5-fold increase in vasopressor administration.
Conclusions
We suggest that that it is possible to reach sufficient sedation levels in cerebrovascular ICU patients by applying volatile isoflurane long-term without a relevant increase in ICP, if baseline ICP values are low or only moderately elevated. However, caution should be exercised in view of isoflurane’s decreasing effect on MAP and CPP. Multimodal neuromonitoring is strongly recommended when applying this off-label sedation method.
Similar content being viewed by others


Introduction
The aims of sedation and analgesia in the intensive care unit (ICU) patient are to achieve freedom from pain, anxiety, agitation, and tolerance of mechanical ventilation [1, 2]. In neurocritical care, additional aims are to prevent seizures, to keep intracranial pressure (ICP) low and to stabilize impaired cerebrovascular autoregulation. Common intravenous sedatives, such as midazolam and propofol, in conjunction with opioids [3], are effective but tend to accumulate and/or have specific side effects. The volatile anesthetic isoflurane, established for general anesthesia in surgery, has recently become applicable outside the operating room (OR) following the introduction of a miniature vaporizer, the AnaConDa® (anesthetic conserving device; SEDANA Medical, Uppsala, Sweden), that can be connected to any ICU respirator [4]. Studies of the AnaConDa® system in mixed ICU populations have suggested advantages [5, 6], but reports in brain-injured patients are lacking. Although still off-label, inhalative ICU sedation is currently spreading all over Europe and has been recommended as an alternative in a recent German consensus guideline [2]. Previous studies on isoflurane have suggested its neuroprotective potential in cerebrovascular disease [7]. More recent reports, however, have raised concerns about neurotoxicity [8, 9]. Furthermore, isoflurane’s reported potential to raise ICP via direct cerebral vasodilation [10–16] has discouraged its use in brain-injured patients who are prone to ICP increases. However, data only exist on the short-term OR but not the more long-term neurological ICU (NICU) setting.
In the light of these uncertainties but intrigued by isoflurane’s potential as an alternative sedative and neuroprotectant, we prospectively investigated isoflurane’s effects on cerebral and systemic hemodynamic parameters in neuromonitored ventilated stroke patients.
Methods
Patient inclusion and sedation management
Approval by our local ethics committee was obtained. All patients or their legal representatives provided written informed consent. Patients admitted to our university hospital NICU between October 2009 and April 2011 were screened for inclusion. Patients were included if they (a) had an acute cerebrovascular event, i.e. ischemic stroke (IS), intracerebral hemorrhage (ICH) or subarachnoid hemorrhage (SAH), (b) had to be ventilated, and (c) were equipped with multimodal neuromonitoring. Patients were excluded if (a) pulmonary gas exchange was severely impaired (as in oxygenation insufficiency, e.g. partial arterial oxygen pressure (PaO2) <60 mmHg despite fractional inspiratory oxygen (FiO2) >0.6 and positive end-expiratory pressure (PEEP) >10 mmHg), (b) they had refractory ICP crises (ICP >25 mmHg longer than 5 min, not responding to osmotherapy), (c) they had a family history of malignant hyperthermia, or (d) they were included in any other trial influencing the sedation regimen. Patients were ventilated in pressure-controlled mode and in a lung-protective fashion (tidal volume <6 ml/kg body weight, avoiding high inspiratory and peak pressures) by Servo respirators (MAQUET, Rastatt, Germany). According to our in-house ICU sedation standard operating procedure, patients were initially put on propofol or on midazolam if propofol induced hypotension. If analgosedation was expected to be necessary for <5 days, intravenous sedation was switched to volatile isoflurane sedation. Technical details of the AnaConDa® device are presented elsewhere [4, 6]. Isoflurane was coadministered until the gas monitor (Scio Four, Dräger; Lübeck, Germany) showed a minimal alveolar concentration (MAC) of 0.5, then the intravenous sedative was stopped. Thereafter, the isoflurane perfusion rate was adjusted to maintain the previous Richmond Agitation Sedation Scale (RASS [17]) score (target range −5 to −4), assessed from 6 h before to 6 h after sedation switch, and according to clinical and ventilation parameters. Patients were kept in the supine position with elevation of the head of the bed at 20° throughout the transition.
Systemic monitoring
Mean arterial pressure (MAP) and heart rate were measured via a radial arterial line with the pressure sensor at the level of the heart. Arterial blood oxygen saturation (SaO2), PaO2 and partial arterial carbon dioxide pressure (PaCO2), pH, base excess and bicarbonate were measured via the same radial arterial line by regular blood gas analysis. Central venous oxygen saturation (ScvO2) was measured via a jugular or subclavian central line. Respiratory rate, PEEP, peak inspiratory pressure, mean inspiratory pressure, fractional inspiratory oxygen (FiO2), and minute volume were directly assessed from the respirator. Pulmonary oxygenation was calculated as PaO2/FiO2. Vital signs, fluid-in and fluid-out, bladder temperature and drug rates were documented at least hourly during the transition period (−6 to +12 h). Safety laboratory parameters including liver enzymes and renal parameters were documented at least every other day.
Neuromonitoring
Intracranial pressure was measured via a parenchymal probe (Raumedic; Münchberg, Germany) or an external ventricular drain (EVD, zero reference level at the ear in a vertical extension device for automatic adjustment of EVD drip chambers or ICP transducers), and cerebral perfusion pressure (CPP) was calculated as MAP − ICP. Regional cerebral oxygen saturation (rSO2) was measured by bifrontal near-infrared spectroscopy (NIRS) using an INVOS 5100 system (Covidien, Mansfield, MA) [18, 19]. Differences between systemic and cerebral arteriovenous oxygen saturation were calculated as SaO2-ScvO2 and SaO2-rSO2, respectively; and fractional tissue oxygen extraction (FTOE, [20] = cerebral oxygen extraction) was calculated as (SaO2 − rSO2)/SaO2. As a surrogate indicator of cerebral blood flow [21], the middle cerebral artery mean flow velocity (MFV) was measured by transtemporal duplex/Doppler sonography (LOGICe; GE Healthcare, München, Germany) and calculated as 1/3(v systolic + 2v diastolic). Cerebrovascular resistance (CVR) was calculated as MAP/MFV.
Fractional tissue oxygen extraction and MFV were assessed 1 h before and 1 h after the sedation switch, because this relatively short time window allowed ventilatory settings and systemic parameters to be kept mostly unchanged, and were measured bilaterally. In cases where only a signal from one side was available, this was then used; in cases where the lesion was bilateral or diffuse, the mean of the bilateral values was used for further analysis. Systemic oxygenation values were also assessed 1 h before and after the sedation switch. ICP, MAP and CPP were measured hourly from 6 h before to at least 12 h after the sedation switch. In-house standard operating procedures for analgosedation, ventilator weaning, transfusion (target hemoglobin >8 g/dl in all patients), normothermia, ventilation, MAP/CPP and ICP management were used. We predefined critical values as follows: SaO2 <90 %, ScvO2 <70 %, rSO2 <50 % or 20 % decrease from baseline, ICP >25 mmHg, MAP <80 mmHg and CPP <60 mmHg.
Statistical analysis
The recorded parameters were analyzed descriptively. According to the scale level of the variables, means and standard deviations or absolute and relative frequencies are given. Measurements before and after the switch were compared using the Student’s t test for paired data. p values were considered significant if >0.05 but descriptive and no adjustment for multiple comparisons was made. Explorative analysis of subgroup differences was done by repeated measures analysis of variance with time as the within-subject factor and diagnosis (or sedation) as the between-subject factor. All analyses were done using Windows Excel for data handling and IBM SPSS Statistics 19 for further statistics.
Results
Patients
During the study period, 742 patients were admitted to our NICU, 146 of whom were equipped with multimodal neuromonitoring, and 132 of whom were ventilated for more than 24 h. Of these patients, 72 were recruited for other interventional trials. Of the remaining 60 patients, 41 either were not cerebrovascular or met other exclusion criteria (see above), leaving 19 patients for recruitment. Therefore, 19 consecutive patients with the admission diagnoses of severe IS (3), SAH (4) or ICH (12) were included, many of whom deteriorated early after admission. The demographic, clinical and sedation details of the included patients are summarized in Table 1. Sedation was induced with propofol in 12 patients and with midazolam (because of propofol-induced hypotension/bradycardia) in 7; initially, 1 patient was directly started on isoflurane. Patients were switched to isoflurane for a mean of 3.5 days (individual durations are shown in Table 1). Sufficient depth of sedation (RASS score before and after the switch −4 to −5) was achieved in most patients by isoflurane alone at concentrations ranging from MAC 0.5 to 0.8 (Table 3). In four patients, however, each previous intravenous sedation could not be completely stopped within 1 h, but had to be continued at a reduced rate for an overlapping period of another 1–2 h.
Adverse effects
No relevant overt safety concerns for patients or staff and no complications (such as malignant hyperthermia) emerged during or after the use of isoflurane. No liver enzyme increases or renal impairment related solely to isoflurane were detected. An autopsy in one patient did not show neuropathological abnormalities other than those associated with the admission diagnosis. Two patients developed anisocoria without changes in ICP or on a control CT scan that disappeared after cessation of isoflurane. In two patients, volatile sedation was stopped earlier than planned because of suboptimal gas resorption due to severe bronchial secretions or because of emergence of relevant intrapulmonary shunts.
Short-term observations (1 h before to 1 h after starting isoflurane)
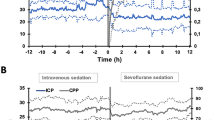
Mean ICP showed an absolute increase of 2.1 mmHg (p = 0.1, Table 2). Mean MAP and CPP both decreased after initiation of isoflurane by 6.5 mmHg (p = 0.076) and 6.3 mmHg (p = 0.119), respectively (Fig. 1; Table 2) despite counteracting measures (see below).

Individual changes in ICP, CPP, MFV and FTOE 1 h before and 1 h after the sedation switch to isoflurane for each non-affected and affected hemisphere (for FTOE and MFV). A full dataset was obtained in 16 patients, data were missing for ICP (and thus CPP) in 1 patient, MFV in 2 patients, and FTOE in 2 patients (partially overlapping) for various technical reasons
There were no relevant changes in mean MFV, PaCO2 (a determinant of cerebral vasoreactivity), or CVR. Oxygen saturation did not change; however, increasing ScvO2 and rSO2 led to reductions in systemic (SaO2 − ScvO2) and cerebral (SaO2 − rSO2) arteriovenous differences. Due to the latter, mean FTOE also decreased from 0.24 before to 0.21 after the switch (95 %CI −0.07 to 0.00; p = 0.031; Fig. 1; Table 2).
Long-term observations (6 h before to 12 h after starting isoflurane)
Changes in the mean values from the pre-start phase (−6 to −1 h) to early (+1 to +6 h) and late (+6 h to +12 h) phases after starting are given in Table 3. ICP remained stable, without necessary additional osmotherapy or augmentation of CSF drainage by EVD. Although MAP was counterbalanced by vasopressors, mean CPP decreased by 9.9 mmHg in the early phase and by 7.5 mmHg in the late phase (Fig. 2). To detect potential differences in pressure dynamics, the three disease entities were also analyzed separately. This did not reveal great deviations from the behavior of the whole group, particularly regarding patients with ICH and SAH. The three patients with IS, however, showed a more pronounced decrease in MAP and thus CPP, and a decrease, as opposed to a slight increase (ICH/SAH), in ICP at 6 and 12 h (see Electronic supplementary material). ICP dynamics were similar in those with previous propofol sedation and those with midazolam sedation, while MAP/CPP decreases appeared more pronounced in the latter (see Table S2a Electronic supplementary material).

Dynamics of selected mean values from 6 h before to 12 h after the switch of sedation to isoflurane (hourly means and standard deviations). ICP intracranial pressure, MAP mean arterial pressure, CPP cerebral perfusion pressure, NA infusion rate of noradrenaline, PaO 2 partial pressure of arterial oxygen, FiO 2 fraction of inspired oxygen, MV minute volume
Peak and mean inspiratory pressures, and thus minute volume, were increased over time to achieve constant oxygenation (PaO2/FiO2). In spite of this, PaCO2 showed an increase and thus pH showed a decrease over the long term, both significant (Table 3; Fig. 2). In terms of drug administration, remifentanil and sufentanil could be reduced. However, noradrenaline concentrations constantly increased with increasing MAC of isoflurane (Fig. 2), and had to be significantly increased about 1.5-fold in both phases after the sedation switch to keep MAP and CPP above tolerable thresholds.
Discussion
Isoflurane at effective sedation doses did not compromise cerebral oxygenation (short-term), blood flow (short-term) or ICP (short-term and long-term) to clinically relevant extents. However, it led to considerable depression of CPP that required substantial counteracting with vasopressors.
In addition to acknowledged general advantages of isoflurane as an anesthetic, it has shown neuroprotective effects when used prior to (preconditioning) [22, 23] or during [24, 25] ischemic insults in a considerable number of experimental studies [7]. Supportive clinical reports are scarce and limited to short-term use during surgery [26, 27]. Long-term administration in neurocritical care was hardly feasible until the advent of AnaConDa®, although there is an early report comparing isoflurane with midazolam for ICU sedation, without detection of adverse effects [28]. In two cases, prolonged use in the NICU via an OR respirator in refractory status epilepticus resulted in effective control of seizure activity but did not convincingly improve outcome [29] and possibly caused basal ganglia MRI abnormalities [9]. Recent experimental studies have even suggested neurotoxic effects including apoptosis induction [8] and amyloid deposition [30]. The greatest concerns are based on OR studies that have shown increases in ICP [10, 11, 15, 16] through isoflurane-associated cerebral vasodilation [12, 13]. Although other studies have not confirmed these findings [31, 32], sedation with isoflurane is currently off-label and discouraged in brain-injured patients who are prone to ICP elevations.
In our study, we did not detect relevant short-term changes in MFV after starting isoflurane. Thus, a direct vasodilatory effect was neither directly observed nor indirectly suggested by a compensatory decrease in CVR (which was absent in the short term). Possibly, the dose-dependent [16, 33] effect might not have been relevant at our sedation doses, which were considerably lower than in the OR setting. Alternatively, vasodilation might have occurred later in the course and was thus not picked up during the momentary measurement 1 h after the sedation switch when residual effects of the potential rCBF depressors propofol and midazolam might have been present. Finally, it might be more revealing to assess peripheral vasodilation in the microcirculation, rather than at the level of the MCA main stem. Recently in 13 patients with SAH equipped with a thermal diffusion probe and transtemporal duplex/Doppler sonography who were switched from propofol to isoflurane, no change in MCA transcranial Doppler velocity was found, but a significant increase of rCBF was found in the microcirculation [34].
The absence of proximal vasodilation might explain why we observed only slight increases in individual ICP values under isoflurane, but not to a clinically relevant extent. Of note, ICP was not externally influenced by measures such as osmotherapy, and thus probably reflects spontaneous fluctuations. It cannot be ruled out, however, that our active measures to control MAP in some cases may have influenced ICP, as is observed in impaired cerebral autoregulation.
MAP was reduced by isoflurane regardless of the underlying disease entity or previous sedation type, with minor differences in the extent. That this decrease appeared more pronounced in patients treated with midazolam than in those treated with propofol probably relates to previous MAP depression by propofol, hence resulting in a smaller difference to the subsequent isoflurane-induced depression. CPP was therefore considerably affected and had to be actively raised by additional noradrenaline. This was not associated with overt side effects here, but potentially exposes the patient to tachyarrhythmia, gastrointestinal disturbance or impairment of the microcirculation. Opioids could be reduced under isoflurane, reflecting its partial analgesic component.
Isoflurane significantly decreased cerebral and systemic oxygen extraction, which might reflect isoflurane’s well-known ability to reduce the metabolic rate and thus cerebral and systemic oxygen consumption. A comparable cerebral metabolic rate/O2-reducing effect of isoflurane has been demonstrated in the (neuro)surgical OR setting [33, 35], but not in cerebrovascular ICU patients on the AnaConDa® system, so far. The observation that FTOE bihemispherically decreased in more than two-thirds of patients appears beneficial with regard to the risk of secondary ischemic damage and high oxygen demand in this patient population. However, the alternative that FTOE reduction might reflect a regional vascular steal effect cannot be ruled out in this study design, even though our results do not support a gross interhemispheric steal.
Liver enzymes and renal parameters were not adversely affected by isoflurane alone. We did not measure fluoride, a potentially nephrotoxic accumulative product of the metabolism of volatile sedatives, but isoflurane produces much less fluoride than sevoflurane, and the latter was recently investigated in a long-term study (7 days) of AnaConDa® ICU sedation and resulted in no renal impairment [5]. In terms of neurotoxicity, one single neuropathological work-up revealed no signs of basal ganglia injuries [9]. Intrapulmonary shunt in two patients was probably caused by blunting of hypoxic pulmonary vasoconstriction that is more common with isoflurane than with propofol [36]. The invasiveness of ventilation was slightly increased over the observation period, probably to compensate for the increase in dead space and CO2 recycling [37] associated with the AnaConDa® device, but did not completely prevent a small long-term rise in PaCO2 (and thus fall in pH), another point of concern in NICU patients with vascular disease. This underlines the importance of tightly controlling PaCO2 when applying volatile sedation with the AnaConDa®. Finally, the atypical occurrence of anisocoria in two patients, not attributable to ICP crises and vanishing after isoflurane cessation, led to substantial alarm and unnecessary transports for CT scanning. Suggested mechanisms with other volatile agents are local pupillomotor effects and dysregulation of sympathetic tone [38].
Our study had several limitations. Above all, it was not a randomized comparison of isoflurane with an alternative sedative to test a hypothesis. Thus, no confirmatory analysis was planned and the reported p values are descriptive only. The patient sample was small and the vascular pathologies quite heterogeneous in nature, location, distribution and extension, thus possibly resulting in considerable differences in cerebral hemodynamic behavior. Given the fact that the majority of patients had ICH, our findings might mainly be applicable to hemorrhagic stroke and not to cerebrovascular disease in general. The chosen time-frames of the observation periods were arbitrary and might not have revealed all conceivable effects of isoflurane. While the short-term observation period allowed fairly good control of most management measures (and was therefore chosen for the more vulnerable measurements by TCD and NIRS), the long-term window was certainly more prone to uncontrollable influences on physiology. Sedation level was assessed by the clinical score RASS only, and although this had been demonstrated to correlate well with bispectral index monitoring (BIS XP) in NICU patients [39], it would have been preferable to have employed more objective means of sedation monitoring in our study, too.
Excluding patients with high initial ICP values from the study was deemed ethically mandatory, but this might have created a caveat with regard to interpretation of our findings, further limiting generalizability and warranting caution. Residual effects of the previously administered sedatives and variations in the opioid dosing during the protocol cannot be ruled out. Finally, neuromonitoring has its own limitations: ICP values can differ considerably with different ways of measurement (e.g., EVD vs. tissue probe), MFV is a useful surrogate for rCBF in healthy subjects, but correlations are weaker in specific brain pathologies [40], and NIRS assesses a tiny frontal cortical area, while oxygenation effects close to the site of lesion might have been more relevant.
Still, this is to our knowledge the first prospective study of AnaConDa®-driven isoflurane sedation in stroke patients under invasive and non-invasive neuromonitoring, generating a considerable amount of novel data. And despite all the warranted caution suggested by our findings, we continue to consider isoflurane a potentially useful alternative for NICU sedation, provided neuromonitoring is in place. Selected patients with good systemic circulatory stability but compromised cerebral microcirculation (e.g., vasospasms in SAH, ischemic penumbra in large vessel occlusion IS) might benefit from volatile sedation. Transient application of isoflurane might provide the reported protective “preconditioning” effect to ameliorate secondary ischemia without risking neurotoxicity potentially associated with longer-term application.
Conclusions
In summary, our findings in this small and selected study population suggest that cerebrovascular NICU patients can be sufficiently sedated with isoflurane administered via an AnaConDa® device, in association with reductions in cerebral oxygen extraction and without relevant increases in ICP, if baseline ICP values are low or only moderately elevated. However, the observation of substantial MAP/CPP reductions and other adverse effects are concerning and warrant caution in this off-label treatment. We strongly recommend neuromonitoring in these patients. More confirmatory research in larger populations is clearly necessary before potential cerebral benefits of volatile sedation can be safely investigated.
References
Sessler CN, Varney K (2008) Patient-focused sedation and analgesia in the ICU. Chest 133:552–565
Martin J, Heymann A, Basell K, Baron R, Biniek R, Burkle H, Dall P, Dictus C, Eggers V, Eichler I, Engelmann L, Garten L, Hartl W, Haase U, Huth R, Kessler P, Kleinschmidt S, Koppert W, Kretz FJ, Laubenthal H, Marggraf G, Meiser A, Neugebauer E, Neuhaus U, Putensen C, Quintel M, Reske A, Roth B, Scholz J, Schroder S, Schreiter D, Schuttler J, Schwarzmann G, Stingele R, Tonner P, Trankle P, Treede RD, Trupkovic T, Tryba M, Wappler F, Waydhas C, Spies C (2010) Evidence and consensus-based German guidelines for the management of analgesia, sedation and delirium in intensive care – short version. Ger Med Sci 8:Doc02
Gommers D, Bakker J (2008) Medications for analgesia and sedation in the intensive care unit: an overview. Crit Care 12(Suppl 3):S4
Soukup J, Scharff K, Kubosch K, Pohl C, Bomplitz M, Kompardt J (2009) State of the art: sedation concepts with volatile anesthetics in critically Ill patients. J Crit Care 24:535–544
Rohm KD, Mengistu A, Boldt J, Mayer J, Beck G, Piper SN (2009) Renal integrity in sevoflurane sedation in the intensive care unit with the anesthetic-conserving device: a comparison with intravenous propofol sedation. Anesth Analg 108:1848–1854
L’Her E, Dy L, Pili R, Prat G, Tonnelier JM, Lefevre M, Renault A, Boles JM (2008) Feasibility and potential cost/benefit of routine isoflurane sedation using an anesthetic-conserving device: a prospective observational study. Respir Care 53:1295–1303
Kitano H, Kirsch JR, Hurn PD, Murphy SJ (2007) Inhalational anesthetics as neuroprotectants or chemical preconditioning agents in ischemic brain. J Cereb Blood Flow Metab 27:1108–1128
Wei H, Liang G, Yang H, Wang Q, Hawkins B, Madesh M, Wang S, Eckenhoff RG (2008) The common inhalational anesthetic isoflurane induces apoptosis via activation of inositol 1,4,5-trisphosphate receptors. Anesthesiology 108:251–260
Fugate JE, Burns JD, Wijdicks EF, Warner DO, Jankowski CJ, Rabinstein AA (2010) Prolonged high-dose isoflurane for refractory status epilepticus: is it safe? Anesth Analg 111:1520–1524
Gordon E, Lagerkranser M, Rudehill A, von Holst H (1988) The effect of isoflurane on cerebrospinal fluid pressure in patients undergoing neurosurgery. Acta Anaesthesiol Scand 32:108–112
Petersen KD, Landsfeldt U, Cold GE, Petersen CB, Mau S, Hauerberg J, Holst P, Olsen KS (2003) Intracranial pressure and cerebral hemodynamic in patients with cerebral tumors: a randomized prospective study of patients subjected to craniotomy in propofol-fentanyl, isoflurane-fentanyl, or sevoflurane-fentanyl anesthesia. Anesthesiology 98:329–336
Holmstrom A, Akeson J (2005) Sevoflurane induces less cerebral vasodilation than isoflurane at the same A-line autoregressive index level. Acta Anaesthesiol Scand 49:16–22
Matta BF, Heath KJ, Tipping K, Summors AC (1999) Direct cerebral vasodilatory effects of sevoflurane and isoflurane. Anesthesiology 91:677–680
Kahveci FS, Kahveci N, Alkan T, Goren B, Korfali E, Ozluk K (2001) Propofol versus isoflurane anesthesia under hypothermic conditions: effects on intracranial pressure and local cerebral blood flow after diffuse traumatic brain injury in the rat. Surg Neurol 56:206–214
Adams RW, Cucchiara RF, Gronert GA, Messick JM, Michenfelder JD (1981) Isoflurane and cerebrospinal fluid pressure in neurosurgical patients. Anesthesiology 54:97–99
Lundar T, Lindegaard KF, Refsum L, Rian R, Nornes H (1987) Cerebrovascular effects of isoflurane in man. Intracranial pressure and middle cerebral artery flow velocity. Br J Anaesth 59:1208–1213
Sessler CN, Gosnell MS, Grap MJ, Brophy GM, O’Neal PV, Keane KA, Tesoro EP, Elswick RK (2002) The Richmond Agitation-Sedation Scale: validity and reliability in adult intensive care unit patients. Am J Respir Crit Care Med 166:1338–1344
Nemoto EM (1999) Cerebral oximetry by near infrared spectroscopy measures cerebral haemoglobin oxygen saturation and the balance between supply and demand, not cerebral blood flow. Br J Neurosurg 13:93–95
Gagnon RE, Macnab AJ, Gagnon FA, Blackstock D, LeBlanc JG (2002) Comparison of two spatially resolved NIRS oxygenation indices. J Clin Monit Comput 17:385–391
Lemmers PM, Toet M, van Schelven LJ, van Bel F (2006) Cerebral oxygenation and cerebral oxygen extraction in the preterm infant: the impact of respiratory distress syndrome. Exp Brain Res 173:458–467
Sorond FA, Hollenberg NK, Panych LP, Fisher ND (2010) Brain blood flow and velocity: correlations between magnetic resonance imaging and transcranial Doppler sonography. J Ultrasound Med 29:1017–1022
Gray JJ, Bickler PE, Fahlman CS, Zhan X, Schuyler JA (2005) Isoflurane neuroprotection in hypoxic hippocampal slice cultures involves increases in intracellular Ca2+ and mitogen-activated protein kinases. Anesthesiology 102:606–615
Kapinya KJ, Prass K, Dirnagl U (2002) Isoflurane induced prolonged protection against cerebral ischemia in mice: a redox sensitive mechanism? Neuroreport 13:1431–1435
Engelhard K, Werner C, Reeker W, Lu H, Mollenberg O, Mielke L, Kochs E (1999) Desflurane and isoflurane improve neurological outcome after incomplete cerebral ischaemia in rats. Br J Anaesth 83:415–421
Sakai H, Sheng H, Yates RB, Ishida K, Pearlstein RD, Warner DS (2007) Isoflurane provides long-term protection against focal cerebral ischemia in the rat. Anesthesiology 106:92–99 (discussion 98–10)
Messick JM Jr, Casement B, Sharbrough FW, Milde LN, Michenfelder JD, Sundt TM Jr (1987) Correlation of regional cerebral blood flow (rCBF) with EEG changes during isoflurane anesthesia for carotid endarterectomy: critical rCBF. Anesthesiology 66:344–349
Kanbak M, Saricaoglu F, Avci A, Ocal T, Koray Z, Aypar U (2004) Propofol offers no advantage over isoflurane anesthesia for cerebral protection during cardiopulmonary bypass: a preliminary study of S-100beta protein levels. Can J Anaesth 51:712–717
Spencer EM, Willatts SM (1992) Isoflurane for prolonged sedation in the intensive care unit; efficacy and safety. Intensive Care Med 18:415–421
Kofke WA, Young RS, Davis P, Woelfel SK, Gray L, Johnson D, Gelb A, Meeke R, Warner DS, Pearson KS et al (1989) Isoflurane for refractory status epilepticus: a clinical series. Anesthesiology 71:653–659
Eckenhoff RG, Johansson JS, Wei H, Carnini A, Kang B, Wei W, Pidikiti R, Keller JM, Eckenhoff MF (2004) Inhaled anesthetic enhancement of amyloid-beta oligomerization and cytotoxicity. Anesthesiology 101:703–709
Fraga M, Rama-Maceiras P, Rodino S, Aymerich H, Pose P, Belda J (2003) The effects of isoflurane and desflurane on intracranial pressure, cerebral perfusion pressure, and cerebral arteriovenous oxygen content difference in normocapnic patients with supratentorial brain tumors. Anesthesiology 98:1085–1090
Artru AA, Lam AM, Johnson JO, Sperry RJ (1997) Intracranial pressure, middle cerebral artery flow velocity, and plasma inorganic fluoride concentrations in neurosurgical patients receiving sevoflurane or isoflurane. Anesth Analg 85:587–592
Kuroda Y, Murakami M, Tsuruta J, Murakawa T, Shiroyama Y (2000) Effects of sevoflurane and isoflurane on the ratio of cerebral blood flow/metabolic rate for oxygen in neurosurgery. J Anesth 14:124–128
Villa F, Iacca C, Molinari AF, Giussani C, Aletti G, Pesenti A, Citerio G (2012) Inhalation versus endovenous sedation in subarachnoid hemorrhage patients: effects on regional cerebral blood flow. Crit Care Med 40:2797–2812
Oshima T, Karasawa F, Okazaki Y, Wada H, Satoh T (2003) Effects of sevoflurane on cerebral blood flow and cerebral metabolic rate of oxygen in human beings: a comparison with isoflurane. Eur J Anaesthesiol 20:543–547
Kellow NH, Scott AD, White SA, Feneck RO (1995) Comparison of the effects of propofol and isoflurane anaesthesia on right ventricular function and shunt fraction during thoracic surgery. Br J Anaesth 75:578–582
Sturesson LW, Malmkvist G, Bodelsson M, Niklason L, Jonson B (2012) Carbon dioxide rebreathing with the anaesthetic conserving device, AnaConDa(R). Br J Anaesth 109:279–283
Tayefeh F, Larson MD, Sessler DI, Eger EI 2nd, Bowland T (1997) Time-dependent changes in heart rate and pupil size during desflurane or sevoflurane anesthesia. Anesth Analg 85:1362–1366
Deogaonkar A, Gupta R, DeGeorgia M, Sabharwal V, Gopakumaran B, Schubert A, Provencio JJ (2004) Bispectral Index monitoring correlates with sedation scales in brain-injured patients. Crit Care Med 32:2403–2406
Brauer P, Kochs E, Werner C, Bloom M, Policare R, Pentheny S, Yonas H, Kofke WA, Schulte am Esch J (1998) Correlation of transcranial Doppler sonography mean flow velocity with cerebral blood flow in patients with intracranial pathology. J Neurosurg Anesthesiol 10:80–85
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is discussed in the editorial available at: doi:10.1007/s00134-012-2711-0.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Bösel, J., Purrucker, J.C., Nowak, F. et al. Volatile isoflurane sedation in cerebrovascular intensive care patients using AnaConDa®: effects on cerebral oxygenation, circulation, and pressure. Intensive Care Med 38, 1955–1964 (2012). https://doi.org/10.1007/s00134-012-2708-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00134-012-2708-8