
Abstract
Objective
Several studies demonstrated that the lungs could produce lactate in patients with acute lung injury (ALI). Because after cardiopulmonary bypass (CPB) some patients develop ALI, the effect of CPB on pulmonary lactate release was investigated.
Design
Prospective observational clinical study.
Setting
Twenty-beds, surgical ICU at a university hospital.
Patients
Sixteen deeply sedated, ventilated and post-cardiac surgery patients, all equipped with a pulmonary artery catheter.
Measurements and results
Lactate concentration was measured using a lactate analyser in simultaneously drawn arterial (A) and mixed venous (V) blood samples. Three measurements per patients were taken at 30-min interval, after body temperature reached 37°C. Concomitantly, measurements of cardiac output were also obtained. Pulmonary lactate release was calculated as the product of transpulmonary A-V lactate and cardiac index. The mean cardiopulmonary bypass duration was 100±44 min (SD), and the aortic cross-clamping time was 71±33 min. After CPB, lactate release was 0.136±0.210 mmol/min m−2. These values were not correlated with cardiopulmonary bypass duration.
Conclusion
The present study shows that in patients receiving mechanical ventilation after CPB, the lung is a source of lactate production. This pulmonary release was not dependent on cardiopulmonary bypass duration.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Pulmonary dysfunction related to acute lung injury (ALI) is one of the complications observed after heart surgery using cardiopulmonary bypass (CPB) [1]. Despite the uncertainty on the exact pathophysiological mechanism leading to post CBP ALI [2], the occurrence of a postoperative hypoxaemic condition has been attributed to a post CPB-systemic inflammatory response syndrome [3].
In patients with ALI and/or adult respiratory distress syndrome, several studies have demonstrated that lactate is released by the lung [4, 5, 6, 7]. Likewise, we have observed that the arterial lactate concentration in the systemic circulation was higher than in the pulmonary artery mixed venous blood in patients after CPB; thus, we hypothesised that during CPB the lung could be a source of lactate production when lung parenchyma has been hypoperfused during extracorporeal circulation.
Based on these observations and hypothesis, the present study was carried out to prospectively investigate, firstly, if a lactate release from the lungs occurs during the early postoperative course after hypothermic CPB, and secondly to determine whether a relationship exists between pulmonary lactate flux and CPB time and/or hypoxaemia [8].
Methods
Our institutional ethical committee approved the study, and all patients gave written consent the day before surgery. Patients scheduled for CABG surgery were prospectively screened. Exclusion criteria were: decreased pre-operative left ventricular ejection fraction (<45% assessed by echocardiography, isotopic or angiographic ventriculography), valve disease, other associated cardiac surgery and/or history of chronic respiratory disease. Post-operatively, patients presenting with haemodynamic instability and bleeding greater than 100 ml/h were also excluded.
Perioperative management
Anti-anginal therapy was discontinued the evening before surgery but per-oral metoprolol was given to patients receiving chronic β-adrenergic blocker therapy. One hour prior to surgery, diazepam and morphine pre-medication were administrated. A 20-G radial artery catheter and a flow-directed modified pulmonary artery catheter capable of continuous monitoring mixed venous oxygen saturation (SVO2; Vigilance Monitor, Baxter Edwards, Irvine, Calif.) were inserted for clinical purpose. Anaesthesia and muscle relaxation were induced and maintained with midazolam, fentanyl and pancuronium bromide. The patient’s trachea was intubated with an endotracheal tube. A nasogastric tube and a Foley urinary catheter were also inserted. Hypothermic (28°C) cardiopulmonary bypass and cold cardioplegic-induced cardiac arrest were used. The cardiopulmonary bypass flow was set at 2 l min−1 m−2 and mean arterial pressure was maintained at 60 mm Hg using vasoactive drugs, as necessary. During cardiopulmonary bypass the lungs were not ventilated but inflated with a continuous positive airway pressure of 2–5 cm H2O. Weaning from cardiopulmonary bypass was achieved with inotropic or vasopressor drug support when required. After surgery, patients were transferred to the surgical intensive care unit. Upon arrival, sedation and analgesia were provided by continuous infusion of midazolam and morphine. The patients were placed on mechanical ventilation (Evita4, Dräger, Lübeck, Germany). Initial ventilator settings in controlled mechanical ventilation (CMV) mode were: tidal volume (8–10 ml kg−1 of body weight) and respiratory rate (12 breath min−1). The targets for arterial pH and PaCO2 were at 7.40 and 5 kPa, respectively. The inspired oxygen fraction (FiO2) was adjusted to obtain an arterial oxygen saturation (SaO2) >92%. Inspiratory to expiratory ratio (I/E) was 1/2. No positive end-expiratory pressure (PEEP) was used. Body temperature, lead II ECG and urine output were monitored throughout the postoperative period. Patients were wrapped with warm blankets to restore normal temperature. The patients were observed for at least 2 h to confirm haemodynamic stability, which was defined as less than 10% variation in haemodynamic variables [heart rate, cardiac output (CO) and mixed venous oxygen saturation (SVO2) continuous monitoring, mean arterial pressure], no clinically relevant bleeding (<100 ml/h) and normal body temperature. A chest radiograph was obtained prior to data collection to define the correct position of the pulmonary artery and central venous catheters, the position of the surgical drains, and also to confirm the absence of cardiopulmonary abnormalities (grossly enlarged mediastinal silhouette, pleural effusion or pneumothorax). A 12-lead ECG was also recorded to verify a regular sinus rhythm. Body position changes, patient care and endotracheal suctioning, if required, were allowed during this period but not during the study protocol. All patients received a continuous intravenous infusion of NaCl 0.9% at rate of 65 ml/h.
Haemodynamic monitoring
All pressure transducers were referenced to mid-chest. The correct position of the catheter tip in West’s zone 3 was checked using the method described by Teboul et al. [9]. SvO2 was calibrated in vivo according to the manufacturer’s instruction by simultaneous measurements of mixed-venous blood on a co-oxymeter (Statprofile Ultra, Nova biomedical, Waltham, Mass.). Mean cardiac output (CO) was estimated by averaging triplicate injections of 10 ml room temperature 0.9% NaCl, delivered randomly during the respiratory cycle, and used to calculate the cardiac index (CI).
Study protocol
During the study protocol, all patients were receiving IV sedation and were not breathing spontaneously. Patients were kept in the supine position. If patients were receiving vasoactive drugs, the administration rate was not changed during the protocol. Three measurements of the following variables were recorded at 30-min intervals in each patient, from the bedside monitor (Agilent technologies M3150A) and Vigilance Monitor (Baxter Edwards, Irvine, Calif.): body temperature; continuous SvO2; continuous CO; mean CO; heart rate; calculated stroke volume; systolic, diastolic and mean arterial and pulmonary arterial pressures; right atrial pressure; and pulmonary artery occlusion pressure. Ventilator settings (respiratory rate, tidal volume, I/E, FiO2, PEEP), peak and mean airway pressures were recorded. The acute lung injury score proposed by Murray et al. was calculated [8].
Lactate measurements
Two blood samples were simultaneously drawn: one from the radial artery catheter and one from the distal port of the pulmonary artery catheter. After discarding the first 3 ml of blood from each sample, arterial and mixed venous blood gases, as well as arterial and mixed venous lactate, were analysed in 2.5-ml samples at 37°C (Statprofile Ultra, Nova biomedical, Waltham, Mass.). Pulmonary lactate release (LR) was calculated using the following formula: (arterial lactate-venous lactate) × CI [4]. Three measurements were done over 90 min in each patient, many hours after the end of the cardiopulmonary bypass, after rectal temperature reached 37°C.
Statistical analysis
The study was designed to detect a mean arterial-venous difference under the alternative hypothesis of 0.05, with a standard deviation of 0.07 based on data from by Routsi et al. [7], and assuming zero difference in arterial and venous lactate under the null hypothesis. Accordingly, the sample size was 16 patients to detect this difference, using a two-sided alpha level of 0.05 and a β-power of 0.8.
All values are expressed as mean±standard deviation (SD). For variables that were measured repeatedly, the mean was calculated using the average over all the measures taken in the same subject, so that each individual only contributed one observation. A two-tailed paired t test was used to compare mixed venous and arterial lactate measurements, cross-sectionally at each time (between subject difference), while ANOVA was used to compare arterial and mixed venous measures longitudinally (within-subject difference). To account for dependence in the data due to repeated measures, regression coefficients for the relationship between pulmonary lactate release and CPB time or the ratio of PaO2 to inspired oxygen fraction (PaO2/FiO2) were obtained using generalised estimating equations (GEE), with robust estimates of the variance [10, 11]. Statistical analyses were run using STATA (version 7.0; Stata Corporation, Texas). A value of p<0.05 was considered statistically significant.
Results
Sixteen patients were included in the study. Demographic and preoperative characteristics are presented in Table 1. The mean cardiopulmonary bypass duration was 100±44 min and the aortic cross-clamp time was 71±33 min. Six patients required catecholamines infusion (37%) with no change in dose during the protocol. Mean ratio of PaO2/FiO2 was 48.8±7 kPa. Mean acute lung injury score (ALI score) was 1.9±0.2. Mean cardiac index was 2.8±0.2 l min–1 m–2.
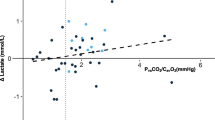
Mean arterial pH was significantly higher than mean mixed venous pH, 7.38±0.03 vs 7.35±0.03 (p<0.01), respectively. Mean arterial lactate concentration was higher than mean mixed venous lactate concentration, 0.83±0.2 vs 0.78±0.17 mmol/l (p=0.028; Fig. 1). Mean LR was 0.136±0.2 mmol min−1 m−2 after CPB (Table 2). There was a suggestion that a higher pulmonary lactate release was associated with a lower PaO2/FiO2 ratio [β-coefficient=−50.53 (95% CI: −107.78, 6.71); p=0.084; Fig. 2]. There was no relationship between cardiopulmonary bypass duration and pulmonary lactate release (p=NS). Moreover, pulmonary lactate release was not correlated to mixed venous oxygen saturation (p=NS)

Box-and-whiskers plot displaying arterial (A) and venous (B) lactate (median, interquartile range) measured every 30 min (T1, T2, T3) after rewarming, in 16 patients after hypothermic cardiopulmonary bypass surgery. A trend in arterial, but not venous, lactate concentration increase was observed over time

Plot displaying the mean PaO2/FiO2 ratio vs lung lactate release (LR), in 16 patients after hypothermic cardiopulmonary bypass surgery. From the regression model, the β-coefficient was −50.53 (95% CI: −107.78, 6.71; p=0.084)
Discussion
The main finding of this study is that following CPB the lungs release lactate into the systemic circulation. Moreover, lung lactate production is not correlated with CPB time and mixed venous saturation, but it tends to be associated with the degree of pulmonary dysfunction, as indicated by the degree of hypoxaemia.
Routsi et al. [7] found that mixed-venous lactate concentration was higher than arterial lactate concentration in patients without ALI and did not undergo CPB; thus, in the absence of ALI, the lung does not produce lactate. To explore the difference between patients without respiratory failure or ALI and those post CPB, we compared our data with those by Routsi et al. [7] (external control; assuming an underlying t distribution with unequal variances [10, 11]). The mean pulmonary lactate release obtained in the present study was 0.136±0.2 mmol min−1 m−2, whereas it was −0.038±0.065 mmol min−1 m−2 in the study by Routsi et al. [7], providing a mean difference of 0.17 (95% CI: 0.061, 0.287; p<0.01). This finding suggests that after CPB even a mild degree of lung dysfunction (hypoxaemia) is associated with pulmonary lactate release.
In the present study, we speculated that the lactate production by the lung was related to the hypoperfused or hypoxic lung tissue producing lactic acidosis; however, the absence of acidosis and the higher systemic arterial pH strongly challenges this hypothesis. Moreover, lung lactate production (which tended to increase over time; Fig. 1) was not correlated with CPB time but inversely correlated with PaO2/FiO2 suggesting a different mechanism of lung lactate production; the latter could be secondary to a systemic inflammatory response induced by CBP [12] and/or surgical trauma (e.g. opening chest) [13] without detectable lung tissue hypoxia. Indeed, cardiac surgery without CPB may also disturbs the respiratory mechanics, as emphasised in a recent work by Roosens et al. [13].
In contrast with the results of the present study, Loer and colleagues [14] showed no evidence of lung ischaemia and/or pulmonary lactate release in patients during hypothermic CPB. The difference between the present study and Loer et al. [14] study could be attributed to a different design. Those authors measure lactate release during the hypothermic stage of CPB. The lung protective condition with hypothermia, in this study, might have disappeared after rewarming. Moreover, there is no reference to experimental models of ALI following aortic cross-clamping and during CPB pulmonary artery blood flow is completed inhibited so that the lungs are perfused solely via bronchial artery; thus, we may expect that lung ischaemia may have occurred and that optimal oxygenation under mechanical ventilation after the end of CPB may have participated in a post-reperfusion lung injury [1]. In the absence of pyruvate measurements (ratio lactate/pyruvate), it is difficult to ascertain the pathogenesis of this lactate production by the lung. Indeed, the study design did not allow us to determine the mechanisms of transpulmonary lactate gradient or the mechanisms underlying the link between pulmonary lactate production and the degree of hypoxaemia.
Some limitations of this work should be acknowledged. Firstly, in view of the small difference between arterial and venous blood lactate concentrations, its clinical relevance could be questioned. Nevertheless, several studies showed a small error in lactate measurement in critically ill patients and validated the accuracy of lactate flux calculation [5, 7, 15].
Secondly, arterial lactate blood samples were drawn from the radial catheter and not from pulmonary vein or the left atrium. Indeed, peripheral arterial lactate concentration could be different from arterial lactate drawn proximal to the lung (pulmonary venous); however, several studies investigating lung lactate production have used a similar methodology and found the same values at both locations [5, 6, 7, 15]. Finally, our sample size was small and ALI occurred in few patients, thus hampering the possibility of explaining lung lactate production via the development of clinically relevant lung damage.
Conclusion
In conclusion, the present study is the first to demonstrate the occurrence of lung lactate production after CPB, as indicated by a positive arterio-venous lactate difference, in patient without history of pulmonary disease; however, further studies are needed to investigate the nature of the relationship between lung lactate release and lung injury or lung tissue hypoxia in patients undergoing surgery involving CPB.
References
Rady MY, Ryan T, Starr NJ (1997) Early onset of acute pulmonary dysfunction after cardiovascular surgery: risk factors and clinical outcome. Crit Care Med 25:1831–1839
Kirklin JK, Westaby S, Blackstone EH, Kirklin JW, Chenoweth DE, Pacifico AD (1983) Complement and the damaging effects of cardiopulmonary bypass. J Thorac Cardiovasc Surg 86:845–857
Wall MH, Royster RL (2000) Pulmonary dysfunction after cardiopulmonary bypass: Should we ventilate the lungs on pump? Crit Care Med 28:1658–1660
Brown SD, Clark C, Gutierrez G (1996) Pulmonary lactate release in patients with sepsis and the adult respiratory distress syndrome. J Crit Care 11:2–8
De Backer D, Creteur J, Zhang H, Norrenberg M, Vincent JL (1997) Lactate production by the lungs in acute lung injury. Am J Respir Crit Care Med 156:1099–1104
Kellum JA, Kramer DJ, Lee K, Mankad S, Bellomo R, Pinsky MR (1997) Release of lactate by the lung in acute lung injury. Chest 111:1301–1305
Routsi C, Bardouniotou H, Delivoria-Ioannidou V, Kazi D, Roussos C, Zakynthinos S (1999) Pulmonary lactate release in patients with acute lung injury is not attributable to lung tissue hypoxia. Crit Care Med 27:2469–2473
Murray JF, Matthay MA, Luce JM, Flick MR (1988) An expanded definition of the adult respiratory distress syndrome. Am Rev Respir Dis 138:720–723
Teboul JL, Besbes M, Andrivet P, Axler O, Douguet D, Zelter M, Lemaire F, Brun-Buisson C (1992) A bedside index assessing the reliability of pulmonary occlusion pressure measurements during mechanical ventilation with positive end-expiratory pressure. J Crit Care 7:22–29
Burton P, Gurrin L, Sly P (1998) Extending the simple linear regression model to account for correlated responses: an introduction to generalized estimating equations and multi-level mixed modelling. Stat Med 17:1261–1291
Zeger SL, Liang KY (1986) Longitudinal data analysis for discrete and continuous outcomes. Biometrics 42:121–130
Liebold A, Keyl C, Birnbaum DE (1999) The heart produces but the lungs consume proinflammatory cytokines following cardiopulmonary bypass. Eur J Cardiothorac Surg 15:340–345
Roosens C, Heerman J, De Somer F, Caes F, Van Belleghem Y, Poelaert JY (2002) Effects of off-pump coronary surgery on the mechanics of the respiratory system, lung and chest wall: comparison with extracorporeal circulation. Crit Care Med 30:2430–2433
Loer SA, Kalweit G, Tarnow J (2000) Effects of ventilation and nonventilation on pulmonary venous blood gases and markers of lung hypoxia in humans undergoing total cardiopulmonary bypass. Crit Care Med 28:1336–1340
Walsh TS, McLellan S, Mackenzie SJ, Lee A (1999) Hyperlactatemia and pulmonary lactate production in patients with fulminant hepatic failure. Chest 116:471–476
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Bendjelid, K., Treggiari, M.M. & Romand, JA. Transpulmonary lactate gradient after hypothermic cardiopulmonary bypass. Intensive Care Med 30, 817–821 (2004). https://doi.org/10.1007/s00134-004-2179-7
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00134-004-2179-7