
Abstract
An analysis of wastewater from Krakow (Poland) for the presence of controlled and uncontrolled stimulant drugs of abuse was performed. Samples were collected from the Plaszow wastewater treatment plant, Krakow, Poland, and prepared by solid phase extraction. The LC-QTOFMS method was applied for identification and quantification of popular stimulants: MDMA, mephedrone, 4-MEC, MDPV and mCPP. Environmental loads of illicit drugs were calculated; the WWTP discharged loads ranging from 3.6 to 6.7 mg day−1 1000 inhabitants−1 of MDMA, 3.6 to 7.1 mg day−1 1000 inhabitants−1 of mephedrone and 4.8 to 5.8 mg day−1 1000 inhabitants−1 of 4-MEC. The results confirmed the growing popularity of new psychoactive substances in Poland.
Similar content being viewed by others
In recent years, a large number of new psychoactive substances (NPS) have been marketed (Adamowicz et al. 2013; Zuba et al. 2013; Reid et al. 2014; Castiglioni et al. 2015; Report 2015). The first ‘head shops’ offering NPS were opened in Poland in 2008 and in 2010, around 1500 stores selling NPS without any control were in operation (Report 2011). Up to May 2009, the most popular class of substances sold as ‘legal highs’ were piperazines, including benzylpiperazine (BZP), 1-[3-trifluoromethyl)phenyl]piperazine (TFMPP), 1-(4-fluorophenyl)piperazine (pFPP), and 1-(3-chlorophenyl)piperazine (mCPP) (Byrska et al. 2010). BZP and mCPP seemed to be the most popular and easy accessible substances for users, therefore the European Monitoring Centre for Drugs and Drug Addiction (EMCDDA) carried out a risk assessment on these substances and noted drug addiction, many intoxications, including lethal poisonings. Based on their reports, BZP was banned in almost every European country (including Poland), whereas mCPP has been actively monitored (Annual Report 2012). After the ban for BZP, the market moved to a new direction; the first derivatives of cathinone were marketed (Zuba and Byrska 2013). Mephedrone (4-methylmethcathinone, MPD) became the substance of preference by users and its popularity has been growing month by month (Zuba 2014). The popularity of mephedrone was reflected in the increase in the number of drug addicts. Due to the growing popularity of NPS, monitoring of drugs of abuse in wastewater had to be expanded in order to cover a broader range of substances. A number of papers on quantification of opioids, cannabis derivatives, codeine, methadone, BZP and mCPP in wastewater were published (Zuccato et al. 2008; Baker et al. 2012; Thomas et al. 2012; Andres-Costa et al. 2014; Bijlsma et al. 2014). It was shown that due to the poor degree of purification in treatment plants, illicit drugs are still present in effluents being discharged to surface water (Kasprzyk-Hordern et al. 2009; van Nuijs et al. 2009; Zuccato and Castiglioni 2009; Mendoza et al. 2014). Due to their properties, they can be toxic to aquatic organisms (Pomati et al. 2007; Rosi-Marshall et al. 2015). Psychoactive substances have been also identified in drinking water, even after the treatment process (Castiglioni et al. 2011; Mendoza et al. 2014). Therefore, monitoring of their presence in different kinds of water is an important issue.
The aims of this pilot study were to investigate the profile of stimulant drugs taken by users in Krakow, and to estimate the environmental loads and consumption. The study covered ‘traditional’ drug of abuse, MDMA, and common novel psychoactive substances, that is mCPP, mephedrone, 4-MEC and MDPV. This is the first study based on the prevalence of stimulant drugs in the Krakow area.
Materials and Methods
Standard solutions of mephedrone and MDPV were purchased from the Australian Government National Measurement Institute (North Ryde, Australia), MDMA from Cerilliant (Round Rock, TX, USA), mCPP from Lipomed AG (Arlesheim, Switzerland), while 4-MEC from LGC Standards Sp. z o.o. (Dziekanów Leśny, Poland). The molecular formulas, physical and chemical properties of the compounds are summarized in Table 1. The isotope labelled standard MDMA-D5 (1.0 mg mL−1 in methanol) was purchased from Cerilliant (Round Rock, TX, USA). HPLC supergradient grade methanol and ammonia (25 %) were obtained from POCH (Gliwice, Poland). Hydrochloric acid (32 %) and formic acid (89–91 %) were purchased from Merck (Darmstadt, Germany). HPLC-grade acetonitrile was purchased from J.T. Baker (Phillipsburg, NJ, USA). Deionized water was obtained by reverse diffusion in a Millipore system (Warsaw, Poland).
Effluent samples were collected from the Plaszow WWTP, Krakow, Poland. It treats approximately 165,000 m3 of urban wastewater per day, which is over 70 % of the total volume of the city’s wastewater. Effluent water was collected after the secondary treatment, which involves primary settling, biological treatment and secondary sedimentation.
Effluent samples were collected in May 2012. Four wastewater samples (5 L each) were collected once a week, on Sunday. Equal aliquots of wastewater were taken every hour over a 24 h period, collected in pre-cleaned polyethylene containers with UV protection and stored at 4°C until the collection process was finished. Then, samples were transported to the laboratory and processed within 12 h. In the first step, before the solid phase extraction, wastewater was filtered using MN GF-4 and MN GF-1 glass fibre filters from Macherey–Nagel (Düren, Germany). Afterwards, samples were acidified to pH 4.5 with 2 M hydrochloric acid.
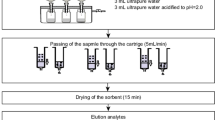
Oasis HLB 3 cc (60 mg/3 mL) extraction cartridges from Waters (Milford, MA, USA) were used in the analytical procedure. SPE was performed using a 12-port vacuum extraction manifold (J.T. Baker, Philipsburg, USA). The extraction cartridges were conditioned by 2 mL MeOH/NH4OH (v/v, 95:5) and 2 mL deionised water adjusted with hydrochloric acid solution to pH 4.5. 500 mL of samples adjusted to pH 4.5 were spiked with 4 µL (200 ng) internal standard solution of concentration 50 µg mL−1 before being passed through SPE cartridges. A flow of 5 mL min−1 water through the cartridge was achieved by applying a mild vacuum to the extraction manifold. The respective cartridges were then washed by 10 mL deionised water (pH 4.5) and completely dried under vacuum. The analytes were eluted twice from the cartridges with 2 mL of MeOH/NH4OH (v/v, 95:5). Extracts were pooled to 5 mL vials and stored in a freezer in temperature −24°C for 24/48 h until analysis. Directly prior to analysis 100 µL (0.025 M) hydrochloric acid were added to extracts. Subsequently, extracts were evaporated to 100 µL by nitrogen stream in a Pierce Reacti-Vap III evaporator and were finally reconstituted with mobile phase to a volume of 1 mL. The supernatants were successively transferred into 2 mL auto sampler vials for analysis by means of LC–ESI-QTOF-MS. In the experiments, all calibration solutions as well as extracts were dissolved in starting mobile phase (0.1 % formic acid in 95 % water/5 % acetonitrile).
LC/MS analyses were carried out using an Agilent Technologies 1200 Series liquid chromatography instrument coupled with a 6520 Accurate-Mass Q-TOF LC/MS detector equipped with electrospray ionization (ESI) manufactured by the same company. The mobile phase was composed of a mixture of 0.1 % (v/v) formic acid in water (A) and in acetonitrile (B). The separation was performed at 35°C using an Ascentic Express C18 column (7.5 cm × 2.1 mm × 2.7 µm) from Supelco. The flow rate was 0.3 mL min−1, and the volume of injection was 10 µL. All analyses were carried out in a gradient mode (shown in relation to content of phase B): 0 min—5 %, 16 min—20 %, 16.2 min—5 %, 21 min—5 %.
The instrument operated with an electrospray ion source (ESI) in positive ionization mode. Nitrogen was used as the drying gas at a temperature of 300°C and a flow rate of 10 L min−1 and the nebulizing gas at a pressure of 45 psi. Capillary voltage was set at 3000 V and skimmer voltage at 65 V. Fragmentor voltage was set at 120 V. The quadrupole was used as an ion guide in MS mode, and for selection of precursor ions with Δm/z = 1.3 in MS/MS mode. Nitrogen was used as the collision gas in MS/MS mode and collision energy was set at 4 V for MDMA and MDMA D5, 8 V for mephedrone and 4-MEC, 16 V for mCPP, 24 V for MDPV. Collision energy for compounds was obtained by using Mass Hunter Optimizer Software, Version B.02.00 (Agilent Technologies).
The mass range was 50–1000 m/z, in both MS and MS/MS modes. Spectra were internally mass-corrected in real time using an automatically introduced reference mass solution containing two compounds: purine ([M + H]+ = 121.0509) and HP-921—hexakis(1H,1H,3H-tetrafluoropropoxy)phosphazine ([M + H]+ = 922.0098).
The concentrations of target compounds were determined by using the main product ion (quantifier) presented in Table 2. Concentrations in the samples were calculated by comparing the peak area ratios of the analytes and surrogate standard to the corresponding ratios in the standard solutions. The stimulant drugs stock solution with a concentration of 10 µg mL−1 in methanol was used for preparation of calibration solutions. Calibrations curves were obtained from this stock solution spiked in mobile phase at 5, 10, 25, 50, 100, 250, 500, 1000 ng mL−1. All calibration solutions contained 200 ng mL−1 of the internal standard, MDMA D5.
Results and Discussion
Extraction efficiency was tested by placing 500 mL deionized water adjusted to pH 4.5 in volumetric flasks and then spiking them with prepared drugs stock solutions. After manual shaking, the samples were extracted by SPE. For each sample three extractions were prepared and each extract was analysed in triplicates. To check for concentration dependencies of the recovery rate and to determine the limit of quantification from the recovery experiment, deionized water samples were spiked to 40, 200 and 600 ng L−1 of the target compounds. All the samples were spiked with labelled internal standard as well as hydrochloric acid to adjust the pH to 4.5. The eluates were concentrated and reconstituted with mobile phase to 1 mL and extracts were analysed by LC–ESI–QTOF–MS. The results presented in Table 3 show that most recovery rates were between 50 % and 100 %.
The recovery rates were quantitated and the relative standard deviations (RSD %) were about 10 %. The applied conditions of the liquid chromatograph allowed for the successful separation of all analytes within 11 min. Before analysis of target compounds, blank measurements were applied for detection of possible impurities. For all analytes, molecular ions were selected as precursor ions. Specific and intense product ions of each target analyte were used for quantification and a secondary product ion was used as qualifier ion for confirmatory purposes.
Details of the specific parameters for detection of the analytes are given in Table 2. Delta retention time in MS/MS mode was 3 min, the time window wherein the fragmentation occurs, i.e. retention time ±1.5 min.
Calibration curves were produced by a weighted (1/x) linear least square regression. Relative peak areas (ratios of the analytes to the surrogate standard) were used for calculations. Correlation coefficients, R, were in the range of 0.9982–0.9997. Limits of detection (LOD) and quantification (LOQ) were calculated on the basis of signal to noise ratios (S/N) of 3 and 10, respectively. All parameters are shown in Table 4.
The analytical results corrected for the average recovery (shown in Table 3) are shown in Table 5. The highest concentration found in wastewater samples corresponds to mephedrone (42.9 ng L−1). The detected concentrations of MDMA and 4-MEC were similar, ranging between 21.6 and 41.3 ng L−1. As the analytes concentrations in wastewater samples were low, the results were recalculated for recoveries at low concentration level (40 ng L−1), but the differences were insignificant.
MDPV and mCPP were not detected in the tested wastewater samples.
Loads (mg 1000 inhabitants−1 day−1) of illicit drugs discharged via effluents into the aquatic system were estimated from concentrations of each compound detected in the effluents (ng L−1) and the daily flow of effluents discharged to the aquatic system. Loads of illicit drugs (MDMA, mephedrone and 4-MEC), discharged by the WWTP ranged from 3.6 to 6.7 mg day−1 1000 inhabitants−1 of MDMA, 3.6 to 7.1 mg day−1 1000 inhabitants−1 of mephedrone and 4.8 to 5.8 mg day−1 1000 inhabitants−1 of 4-MEC.
The hazard quotient (HQ) method was used to screen the toxicological risk level (Mendoza et al. 2014). The lack of aquatic ecotoxicological data for psychoactive compounds makes it difficult to conduct proper environmental risk assessment. For MDMA, Mendoza et al. (Mendoza et al. 2014) published a PNEC of 0.216 µg L−1 based on the lowest median lethal (effective) concentration (L(E)50) in algae, cladocerans and fish divided by an assessment factor of 1000. PNEC derived by the Ecological Structure Activity Relationships (ECOSAR) modeling. For mephedrone and 4-MEC, no PNECs could be found in available literature. The calculated HQs for MDMA, which are well below 1, up to 0.188, mean that, as far as data are available, the environmental risk is low but a potential adverse effect could be expected for MDMA. The values of HQs estimated in this work for an environmental risk assessment are in agreement with data reported by Mendoza et al. (Mendoza et al. 2014) and Bijlsma et al. (Bijlsma et al. 2014). These data were calculated in the effluent of the WWTP, so this is the worst case scenario because effluents are diluted further in a surface water.
References
Adamowicz P, Zuba D, Sekula K (2013) Analysis of UR-144 and its pyrolysis product in blood and their metabolites in urine. Forensic Sci Int 233:320–327. doi:10.1016/j.forsciint.2013.10.005
Andres-Costa MJ, Rubio-Lopez N, Morales Suarez-Varela M, Pico Y (2014) Occurrence and removal of drugs of abuse in Wastewater Treatment Plants of Valencia (Spain). Environ Pollut 194:152–162. doi:10.1016/j.envpol.2014.07.019
Baker DR, Očenášková V, Kvicalova M, Kasprzyk-Hordern B (2012) Drugs of abuse in wastewater and suspended particulate matter — Further developments in sewage epidemiology. Environ Int 48:28–38. doi:10.1016/j.envint.2012.06.014
Bijlsma L, Serrano R, Ferrer C, Tormos I, Hernandez F (2014) Occurrence and behavior of illicit drugs and metabolites in sewage water from the Spanish Mediterranean coast (Valencia region). Sci Total Environ 487:703–709. doi:10.1016/j.scitotenv.2013.11.131
Byrska B, Zuba D, Stanaszek R (2010) Determination of piperazine derivatives in “Legal Highs”. Probl Forensic Sci 81:101–113
Castiglioni S, Zuccato E, Fanelli R (2011) Illicit Drugs in the Environment: Occurrence, Analysis, and Fate Using Mass Spectrometry. John Wiley & Sons Inc, Hoboken
Castiglioni S, Borsotti A, Senta I, Zuccato E (2015) Wastewater analysis to monitor spatial and temporal patterns of use of two synthetic recreational drugs, ketamine and mephedrone, in Italy. Environ Sci Technol 49:5563–5570. doi:10.1021/es5060429
Kasprzyk-Hordern B, Dinsdale RM, Guwy AJ (2009) The removal of pharmaceuticals, personal care products, endocrine disruptors and illicit drugs during wastewater treatment and its impact on the quality of receiving waters. Water Res 43:363–380. doi:10.1016/j.watres.2008.10.047
Mendoza A, Rodríguez-Gil JL, González-Alonso S, Mastroianni N, López de Alda M, Barceló D, Valcárcel Y (2014) Drugs of abuse and benzodiazepines in the Madrid Region (Central Spain): seasonal variation in river waters, occurrence in tap water and potential environmental and human risk. Environ Int 70:76–87. doi:10.1016/j.envint.2014.05.009
Pomati F, Cotsapas CJ, Castiglioni S, Zuccato E, Calamari D (2007) Gene expression profiles in zebrafish (Danio rerio) liver cells exposed to a mixture of pharmaceuticals at environmentally relevant concentrations. Chemosphere 70:65–73. doi:10.1016/j.chemosphere.2007.07.048
Reid MJ, Baz-Lomba JA, Ryu Y, Thomas KV (2014) Using biomarkers in wastewater to monitor community drug use: a conceptual approach for dealing with new psychoactive substances. Sci Total Environ 487:651–658. doi:10.1016/j.scitotenv.2013.12.057
Report (2011) Chief Sanitary Inspectorate, Polish new psychoactive substances report, Warsaw
Report ED (2015) European Drug Report, Trends and Developments, European Monitoring Centre for Drugs and Drug Addiction (EMCDDA)
Rosi-Marshall EJ, Snow D, Bartelt-Hunt SL, Paspalof A, Tank JL (2015) A review of ecological effects and environmental fate of illicit drugs in aquatic ecosystems. J Hazard Mater 282:18–25. doi:10.1016/j.jhazmat.2014.06.062
Thomas KV, Bijlsma L, Castiglioni S, Covaci A, Emke E, Grabic R, Hernández F, Karolak S, Kasprzyk-Hordern B, Lindberg RH, Lopez de Alda M, Meierjohann A, Ort C, Pico Y, Quintana JB, Reid M, Rieckermann J, Terzic S, van Nuijs ALN, de Voogt P (2012) Comparing illicit drug use in 19 European cities through sewage analysis. Sci Total Environ 432:432–439. doi:10.1016/j.scitotenv.2012.06.069
van Nuijs AL, Pecceu B, Theunis L, Dubois N, Charlier C, Jorens PG, Bervoets L, Blust R, Neels H, Covaci A (2009) Cocaine and metabolites in waste and surface water across Belgium. Environ Pollut 157:123–129. doi:10.1016/j.envpol.2008.07.020
Zuba D (2014) New psychoactive substances—a contemporary challenge for forensic toxicologists. Probl Forensic Sci 100:359–385
Zuba D, Byrska B (2013) Prevalence and co-existence of active components of ‘legal highs’. Drug Test Anal 5:420–429. doi:10.1002/dta.1365
Zuba D, Sekula K, Buczek A (2013) 25C-NBOMe–new potent hallucinogenic substance identified on the drug market. Forensic Sci Int 227:7–14. doi:10.1016/j.forsciint.2012.08.027
Zuccato E, Castiglioni S (2009) Illicit drugs in the environment. Philos Trans R Soc 367:3965–3978. doi:10.1098/rsta.2009.0107
Zuccato E, Chiabrando C, Castiglioni S, Bagnati R, Fanelli R (2008) Estimating community drug abuse by wastewater analysis. Environ Health Perspect 116:1027–1032. doi:10.1289/ehp.11022
Acknowledgments
This work was supported by AGH University Grant No 11.11.210.244. The authors thank the staff of “Municipal Waterworks and Sewer Enterprise in Kraków” for their support in the collecting of samples.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Styszko, K., Dudarska, A. & Zuba, D. The Presence of Stimulant Drugs in Wastewater from Krakow (Poland): A Snapshot. Bull Environ Contam Toxicol 97, 310–315 (2016). https://doi.org/10.1007/s00128-016-1869-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00128-016-1869-5