
Abstract
The road traffic impact on the concentrations of 13 fractions of particulate matter (PM) and their components was assessed. PM was sampled at two points in Katowice (southern Poland), a background point beyond the effects of road traffic, and a near-highway traffic point. The samples were analyzed for organic and elemental carbon, 8 water-soluble ions, 24 elements, and 16 polycyclic aromatic hydrocarbons (PAHs). The traffic emissions (mainly particles from car exhaust) enriched the ultrafine, submicron, and fine PM particles with elemental carbon. The traffic-caused re-suspension of the road and soil dust affecting the concentrations and chemical composition of the coarse PM fraction. However, for each PM fraction, the carcinogenic equivalent ratios, assumed as a measure of the hazard from 16 PAHs in this paper, were similar at the two sampling points. The traffic emissions from the highway appeared to have a weaker influence on the concentrations and chemical composition of PM in a typical urban area of southern Poland than elsewhere in Europe.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
The growth of road traffic causes the growth of health hazards from atmospheric aerosols (Schwartz 1997; Han and Naeher 2006). However, attributing this effect only to the traffic-related elevation of particulate matter (PM) concentrations, although most obvious, is a simplification–in great part the threat is due to the specific influence of traffic emissions on the chemistry of PM (Han and Naeher 2006; Daher et al. 2014). In general, the ambient concentrations of PM-bound carbonaceous matter (including polycyclic aromatic hydrocarbons, elemental and organic carbon) are higher at crossroads and highways than in areas beyond the traffic influence (e.g. Harrison et al. 2004; Hueglin et al. 2005; Slezakova et al. 2010). The concentrations of PM-bound sulfates, nitrates, ammonium, chlorates, soil matter, and some elements at traffic-affected sites apparently differ from those in rural, urban, or suburban areas where the traffic effects are weaker (Harrison et al. 2004; Hueglin et al. 2005; Daher et al. 2014).
The goal of the work was to determine the differences in the chemical composition between various fractions of PM and to evaluate the road traffic influence on these differences in a typical urban area in southern Poland. PM was sampled in Katowice, at two sites differing in the traffic contributions to the air pollution. The concentrations, chemical composition, and exactness of the mass reconstruction (chemical mass closure) of each of thirteen following PM fractions: PM0.03-0.06, PM0.06-0.108, PM0.108-0.17, PM0.17-0.26, PM0.26-0.4, PM0.4-0.65, PM0.65-1, PM1-1.6, PM1.6-2.5, PM2.5-4.4, PM4.4-6.8, PM6.8-10, PM10-40 (subscript ranges are the intervals of the particle aerodynamic diameters Dp in µm) at these two sites are discussed in the paper.
Materials and Methods
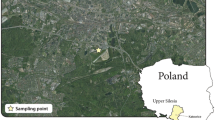
Particulate matter was sampled simultaneously at two points in Katowice, a city in southern Poland with approximately 2,000,000 population. One of the points, UB (Fig. 1), was an urban background sampling point (2008/50/EC Directive), and was located beyond the effects of industrial and traffic emissions (the nearest road was 150 m away, and traffic density was less than 2,500 cars per 24 h). The area was a living district surrounded by greenbelts, about 2,000 m from the city center. The meteorological conditions at the point were typical of the region. The second point, HW (Fig. 1), was a traffic point located on the shoulder of the highway A4, about 1,500 m south of the city center and 1,200 m northwest of the point UB. Average traffic density at HW was 30,000 cars per 24 h. To the north of HW, there was a living quarter. To the south, there was an open area of the A4 highway, and further behind A4, about 300 m from the point, there were some public utilities and detached houses of another living quarter. PM was sampled with the use of two thirteen-stage low pressure DLPI impactors (Dekati Ltd, Kangasala, FL).

Location of the measuring sites in Katowice
In Katowice, especially in winter, traffic PM emissions can be obscured by emissions of PM from combustion of fossil fuels or biomass for residential heating (Rogula-Kozłowska et al. 2013; Rogula-Kozłowska and Klejnowski 2013). Therefore, the sampling period was selected beyond the major heating season, and the samples were taken within mid-March to mid-June of 2012. There were nine, about one-week long (from 142 to 173 h), pairwise sample-takings.
The mass of the collected PM was determined by weighing the substrates before and after the exposure; a MYA 5.3Y.F micro balance (RADWAG; Radom, PL) was used (1 µg resolution). Before each weighing, the substrates were conditioned for 48 h in the weighing room (relative air humidity 45 % ± 5 %, air temperature 20 ± 2°C). The substrates and impactors were prepared for exposure in a laminar chamber. After weighing, the exposed substrates were stored in a freezer until analysis.
Two kinds of Whatman (GE Healthcare Bio-Sciences Corp.; Piscataway, NJ, USA) substrate filters were used. Alternating between the sample-takings, quartz fiber filters (QMA, ø25 mm, CAT No. 1851-025) and nylon membrane filters (0.2 µm, ø25 mm, Cat No. 7402-002) were used; the former four and the latter five times. Always the same type substrates were used on all impactor stages, with the same type simultaneously used at both sampling points. Altogether, nine samples (four on quartz and five on nylon filters) of each of the 13 PM fractions were taken at each point over the entire measuring period.
At each point, four samples of each fraction were taken on quartz filters. Two equal (1.5 cm2) fragments were cut out from each quartz filter just before the analysis. The PM on one of them was analyzed for organic carbon (OC) and elemental carbon (EC). The remaining fragments, four for each of the 13 fractions and each point, were used to make fraction samples. A fraction sample for a point was made by putting together the four filter fragments containing this PM fraction sample from this point. Each of these 26 fraction samples was analyzed for 16 PAHs.Footnote 1
Five samples of each fraction were taken on nylon filters at each sampling point. The PM on each nylon filter was analyzed for elemental composition first (i.e. for Al, Si, Sc, Ti, V, Cr, Mn, Fe, Co, Ni, Cu, Zn, As, Se, Br, Rb, Sr, Mo, Ag, Cd, Sb, Te, Ba, Pb), then it was extracted in water and concentrations of the main ions (Cl−, NO3 −, SO4 2−, Na+, NH4 +, K+, Ca2+, Mg2+) were determined in the extracts.
The OC and EC contents of dust were determined with the use of a Lab OC–EC Aerosol Analyzer (Sunset Laboratories Inc.; Portland, OR, USA). Sixteen PAHs1 in the PM fractions were determined on a Clarus 500 gas chromatograph (PerkinElmer; Waltham, MA, USA). The analytical procedures, equipment, and quality control for OC, EC, and PAH in PM are described in Rogula-Kozłowska and Klejnowski (2013) and Rogula-Kozłowska et al. (2013).
The elemental composition of PM was determined by means of energy dispersive X-ray fluorescence (EDXRF). An Epsilon 5 instrument (PANalytical B.V.; Almelo, NL), calibrated with thin-layer single-element standards (Micromatter; Vancouver, BC,CA) was used to measure total concentrations of the elements. To control the performance of the analytical procedure, samples of reference material (SRM2873, NIST, Gaithersburg, MD, USA) were measured weekly. The recoveries were between 85 % and 120 % of the certified values (except 52 % and 39 % recoveries of V and Co) and the X-ray tube and detector drifts were monitored monthly. The detection limits were from 0.18 ng/cm2 (Se) to 19.6 ng/cm2 (Si).
The water extracts of PM were made by ultrasonizing the substrates containing the samples in 25 cm3 of de-ionized water for 60 min at 15°C, then shaking for about 12 h (18°C, 60 r/min). The ion content of extracts was determined using an ion chromatograph (Metrohm AG; Herisau, CH). The method was validated against the CRM Fluka products nos. 89316 and 89886; the standard recoveries were 92 %–109 % of the certified values, and the detection limits were: 10 ng/cm3 for NH4 +, 18 ng/cm3 for Cl−, SO4 2− and K+, 27 ng/cm3 for NO3 − and Na+, and 36 ng/cm3 for Ca2+ and Mg2+.
Results and Discussion
The average concentrations for the sampling period are presented in Table 1. In Katowice, the core PM mass consisted of particles with Dp between 0.17 and 1.6 µm (PM0.17-1.6). They contributed 72 % to the total PM mass at UB and 64 % at HW (Table 1). The density function of the PM mass size distribution assumes the absolute maximum between 0.4 and 1.0 µm at both sampling points (Fig. 2). It is clear that the concentrations of PM0.17-1.6 were not significantly affected by road traffic.

Mass size distribution of PM and selected components of PM at urban background (UB) and at the highway (HW)
The concentrations of PM0.17-1.6 and of its chemical components did not differ between the points except for (usually) traffic-related Fe, Co, Ni, and Mo (Pant and Harrison 2013), and for EC.
In general, the fractionated EC concentrations were higher at HW than at UB. Within PM0.17-1.6, the concentrations of PM0.17-0.26-, PM0.26-0.4-, PM0.4-0.65-, PM0.65-1-, PM1-1.6-bound EC were from 40 % to >300 % higher at HW than at UB. The concentrations of PM0.03-0.06, PM0.06-0.108, PM0.108-0.17-related EC were from 4 to 8 times greater at HW than at UB. The striking difference in the behavior of the concentrations of PM-related EC between HW and UB can be seen by comparing the density functions of the mass distribution (relative to particle size) of EC bound to the nuclei and accumulation modes at HW and UB (Fig. 2).
Also, the concentrations of PM0.03-0.17 and of PM0.03-0.17-bound OC were higher at HW than at UB (averages of the concentrations of PM0.03-0.06, PM0.06-0.108, PM0.108-0.17, and of the related OC were higher by 84 % and 54 %, respectively). The concentrations of PM0.03-0.17-related Fe, Cu, and Zn were also elevated at HW. The density functions for some PM-related metals (e.g. Zn, Fig. 2) had maxima in the interval 0.03-0.17 µm at HW; these maxima did not exist at UB. Great differences in the concentrations of PM0.03-0.06-, PM0.06-0.108-, and PM0.108-0.17-bound EC, OC, Fe, Cu, and Zn between the points are due to the chemical composition of car exhaust: car exhaust consists mainly of ultrafine particles of EC, OC (incomplete fuel combustion), substances synthesized from residues of fuel combustion, and lubricant or fuel additives (metals and their compounds, e.g.) (Geller et al. 2006; Maricq 2007).
Surprisingly, despite the significant PAH content of exhaust OC, the ambient concentrations of fine particle-bound ∑PAH did not differ significantly between the points except for the PM0.06-0.108-bound ∑PAH, higher at HW than at UB by 90 %. The density function of the PM-bound ∑PAH distribution had the maximum in 0.06-0.108 µm. However, some from among the 16 PAHs had significantly higher concentrations at HW than at UB. The concentration of PM-bound BaP at HW was almost twice that for UB (4.3 and 2.3 ng/m3, respectively). The significantly higher PM0.03-1.6-bound BaP concentration at HW than at UB allows for consideration of BaP as a marker for exhaust emissions in urban areas of Upper Silesia in the periods of lower municipal emissions.
The concentrations of coarser PM (Dp > 4.4 µm) and of some its components (OC, EC, Na+, K+, Cl−, Ca2+, Al, Si, Fe, Cu, Zn, Mo) also were higher at HW. For example, the average concentrations of PM4.4-6.8-, PM6.8-10-, and PM10-40-related Na+ and K+ were almost four times greater at HW than at UB, while that for Ca2+ was approximately two-fold greater.
The PM mass size distribution was bimodal at UB. At HW, in 6.8-10 µm, the third peak for its density function appeared. Also the maxima of the density functions for Na+, K+, Cl−, and Ca2+ occurred in this interval. The coarse ambient particles containing OC, EC, Na+, K+, Cl−, Ca2+, Al, Si, Fe, Cu, Zn, Mo are probably a mixture of the particles of worn tires, brake linings, chassis, other car parts, road surface, soil, etc. (Wahlin et al. 2006; Thorpe and Harrison 2008; Pant and Harrison 2013). Such particles, involved in the process of alternate lifting (by passing cars, wind, etc.) and deposition, can stay within the road vicinity for a long time.
The chemical mass closure of PM was checked at both points. There were six groups of identified chemical PM components, and the seventh one, unidentified matter (UM); their masses together made the total PM mass. The mass [EC] of elemental carbon (EC) was assumed to be the analytically determined mass [EC]A of elemental carbon: [EC] = [EC]A. The mass [OM] of organic matter (OM, all PM-bound organic compounds) was assumed to be 1.4 of the analytically determined mass [OC]A of organic carbon (OC): [OM] = 1.4[OC]A. Secondary inorganic aerosol, SIA, consisted of SO4 2−, NO3 −, NH4 +; [SIA] = [SO4 2−]A + [NO3 −]A + [NH4 +]A. [NaCl] = [Cl−]A + [Na+]A.
The rest of the chemical components of PM listed in Table 1, i.e. the analytically determined PM-bound chemical elements (including K+, Ca2+, and Mg2+), were used to define crustal matter (CM) and trace elements (TE). The contents of CM and TE depended on both the PM fraction and the measuring point. These PM-bound elements were divided into two groups based on their enrichment factors EF (Rogula-Kozłowska et al. 2013; 2014). EF expresses how anthropogenic an element is: the closer an element’s EF value is to 1, the smaller the anthropogenic contribution to the element’s concentration in air.
The elements with EF ≤ 20 were assumed crustal (of natural origin). The chemical compounds containing an element with EF ≤ 20 that were known to contribute significantly to the PM mass were included in CM; if there were no such compounds (known) for an element, or the element was probable to occur in numerous compounds, only the element was included in CM. CM included CO3 2−, SiO2, Al2O3, Mg2+, Ca2+, K2O (except PM0.4-0.65- and PM0.06-0.108-bound K2O at UB), FeO and Fe2O3 (except PM0.4-1.6-bound FeO and Fe2O3 at both points), Rb (except PM0.17-1.6-bound Rb at both points), Sr (except PM0.4-1.6- and PM0.03-0.26-bound Sr at HW, and PM0.03-1-bound Sr at UB), PM2.5-6.8-bound Ba, and Ba from particles with Dp greater than 10 µm. Except for CO3 2−, the masses of the compounds (oxides) in CM (SiO2, Al2O3, K2O, FeO and Fe2O3) were computed stoichiometrically from the masses of their component elements determined analytically; Fe was assumed to be distributed equally between FeO and Fe2O3. [CO3 2−] was computed from [Ca2+]A and [Mg2+]A (Marcazzan et al. 2001).
The chemical elements with EF > 20 were assumed to be anthropogenic; these many-source origin elements occurred in many PM-bound compounds that were not determined because of their multiplicity. These elements alone were put in TE. TE consisted of K+ (only for PM0.4-0.65 and PM0.06-0.108 at UB), Sc, Ti, V, Cr, Mn, Fe (Fe at both points for PM0.4-1.6 only), Co, Ni, Cu, Zn, As, Se, Br, Rb (Rb at both points for PM0.17-1.6 only), Sr (only for PM0.4-1.6 and for PM0.03-0.26 at HW and for PM0.03-1 at UB), Mo, Ag, Cd, Sb, Te, Ba (Ba only for PM0.03-2.5 and PM6.8-10 at both points), and Pb. The mass [TE] of TE was assumed to be the sum of all the analytically determined masses of the elements from TE.
The mass of unidentified matter [UM] was the deficient mass; it was the difference between the gravimetrically determined mass of PM and [SIA] + [EC] + [OM] + [NaCl] + [CM] + [TE].
In Fig. 3, the mass distributions of the PM component groups (relative to particle size) at both sites are presented. Within particular PM fractions, the identified matter accounted for the mass in 52 %–98 % at HW and in 55 %–96 % at UB. For all fractions except PM4.4-6.8, the mass of identified compounds was higher at UB than at HW.

Mass size distribution of the PM component groups (a) and the mass reconstruction of selected PM fractions (b) at traffic affected HW and at urban background UB (EC elemental carbon, OM organic matter, SIA secondary inorganic aerosol, NaCl sum of Na+ and Cl−, CM crustal matter, TE trace elements, UM unidentified matter)
The shares of UM in twelve PM fractions were greater, and in ultrafine PM much greater, at HW than at UB (Fig. 3). At HW, the UM shares in PM0.06-0.65 and in particles with Dp > 10 µm were greatest; whereas at UB, they were greatest in PM0.108-0.65 and PM1.6-4.4. UM consists of the organic compounds and nitrates that evaporate during transportation and storage of PM samples, unidentified compounds, and water (Tsyro 2005). Also, inaccurate identification of compounds in OM, CM and TE affects UM. For example at HW, considering the high UM content of 12 fractions, [OM] should probably have been computed by multiplying [OC]A by a coefficient greater than 1.4 (Turpin and Lim 2001).
The EC concentrations were higher at HW, and the EC mass distribution differed from that at UB (Table 1; Fig. 2). Therefore, the EC contributions to particular PM fractions, especially to finer ones, at HW were higher than at UB (Fig. 3). The fractional mass contributions of OM to PM at both points were close, and only for the sub-fractions of PM2.5-6.8 were they slightly higher (by 5 % in average) at HW.
Although the concentrations of the components of SIA (SO4 2−, NO3 −, NH +;4 Table 1) at both points were comparable, the distributions of SIA and its mass contributions to the fractions differed slightly between the points. The contributions to PM0.03-0.108 and coarser particles (Dp > 4.4 µm) were on average greater by 4 and 5 % at UB, respectively.
NaCl was more abundant at HW than at UB only in PM4.4-10. This was due to the previously mentioned differences in the ambient concentrations of Na+ and Cl− between the points. Similarly, the elevated concentrations of the components of CM, especially of K+, Ca2+, Al, Si, and Fe, caused a slightly greater share of CM in PM1.6-10 at HW (CM contribution to the PM mass at UB was greater than at HW by no more than 2 %). The mass contribution of TE to PM0.65-1.6 was greater by about 2 % at HW than at UB. The NaCl, CM, and TE contributions to fine, especially to ultrafine, PM were higher at UB than at HW because ultrafine PM had noticeably lower concentrations at UB (Fig. 3). NaCl and TE contain elements coming from energy production, traffic or industry at UB, and mainly from traffic (both exhaust and non-exhaust emissions) at HW (Pant and Harrison 2013; Kumar et al. 2013; Rogula-Kozłowska et al. 2014). Also CM in ultrafine PM, despite being “crustal,” probably contains particles coming from exhaust gases and furnaces, including particles containing metal oxides from condensation (nuclei mode) (Geller et al. 2006; Maricq 2007).
The differences in the chemical composition of PM between the points were mainly in ultrafine and coarse PM. The distributions of the mass among the component groups differed extremely for PM0.1 (in fact PM0.03-0.108) between the two points (Fig. 3). PM1 and PM2.5 (in fact PM0.03-1 and PM0.03-2.5) differed between the points only in their EC content; PM10—in the EC and, greater by about 1 % at HW, CM contents (Fig. 3). The concentration of PM0.1 was two times greater at HW than at UB (1.5 and 0.7 µg/m3), while the concentrations of PM2.5 and PM10 at HW were greater by no more than 5 % (for PM10 they were 28.6 µg/m3 at HW and 27.3 µg/m3 at UB).
So, although the number of cars per person grows closer to the European average, traffic emissions are not as significant a PM source in the Silesian Agglomeration as they are in other European regions (e.g. Viana et al. 2008). In Katowice, PM0.17-1.6 contributes most to the PM mass, and traffic does not affect this PM fraction much. The concentrations and the chemical composition of this PM fraction within the Silesian Agglomeration are formed by municipal emissions, big power plants, and industry. The presented results show that human health is not more at risk near a big highway than some distance away. At both sampling points the carcinogenic equivalent ratios (CEQ) for particular PM fractions were close, both CEQ for total PM were equal to 4.2 µg/m3 (Table 1). As elsewhere in Poland, the concentration, chemical composition, and toxicity of PM in the Silesian Agglomeration depend on the season of the year; i.e., upon meteorological conditions and emissions from heat and power production facilities (Pastuszka et al. 2003, Majewski et al. 2011; Rogula-Kozłowska and Klejnowski 2013; Rogula-Kozłowska et al. 2014).
The present pioneering research shows that some differences in the chemical characteristics of PM4.4-10 between two sites in such a specific area as the Silesian Agglomeration can be found only by investigating particular fractions of PM4.4-10. However, the experiment should rather be considered as a pilot research and should be repeated on a wider scale at other traffic-affected sites, such as crossroads in a city center, street canyons, etc., to generalize the conclusions.
Notes
The group of 16 PAHs included: naphthalene (Na), acenaphthene (Ace), acenaphthylene (Acy), anthracene (An), benzo[a]anthracene (BaA), benzo[a]pyrene (BaP), benzo[b]fluoranthene (BbF), benzo[k]fluoranthene (BkF), benzo[g,h,i]perylene (BghiP), chrysene (Ch), dibenzo[a,h]anthracene (DBA), fluoranthene (Fl), fluorene (F), phenanthrene (Ph), pyrene (Py) and indeno[1,2,3-cd]pyrene (IP).
References
Daher N, Saliba N, Shihadeh AL, Jaafar M, Baalbaki R, Shafer MM, Schauer JJ, Sioutas C (2014) Oxidative potential and chemical speciation of size-resolved particulate matter (PM) at near-freeway and urban background sites in the greater Beirut area. Sci Total Environ 470–471:417–426
Geller MD, Ntziachristos L, Mamakos A, Samaras Z, Schmitz DA, Froines JR, Sioutas C (2006) Physicochemical and redox characteristics of particulate matter (PM) emitted from gasoline and diesel passenger cars. Atmos Environ 40:6988–7004
Han X, Naeher LP (2006) A review of traffic-related air pollution exposure assessment studies in the developing world. Environ Int 32:106–120
Harrison RM, Jones AM, Lawrence RG (2004) Major component composition of PM10 and PM2.5 from roadside and urban background sites. Atmos Environ 38:4531–4538
Hueglin C, Gehrig R, Baltensperger U, Gysel M, Monn C, Vonmont H (2005) Chemical characterization of PM2.5, PM10 and coarse particles at urban, near-city and rural sites in Switzerland. Atmos Environ 39:637–651
Kumar P, Pirjola L, Ketzel M, Harrison RM (2013) Nanoparticle emissions from 11 non-vehicle exhaust sources—a review. Atmos Environ 67:252–277
Majewski G, Kleniewska M, Brandyk A (2011) Seasonal variation of particulate matter mass concentration and content of metals. Pol J Environ Stud 20:417–427
Marcazzan GM, Vaccaro S, Valli G, Vecchi R (2001) Characterisation of PM10 and PM2.5 particulate matter in the ambient air of Milan (Italy). Atoms Environ 35:4639–4650
Maricq MM (2007) Chemical characterization of particulate emission from diesel engines: a review. Aerosol Sci 38:1079–1118
Pant P, Harrison RM (2013) Estimation of the contribution of road traffic emissions to particulate matter concentrations from field measurements: a review. Atmos Environ 77:78–97
Pastuszka JS, Wawroś A, Talik E, Paw UKT (2003) Optical and chemical characteristics of the atmospheric aerosol in four towns in southern Poland. Sci Total Environ 309:237–251
Rogula-Kozłowska W, Klejnowski K (2013) Submicrometer aerosol in rural and urban backgrounds in southern Poland: primary and secondary components of PM1. Bull Environ Contam Toxicol 90:103–109
Rogula-Kozłowska W, Kozielska B, Klejnowski K, Szopa S (2013) Hazardous compounds in urban PM in the central part of Upper Silesia (Poland) in winter. Arch Environ Prot 39:53–65
Rogula-Kozłowska W, Klejnowski K, Rogula-Kopiec P, Ośródka L, Krajny E, Błaszczak B, Mathews B (2014) Spatial and seasonal variability of the mass concentration and chemical composition of PM2.5 in Poland. Air Qual Atmos Health 7:41–58
Schwartz J (1997) Health effects of air pollution from traffic: ozone and particulate matter. In: Fletcher T, McMichael AJ (eds) Health at the crossroads. Transport policy and urban health. Wiley, New York, pp 61–82
Slezakova K, Castro D, Pereira MC, Moralis S, Delerue-Matos C, Alvim-Ferraz MC (2010) Influence of traffic emissions on the carcinogenic polycyclic aromatic hydrocarbons in outdoor breathable particles. J Air Waste Manag Assoc 60:393–401
Thorpe A, Harrison RM (2008) Sources and properties of non-exhaust particulate matter from road traffic: a review. Sci Total Environ 400:270–282
Tsyro SG (2005) To what extent can aerosol water explain the discrepancy between model calculated and gravimetric PM10 and PM2.5. Atmos Chem Phys 5:515–532
Turpin BJ, Lim H-J (2001) Species contributions to PM2.5 mass concentrations: revisiting common assumptions for estimating organic mass. Aerosol Sci Tech 35:602–610
Viana M, Kuhlbusch TAJ, Querol X, Alastuey A, Harrison RM, Hopke PK, Winiwarter W, Vallius M, Szidat S, Prevot ASH, Hueglin C, Bloemen H, Wahlin P, Zecchi R, Kasper-Giebl A, Maenhaut W, Hitzenberger R (2008) Source apportionment of particulate matter in Europe: a review of methods and results. J Aerosol Sci 39:827–849
Wahlin P, Berkowicz R, Palmgren F (2006) Characterisation of traffic-generated particulate matter in Copenhagen. Atmos Environ 40:2151–2159
Acknowledgments
The work was realized within the projects Nos. C.1.2. and N N523 564038, the former financed by the Institute of Environmental Engineering, PAS, the latter–by the Polish Ministry of Science and Higher Education.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article

Cite this article
Rogula-Kozłowska, W. Traffic-Generated Changes in the Chemical Characteristics of Size-Segregated Urban Aerosols. Bull Environ Contam Toxicol 93, 493–502 (2014). https://doi.org/10.1007/s00128-014-1364-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00128-014-1364-9