
Abstract
Aims/hypothesis
Accumulating evidence suggests that leucocytes play a critical role in diabetes-induced vascular lesions and other abnormalities that characterise the early stages of diabetic retinopathy. However, the role of monocytes has yet to be fully investigated; therefore, we used Ccr2−/− mice to study the role of CCR2+ inflammatory monocytes in the pathogenesis of diabetes-induced degeneration of retinal capillaries.
Methods
Experimental diabetes was induced in wild-type and Ccr2−/− mice using streptozotocin. After 2 months, superoxide levels, expression of inflammatory genes, leucostasis, leucocyte- and monocyte-mediated cytotoxicity against retinal endothelial cell death, retinal thickness and visual function were evaluated. Retinal capillary degeneration was determined after 8 months of diabetes. Flow cytometry of peripheral blood for differential expression of CCR2 in monocytes was assessed.
Results
In nondiabetic mice, CCR2 was highly expressed on monocytes, and Ccr2−/− mice lack CCR2+ monocytes in the peripheral blood. Diabetes-induced retinal superoxide, expression of proinflammatory genes Inos and Icam1, leucostasis and leucocyte-mediated cytotoxicity against retinal endothelial cells were inhibited in diabetic Ccr2-deficient mice and in chimeric mice lacking Ccr2 only from myeloid cells. In order to focus on monocytes, these cells were immuno-isolated after 2 months of diabetes, and they significantly increased monocyte-mediated endothelial cell cytotoxicity ex vivo. Monocytes from Ccr2-deficient mice caused significantly less endothelial cell death. The diabetes-induced retinal capillary degeneration was inhibited in Ccr2−/− mice and in chimeric mice lacking Ccr2 only from myeloid cells.
Conclusions/interpretation
CCR2+ inflammatory monocytes contribute to the pathogenesis of early lesions of diabetic retinopathy.
Graphical abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.

Introduction
Adhesion of circulating leucocytes to the wall of retinal capillaries (leucostasis) is increased in diabetes. Inhibition of that interaction by deletion of intercellular adhesion molecule 1 (ICAM1) or its ligand, or by expression of neutrophil inhibitory factor (NIF) inhibits diabetes-induced retinal leucostasis, capillary leakage and degeneration of retinal capillaries [1,2,3,4,5]. Selective deletion of proteins involved in oxidative stress and inflammation solely from myeloid-derived cells likewise inhibits the retinal oxidative stress and capillary degeneration [5].
Neutrophils are one leucocyte subtype, and they have been previously implicated in the development of diabetes-induced retinal vascular lesions [6]. Mice with induced diabetes and lacking granulocyte colony-stimulating factor receptor were neutrophil-deficient, and showed significant reduction in diabetes-induced retinal oxidative stress, inflammation, and degeneration of retinal capillaries [5]. Whether or not neutrophils are the only blood cell contributing to the development of retinopathy has not been determined. Monocytes represent another major subset of leucocytes, and they have also been found to play major roles in oxidative stress and inflammation in a variety of diseases [7,8,9,10], in part via release of proteases and oxygen-derived free radicals [11].
In mice, like in humans, monocytes represent a heterogeneous group of cells and are commonly divided into classical monocytes (previously called inflammatory monocytes) and nonclassical monocytes (previously called patrolling monocytes) [12,13,14]. CC chemokine receptor 2 (CCR2) is expressed predominantly by monocytes, and especially by classical inflammatory monocytes. Whole-body deletion of Ccr2 decreases the abundance of classical monocytes (CCR2+) in the blood (a reduction of >84%) [12, 15, 16]. On the other hand, nonclassical monocytes express low levels of CCR2 (CCR2-) and high levels of CX3C motif chemokine receptor 1 (CX3CR1) [17]. CCR2 mediates mobilisation of the classical monocyte subset from bone marrow, subsequent migration into sites of injury [15, 18,19,20], firm adhesion of the cells to the endothelium and extravasation in vivo [21]. The beneficial effect of a CCR2/5 inhibitor on vascular permeability in rodent models of diabetes has been reported previously [22].
The aim of this study was to determine the role of CCR2+ inflammatory monocytes in the pathogenesis of diabetes-induced degeneration of retinal capillaries.
Methods
All procedures involving animals were performed in strict accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals, the Association for Research in Vision and Ophthalmology (ARVO) Statement for the Use of Animals in Ophthalmic and Vision Research, and with authorisation of the Institutional Animal and Care Use Committees (IACUC) at Case Western Reserve University, and University of California, Irvine.
Mice
Wild-type (WT) C57Bl/6J and Ccr2 knockout (C57Bl/6 background) (Ccr2−/−) mice were obtained from the Jackson Laboratory (Bar Harbor, ME, USA). In all studies, male mice (2–3 months old) were randomly assigned to become diabetic or remain as nondiabetic controls. Diabetes was induced by i.p. injection of a freshly prepared solution of streptozotocin in citrate buffer (pH 4.5) at 60 mg/kg of body weight for 5 consecutive days. Hyperglycaemia was verified at least three times during the second week after streptozotocin administration, and mice having three consecutive measurements of fasting blood glucose >15.3 mmol/l were classified as being diabetic. Insulin was given as needed to prevent weight loss without preventing hyperglycaemia and glucosuria (0–0.2 units of neutral protamine Hagedorn (NPH) insulin s.c 0–3 times per week). All animals were maintained on a standard 12 h light (∼10 lux)−dark cycle and were provided standard rodent chow (Purina TestDiet 5001; TestDiet Richmond, IN, USA) and water ad libitum. Blood glucose and HbA1c were measured as reported previously. Body weight was measured weekly. Animals were euthanised and eyes collected at 2 months of diabetes (4–5 months of age) to assess retinal function and biochemistry, and at 8 months of diabetes (10 months of age) to assess retinal capillary degeneration.
Chimeric mice were generated as previously described [5]. Briefly, recipient mice (nondiabetic or diabetic for 2 weeks) were irradiated with two doses of 600 rads, 3 h apart, and subsequently injected intravenously with 3–5 million bone marrow cells from donor mice. Chimeras lacking Ccr2 from only their marrow-derived cells were generated by transplanting marrow from Ccr2−/− donors into irradiated WT (C57Bl/6J) hosts (identified as Ccr2−/−→WT). Diabetes was induced prior to irradiation to make sure that irradiation and resulting immune cell damage did not interfere with the induction of diabetes. To control for potential effects of irradiation, nondiabetic and diabetic WT mice were irradiated and transplanted with marrow cells from analogous WT donors (WT→WT).
Retinal imaging and visual function
We studied retinal structure and function of Ccr2−/− and WT mice using spectral domain optical coherence tomography (SD-OCT; the 840HHP SD-OCT system, Bioptigen, USA), and electroretinogram (ERG; Diagnosys Celeris rodent ERG device, Diagnosys, USA) recordings [23,24,25]. Spatial frequency threshold and contrast sensitivity were measured at 2 months of diabetes (5 months of age) with the Virtual Optokinetic system as previously described [26,27,28]. Briefly, the minimum spatial frequency capable of driving head tracking was determined as the spatial frequency threshold. The experimenter was masked as to the identity of the experimental animals. The contrast sensitivity was measured at spatial frequency of 0.064 cycles/degree.
Blood phenotype
Mice were euthanised with CO2, and blood was collected by cardiac puncture into EDTA-containing tubes (100 mmol/l EDTA). After lysing erythrocytes with RBC lysis buffer (eBiosciences, San Diego, USA) and washing with PBS, antibodies and viability dye were incubated with white blood cells in FACS buffer (PBS with 0.5% BSA and 2 mmol/l EDTA) for 20 min at 4°C. The following antibodies were used (Biolegend, San Diego, USA): CD45-FITC (30-F11 clone), Ly6G-Brilliant Violet 510 (1A8 clone), Ly6C-PE-Cy7 (HK1.4 clone), CD11b-APC (M1/70 clone), F4/80-PE (BM8 clone), CCR2-Brilliant Violet 421 (SA203G11 clone) and CD14-PE-Dazzle 594 (Sa14-2 clone). Viability dye (Fixable Viability Dye eFluor 780, eBiosciences) was added to distinguish live cells. Cells were washed in FACS buffer and fixed for 20 min at 4°C (Perm/Fix buffer, BD Biosciences, USA) before analysis on a Novocyte cytometer (Acea, USA).
Lucigenin assay of superoxide
Retinal superoxide was measured chemically with lucigenin (bis-N-methylacridinium nitrate) [29]. Freshly isolated retinas were pre-incubated in 200 μl of Krebs-Hepes buffer (pH 7.2) with 5 or 30 mmol/l glucose for 10 min at 37°C in 5% CO2. Luminescence indicating the presence of superoxide was measured 5 min after addition of lucigenin (5 mmol/l). Luminescence intensity is reported in arbitrary units/mg of protein.
Quantitative reverse transcription-PCR
Both retinas from each mouse were combined (total of four to six mice per group) and total RNA was isolated with RNeasy Mini kit (Qiagen, USA). Total RNA (0.5 μg) was converted to cDNA by SuperScript III Reverse Transcriptase (Invitrogen from ThermoFisher Scientific, USA) and used for quantitative reverse transcription--PCR (qRT-PCR) conducted on C1000 Touch Thermal Cycler (Bio-Rad, USA). β-actin (also known as Actb) was used as a housekeeping gene. PCR reactions were performed in triplicate and normalised to β-actin.
Leucostasis
Leucostasis was determined as previously described [30]. Briefly, at 2 months of diabetes, blood was removed from the vasculature of anaesthetised animals by extensive perfusion with PBS via a heart catheter. Subsequently animals were perfused with fluorescein coupled Concanavalin A lectin (20 μg/ml in PBS; Vector Laboratories, USA). Flat-mounted retinas were imaged via fluorescence microscopy and the number of leucocytes adherent to the vascular wall was counted.
Leucocyte- and monocyte-mediated endothelial cell cytotoxicity ex vivo
Leucocyte-mediated endothelial cell death was determined as previously described [5]. Briefly, mouse retinal endothelial cells (mRECs; Cells Biologics, USA) [31] were grown in DMEM containing 10% FBS and 5.5 or 25 mmol/l glucose. When mRECs were 80% confluent (500,000 cells), leucocytes (100,000; purified from blood with RBC lysis buffer) were added to the mRECs and incubated for 24 h. After 24 h, mRECs were gently rinsed with PBS to remove non-adherent leucocytes, incubated with trypsin for 2 min and washed twice in PBS. The viability of mRECs was measured by trypan blue exclusion with a haemocytometer. Sample identity was masked during counting.
Monocytes were isolated from bone marrow because of the significantly higher yield when using bone marrow compared with peripheral blood (up to 10×106 vs 0.2×106 monocytes per mouse, respectively) [32]. Monocytes were immuno-isolated using EasySep Mouse monocyte isolation kit (StemCell Technologies, USA) following manufacture’s instruction (the purity is up to 94%). Monocyte-mediated endothelial cell cytotoxicity was performed as shown above for leucocytes.
Diabetes-induced retinal vascular histopathology
After 8 months of diabetes (10 months of age), mice were euthanised and the eyes were then enucleated and fixed in formalin. Retinal vasculature was isolated as previously described [30]. The fixed retina was isolated, rinsed in running water overnight, and then digested with 40 U/ml elastase (Calbiochem, San Diego, USA), 5 mmol/l EDTA, 100 mmol/l sodium phosphate and 150 mmol/l NaCl pH 6.5 at 37°C for 2–3 h. When totally cleaned of neural cells, the isolated vasculature was laid out on a glass microscope slide, dried overnight, stained with haematoxylin and periodic acid−Schiff, dehydrated and mounted with a glass coverslip. Degenerated (acellular) capillaries were quantified in 6–7 field areas corresponding to the mid-retina (×200 magnification) in a masked manner. Acellular capillaries reported per square mm of retinal area were identified as capillary-sized vessel tubes having no nuclei along their length.
Statistical analysis
Data are expressed as mean ± S.D., except for retinal thickness and ERG measurement that are expressed as mean ± SEM. Statistical analyses were performed with ANOVA followed by Fisher’s test (StatView for Windows software version 5.0.1; SAS Institute, Cary, NC, USA), except for ERG data which was analysed by two-way repeated measures of variance. *p≤0.05; **p≤0.01; and ***p≤0.001.
Results
Animals
There was no difference with respect to body weight or blood glucose levels between nondiabetic members of the strains studied. Blood glucose was elevated in all animals assigned to diabetic groups, and the severity of diabetes was not different among the diabetic groups (Table 1).
The circulating CCR2+ monocyte population is absent in nondiabetic and diabetic Ccr2 −/− mice
It has been reported that Ccr2 deficiency results in monocyte retention in bone marrow and subsequent depletion from peripheral blood [15]. To determine the effect of Ccr2 deficiency and diabetes on the subpopulation of monocytes, flow cytometry was performed on whole peripheral blood. Erythrocytes were lysed, and white blood cell suspensions were stained with subset-specific antibodies (CD11b+Ly6GlowLy6Chigh for classical monocytes, and CD11b+Ly6GhighLy6Chigh for neutrophils). We subsequently used CCR2 expression to differentiate between CCR2high and CCR2low cells. The gating strategy of the flow cytometry data is shown in electronic supplementary material (ESM) Fig. 1. In nondiabetic WT mice, most monocytes were CCR2high (corresponding to classical monocytes). In contrast, this population of cells was completely absent in nondiabetic Ccr2−/− mice (Fig. 1a,b). In WT mice, the induction of diabetes resulted in the reduction of classical monocytes (Fig. 1a,b), and the majority of neutrophils were CCR2low (ESM Fig. 2). Diabetes did not significantly affect the distribution of neutrophils in both strains of mice (ESM Fig. 2). These results indicate that the circulating leucocytes of Ccr2−/− mice are deficient in classical monocytes and can therefore be used as a model to study the role of classical monocytes in the development of diabetic retinopathy.

Effect of Ccr2 deficiency and diabetes on monocytes. Representative flow cytometry dot plots of cell suspensions from the blood of nondiabetic and diabetic WT and Ccr2−/− mice (a). FSC and SSC identified total leucocytes, CD11b+ identified myeloid cells and CD11b, Ly6G and Ly6C staining identified monocytes as CD11b+Ly6GlowLy6Chigh. CCR2 further identified monocytes as CCR2high or CCR2low monocytes. Numbers in the boxed areas indicate per cent of CD11b+, CCR2highLy6GlowLy6Chigh (CCR2+ M) or CCR2lowLy6GlowLy6Chigh (CCR2− M). Absolute numbers of monocytes are presented as per cent of total number of myeloid cells (b). Mean ± SD. *p≤0.05, ***p≤0.001. N, nondiabetic; D, diabetic
Neither the absence of CCR2+ monocytes nor 2 months of diabetes affects outer nuclear layer thickness
In vivo examination of mice by high-resolution SD-OCT was used to determine the effect of diabetes and the absence of Ccr2 on retinal structure and thickness. OCT analysis after 2 months of diabetes (4–5 months of age) indicated that neither diabetes nor the deletion of Ccr2 caused a significant increase or decrease of outer nuclear layer (ONL) thickness (Fig. 2a,b). We also measured the thickness of total retina along with nerve fibre layer and ganglion cell layer (NFL+GCL) thickness at 300 μm from the optic nerve. The results showed that total retinal thickness (0.24±0.02, 0.23±0.03, 0.23±0.02 and 0.23±0.02 in nondiabetic WT, diabetic WT, nondiabetic Ccr2−/− and diabetic Ccr2−/− mice, respectively) and NFL+GCL thickness (0.04±0.01, 0.03±0.01, 0.03±0.01 and 0.03±0.01 in nondiabetic WT, diabetic WT, nondiabetic Ccr2−/− and diabetic Ccr2−/− mice, respectively) were not significantly different between all four groups. However, it is worth mentioning that the thickness of inner retina of diabetic animals is controversial; we and others did not detect significant reduction in the inner retina [24, 33,34,35], whereas others have presented evidence showing significant reduction in the thickness of the inner retina in diabetes [36].

Effect of Ccr2 deletion and diabetes on outer nuclear layer (ONL) thickness. SD-OCT images (a) and quantification of data (spider web and histogram; b and c, respectively) show that the deletion of Ccr2 (solid grey lines and grey bars) alone or in conjunction with 2 months of diabetes (grey dashed lines and dotted bars) resulted in no loss of retinal photoreceptors (ONL thickness) compared with nondiabetic (solid black lines and black bars) or diabetic (black dashed lines and striped bars) WT mice. Scale bar, 50 μm. Data are presented as mean ± SD, n=8 mice (16 retinas) per group
Inflammatory monocytes mediate diabetes-induced oxidative stress and upregulation of inflammatory genes in the retina
Oxidative stress has been implicated in the development of diabetic retinopathy [37,38,39,40,41,42]. To determine if CCR2+ monocytes contribute to oxidative stress in diabetic retinopathy, retinal superoxide was measured chemically with the lucigenin method. Compared with nondiabetic WT controls, superoxide levels in the retina were significantly increased in WT mice after 2 months of diabetes. In contrast, retinal levels of superoxide were significantly reduced in diabetic Ccr2−/− mice compared with diabetic WT mice (Fig. 3). Two months of diabetes also significantly increased the expression of inflammatory genes Inos (also known as Nos2) and Icam1 in the retina of WT mice, whereas the diabetic Ccr2−/− animals showed significantly inhibited expression of both of these proinflammatory genes in the retina compared with diabetic WT mice (Fig. 4).

Effect of diabetes and the loss of Ccr2 on retinal superoxide levels after 2 months of diabetes (4–5 months of age). Superoxide levels in retinas from nondiabetic and diabetic Ccr2−/− mice and WT mice are shown. Mean ± SD (n=4–8 per group). ***p≤0.001. N, nondiabetic; D, diabetic

Effect of diabetes and the deletion of Ccr2 on proinflammatory gene expression in the retina. Genetic deletion of Ccr2 mitigates diabetes-induced upregulation of Inos and Icam1. Duration of diabetes was 2 months at the time of this assay. Data are expressed relative to the expression of actin. Data are presented as a per cent of the value of nondiabetic WT controls (n=4–10). **p≤0.01; ***p≤0.001. N, nondiabetic; D, diabetic
CCR2+ monocytes mediate a diabetes-induced increase in leucostasis in retinal capillaries
WT mice showed the expected diabetes-induced increase in leucostasis in the retina. In contrast, leucocyte adhesion to retinal capillaries was significantly inhibited in diabetic Ccr2-deficient mice (Fig. 5).

Effect of diabetes and the loss of Ccr2 on retinal leucostasis. (a) Diabetes increases leucostasis in the retina of WT mice, but leucocyte adhesion to retinal capillaries was significantly inhibited in diabetic Ccr2−/− mice. Leucostasis in retinal microvessels was determined by injection of fluorescein coupled concanavalin A lectin. (b) A representative image of leucostasis in the retina of a diabetic mouse (white arrows). Scale bar, 100 μm. Total duration of diabetes was 2 months. Data are presented as mean ± SD, n=6 per group. *p≤0.05. N, nondiabetic; D, diabetic
Deficiency of Ccr2 inhibits leucocyte- and monocyte-mediated cytotoxicity against retinal endothelial cells
Ex vivo incubation of leucocytes isolated from WT mice after 2 months of diabetes resulted in more retinal endothelial cell cytotoxicity than leucocytes isolated from nondiabetic WT controls. In contrast, leucocytes harvested from diabetic Ccr2−/− mice killed significantly fewer endothelial cells when compared with diabetic WT mice (Fig. 6a).

Effect of diabetes and the deletion of Ccr2 on leucocyte- and monocyte-mediated endothelial cell cytotoxicity. The data are expressed as per cent of corresponding nondiabetic mice. Leucocyte-mediated cytotoxicity towards retinal endothelial cells increased in diabetic WT mice but is significantly inhibited in diabetic Ccr2−/− mice (a). Similarly, monocyte-mediated cytotoxicity towards retinal endothelial cells was increased in diabetic WT mice and significantly inhibited in diabetic Ccr2−/− mice (b). Total duration of diabetes was 2 months. Data are presented as mean ± SD, n=5–13 per group. *p≤0.05, **p≤0.01, ***p≤0.001. N, nondiabetic; D, diabetic
Although data from others [12, 15, 16] and us (Fig. 1) indicates that CCR2 is expressed predominantly on monocytes, we immuno-isolated monocytes to directly evaluate the effect of CCR2+ monocytes on endothelial cell cytotoxicity in diabetes. Ex vivo incubation of purified monocytes isolated from WT mice after 2 months of diabetes resulted in significantly more endothelial cell death compared with monocytes harvested from nondiabetic WT controls. In contrast, monocytes isolated from diabetic Ccr2−/− mice resulted in significantly less endothelial cell death than monocytes harvested from diabetic WT mice (Fig. 6b). This data suggests that CCR2+ monocytes (classical monocytes) contribute to endothelial cell death in diabetic retinas.
Inflammatory monocytes mediate diabetes-induced increase in retinal capillary degeneration
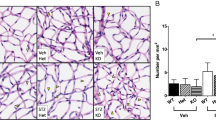
Retinal capillary degeneration is one of the most clinically meaningful endpoints of diabetic retinopathy that develop in rodents [24, 43, 44]. As previously reported, diabetes of 8 months in duration significantly increased the number of degenerated capillaries in retinas of WT mice compared with that in nondiabetic WT controls (Fig. 7). In contrast, diabetic mice deficient in Ccr2 were protected from the retinal capillary degeneration compared with diabetic WT controls (Fig. 7). Since retinal capillary degeneration in diabetic Ccr2−/− mice was significantly higher than that in nondiabetic Ccr2−/− mice, however, the data suggest that CCR2+ monocytes are not the only determinants of retinal capillary degeneration in diabetes.

Effects of streptozotocin-induced diabetes on capillary degeneration in retinas from WT and Ccr2−/− mice. WT mice diabetic for 32 weeks developed significantly more retinal capillary loss than nondiabetic controls (a). Ccr2 deletion resulted in significantly less retinal capillary loss in diabetes compared with that in diabetic WT mice, but was slightly increased when compared with nondiabetic Ccr2−/− mice. Data are graphed as degenerated capillaries per unit area of retina (b). White arrows indicate acellular capillaries. Scale bar, 50 μm. Data are presented as mean ± SD, n=6 per group. *p≤0.05, **p≤0.01, ***p≤0.001. N, nondiabetic; D, diabetic
Diabetes-induced increase in retinal superoxide, leucocyte-mediated endothelial cell cytotoxicity and retinal capillary degeneration is mediated by CCR2+ monocytes
Although CCR2 is expressed in myeloid cells in nondiabetic animals, it is conceivable that diabetes might result in induction of the receptor elsewhere. To further test the contribution of Ccr2-containing myeloid cells in the development of the early stages of diabetic retinopathy, we generated chimeric mice lacking Ccr2 solely from bone marrow-derived cells. Thirty weeks after the induction of diabetes, diabetic control mice (nonirradiated WT mice and WT→WT mice) showed the expected significant increase in retinal superoxide, leucocyte-mediated killing of endothelial cells, and capillary degeneration (Fig. 8a-c), showing that the irradiation itself did not alter the diabetes-induced pathogenic process that contributes to diabetic retinopathy. In contrast, retinal levels of superoxide, ex vivo killing of retinal endothelial cells by leucocytes, and capillary degeneration were significantly inhibited in diabetic Ccr2−/− chimeras (Ccr2−/−→ WT) (Fig. 8a-c).

Diabetes-induced retinal superoxide (a), leucocyte-mediated endothelial cell (EC) cytotoxicity (b) and degeneration of retinal capillaries (c) are significantly inhibited in chimeric mice lacking Ccr2 (Ccr2→WT), but not in WT→WT chimeric mice. Total duration of diabetes was 30 weeks. Data are presented as mean ± SD, n=4–10 per group. Green symbols indicate WT mice and red symbols indicate Ccr2−/− mice. Solid symbols indicate nondiabetic mice and empty symbols indicate diabetic mice. *p≤0.01, **p≤0.01, ***p≤0.001. RLU, relative luminescence units
CCR2+ monocytes mediate diabetes-induced reductions of ERG b-wave and spatial frequency threshold, but do not affect contrast sensitivity
Diabetes impairs visual function as assessed by ERG or psychophysical tests, including visual acuity and contrast sensitivity. In this study, we used ERG, spatial frequency threshold and contrast sensitivity to investigate the role of CCR2+ monocytes in diabetes-induced visual dysfunction. Consistent with previous studies, 2 months of diabetes significantly decreased ERG b-wave amplitudes (Fig. 9b), spatial frequency threshold (Fig. 9c) and contrast sensitivity (Fig. 9d) in WT mice. Ccr2 deficiency inhibited the diabetes-induced defects in b-wave amplitude and partially inhibited spatial frequency threshold but had no significant effect on contrast sensitivity (Fig. 9).

Visual function in nondiabetic and diabetic WT and Ccr2−/− mice. (a, b) ERG a-wave and b-wave amplitudes recorded at 8–10 weeks of diabetes (5 months of age) at increasing light intensities. ERG b-wave amplitudes were significantly reduced in diabetic WT mice, and Ccr2 deletion further inhibited diabetes-induced b-wave amplitudes in diabetic Ccr2−/− mice. (c) Spatial frequency threshold was significantly reduced in diabetic WT mice, and the lack of Ccr2 partially inhibited diabetes-induced spatial frequency threshold. (d) Contrast sensitivity was impaired in diabetic WT and Ccr2−/− mice. Data shown as mean ± SEM (n=16–20 eyes). *p≤0.05; ** p≤0.01; ***p≤0.001. N, nondiabetic; D, diabetic
Discussion
Leucocytes play a crucial role in diabetes-induced retinal capillary loss [4, 45, 46]. However, leucocytes comprise a diverse group of cells that originate from bone marrow but differ greatly with regard to life span and action. Determining the leucocyte subtypes involved in the pathogenesis of diabetic retinopathy may help design a novel therapeutic approach to inhibit the retinopathy.
Monocytes, which are a major subpopulation of leucocytes, are part of the innate immune system, where they play a critical role in surveying peripheral tissues and maintaining endothelial cells integrity. However, in some instances, they can contribute to disease development and progression [47,48,49]. In both humans and mice, monocytes are heterogeneous and have been further divided into several subsets differentially expressing chemokine receptors. CCR2 has been shown to be expressed mainly by classical monocytes (CCR2high) [12, 15, 16, 50, 51] (the nonclassical subset does not express CCR2; CCR2low), although other leucocyte cells have also been reported to express CCR2 to a lesser extent. The infiltration of inflammatory monocytes was shown to be CCR2-dependent in inflammatory diseases, including several retinal disorders such as retinal injury [8], atrophic age-related macular degeneration [10] and photoreceptor degeneration in models of retinitis pigmentosa [7]. CCR2 is the chemokine receptor for monocyte chemoattractant protein-1 (MCP-1; also known as C-C chemokine ligand 2, CCL2) [50].
It has been shown that the entrance of monocytes into the retina relies on activation of the MCP-1/CCR2 axis [10, 52,53,54]. In this regard, Sennlaub et al showed that Ccl2 deficiency (either in Ccl2−/− mice or as a result of treatment with a pharmacologic inhibitor [RS 102895]) inhibits inflammatory monocyte recruitment to the retina [10]. With regard to diabetic retinopathy, Rangasamy et al showed that the expression of MCP-1 was increased in the retina of diabetic rodents [9], that this expression was accompanied by greater-than-normal numbers of perivascular monocytes in the retina, and that the deletion of Mcp-1 (also known as Ccl2) resulted in significant reduction of monocyte infiltration [9]. Whether or not monocytes contribute to the development of the retinopathy, however, has not been clear. In this study, we focused on the effect of CCR2+ monocytes on molecular changes that are characteristic of early stages of diabetic retinopathy (such as oxidative stress and inflammation) and retinal capillary degeneration.
Consistent with previous reports, under steady state condition the majority of peripheral blood monocytes express CCR2 (more than 90%, Fig. 1) [12, 15]. Most neutrophils do not express CCR2 under normal conditions (ESM Fig. 2) [55]. Mice genetically deficient in Ccr2 had substantially fewer CCR2+ monocytes in the peripheral blood [15, 55], however, neutrophil number was not affected. These results suggest that Ccr2−/− mice could be used as a mouse model to study the role of CCR2+ monocytes in the development of diabetic retinopathy.
In the early stages of diabetic retinopathy, an increased number of leucocytes adhere to retinal blood vessels in both diabetic patients and animals [4, 11, 56], potentially contributing to the retinal capillary occlusion that has been observed in diabetes. Consistent with previous reports, we found that diabetic WT mice showed a significant increase in leucostasis in retinal microvessels, and leucocytes from those diabetic WT mice caused more cytotoxicity to retinal endothelial cells. In contrast, leucocytes isolated from diabetic Ccr2−/− mice and from chimeric mice lacking Ccr2 only from bone marrow did not cause this leucocyte-mediated endothelial cell death. Since nonclassical monocytes are not significantly affected by Ccr2 deletion (Fig. 1), we suggest that CCR2+ monocytes play an important role in retinal leucostasis and leucocyte-mediated endothelial cell cytotoxicity in diabetic retinopathy.
In order to further investigate if diabetes-induced leucostasis and leucocyte-mediated endothelial cell death could be attributed to CCR2+ monocytes, we immuno-isolated monocytes from bone marrow using magnetic bead depletion. Co-culture of mRECS with immuno-isolated monocytes from diabetic WT mice expressing CCR2 resulted in a significant increase in monocyte-mediated endothelial cell cytotoxicity, whereas endothelial cell death was inhibited when cultured with monocytes from diabetic Ccr2-deficient mice. These results further demonstrate that CCR2+ monocytes play an important role in diabetes-induced leucocyte-mediated endothelial cell cytotoxicity.
Oxidative stress and retinal expression of proinflammatory mediators have been implicated in the development of diabetic retinopathy, and inhibition of retinal oxidative stress in diabetes by antioxidants or overexpression of antioxidant enzymes has been reported to preserve the retinal vasculature in diabetes [41, 57, 58]. Likewise, the inhibition of inflammation has been shown to preserve the retinal vasculature despite hyperglycaemia [46, 59,60,61], and deletion or inhibition of certain inflammatory proteins or cytokines such as ICAM1, inducible nitric oxide synthase (iNOS) and IL1β inhibited diabetes-induced degeneration of retinal capillaries in diabetic animals [62,63,64]. Our data showed that retinal superoxide and expression of inflammatory genes were inhibited in Ccr2−/− mice, suggesting that inflammatory monocytes are implicated in these diabetes-induced abnormalities.
We previously demonstrated a critical role of neutrophils in the development of the early stages of diabetic retinopathy [5, 65]. The finding now that both neutrophils and monocytes contribute to early diabetic retinopathy implicates innate immunity in this disease process. Both neutrophils and monocytes are part of the innate immune system, the first line of defence against toxins, and consist of physical, chemical and cellular defences against various aggressions to tissues. Several reports have shown that both cells are absolutely required for an adequate immune response, and the depletion of one cell population affects the infiltration and/or the function of the other cell [55, 66,67,68,69,70]. For instance, neutrophils attract classical monocytes and facilitate their transmigration, and depletion of neutrophils resulted in decreased classical monocyte infiltration [70]. Likewise, monocytes promote neutrophil accumulation [55]. This new vantage point offers new potential therapeutic targets at which retinopathy might be inhibited.
A CCR2/5 inhibitor has been administered to patients with diabetic macular oedema (DME) [22], but results of that trial indicated that the drug was inferior to monthly ranibizumab (a blood vessel growth inhibitor) with respect to improvement of best corrected visual acuity. The reason for the differing conclusions from this patient study and our pre-clinical study are not clear, but it is possible that the duration of drug administration to patients was too short to demonstrate an effect (only 12 weeks). In addition, it seems likely that the degree of CCR2 inhibition played an important role; Ccr2 expression in our Ccr2−/− mice was totally inhibited, whereas the drug administered to patients only blocked the receptor by an unknown amount. Perhaps CCR2 inhibition has a stronger effect on capillary degeneration (studied in the present report) than it has on capillary permeability (studied in the clinical report). Additional studies will be required to determine if more potent inhibitors of CCR2 offer meaningful clinical benefit to patients with diabetes.
Our major findings showed that the whole-body deletion of Ccr2 (Ccr2−/− mice) and chimeric mice lacking Ccr2 only from bone marrow-derived cells in mice resulted in the inhibition of diabetes-induced increases of retinal superoxide, upregulation of proinflammatory genes (Inos and Icam1), leucostasis, leucocyte- and monocyte-mediated cytotoxicity against retinal endothelial cells, and most importantly, retinal capillary degeneration. The absence of inflammatory monocytes also mitigated diabetes-induced visual dysfunction, notably in ERG b-wave. These results demonstrate that monocytes (and innate immunity) contribute to at least the vascular lesions of early diabetic retinopathy.
Data availability
The data generated and presented in the present study are available from the corresponding author on reasonable request.
Abbreviations
- CCR2:
-
CC chemokine receptor 2
- ERG:
-
Electroretinogram
- GCL:
-
Ganglion cell layer
- ICAM1:
-
Intercellular adhesion molecule 1
- mREC:
-
Mouse retinal endothelial cell
- NFL:
-
Nerve fibre layer
- SD-OCT:
-
Spectral domain optical coherence tomography
- WT:
-
Wild-type
References
Joussen AM, Murata T, Tsujikawa A, Kirchhof B, Bursell SE, Adamis AP (2001) Leukocyte-mediated endothelial cell injury and death in the diabetic retina. Am J Pathol 158(1):147–152. https://doi.org/10.1016/S0002-9440(10)63952-1
Joussen AM, Poulaki V, Le ML et al (2004) A central role for inflammation in the pathogenesis of diabetic retinopathy. FASEB J 18:1450–1452. https://doi.org/10.1096/fj.03-1476fje
Liu H, Tang J, Du Y et al (2019) Transducin1, phototransduction and the development of early diabetic retinopathy. Investig Ophthalmol Vis Sci 60(5):1538–1546. https://doi.org/10.1167/iovs.18-26433
Veenstra AA, Tang J, Kern TS (2013) Antagonism of CD11b with Neutrophil Inhibitory Factor (NIF) inhibits vascular lesions in diabetic retinopathy. PloS One 8(10):e78405. https://doi.org/10.1371/journal.pone.0078405
Li G, Veenstra AA, Talahalli RR et al (2012) Marrow-derived cells regulate the development of early diabetic retinopathy and tactile allodynia in mice. Diabetes 61:3294–3303. https://doi.org/10.2337/db11-1249
Li G, Tang J, Du Y, Lee CA, Kern TS (2011) Beneficial effects of RAGE-Ig fusion protein on early diabetic retinopathy and tactile allodynia. Mol Vis 17:3156–3165
Guo C, Otani A, Oishi A et al (2012) Knockout of ccr2 alleviates photoreceptor cell death in a model of retinitis pigmentosa. Exp Eye Res 104:39–47. https://doi.org/10.1016/j.exer.2012.08.013
Ma W, Zhang Y, Gao C, Fariss NR, Tam J, Wong TW (2017) Monocyte infiltration and proliferation reestablish myeloid cell homeostasis in the mouse retina following retinal pigment epithelial cell injury. Sci Rep 7:8433. https://doi.org/10.1038/s41598-017-08702-7
Rangasamy S, McGuire GP, Nitta Franco C, Monickaraj F, Oruganti RS, Das A (2014) Chemokine mediated monocyte trafficking into the retina: role of inflammation in alteration of the blood-retinal barrier in diabetic retinopathy. PloS One 9:e108508. https://doi.org/10.1371/journal.pone.0108508
Sennlaub F, Auvynet C, Calippe B et al (2013) CCR2(+) monocytes infiltrate atrophic lesions in age-related macular disease and mediate photoreceptor degeneration in experimental subretinal inflammation in Cx3cr1 deficient mice. EMBO Mol Med 5:1775–1793. https://doi.org/10.1002/emmm.201302692
Schroder S, Palinski W, Schmid-Schonbein GW (1991) Activated monocytes and granulocytes, capillary nonperfusion, and neovascularization in diabetic retinopathy. Am J Pathol 139(1):81–100
Geissmann F, Jung S, Littman RD (2003) Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity 19:71–82. https://doi.org/10.1016/S1074-7613(03)00174-2
Guilliams M, Mildner A, Yona S (2018) Developmental and functional heterogeneity of monocytes. Immunity 49:595–613. https://doi.org/10.1016/j.immuni.2018.10.005
Palframan TR, Jung S, Cheng G et al (2001) Inflammatory chemokine transport and presentation in HEV. J Exp Med 194:1361–1374. https://doi.org/10.1084/jem.194.9.1361
Fujimura N, Xu B, Dalman J, Deng H, Aoyama K, Dalman LR (2015) CCR2 inhibition sequesters multiple subsets of leukocytes in the bone marrow. Sci Rep 5:11664. https://doi.org/10.1038/srep11664
Mack M, Cihak J, Simonis C et al (2001) Expression and characterization of the chemokine receptors CCR2 and CCR5 in mice. J Immunol 166:4697–4704. https://doi.org/10.4049/jimmunol.166.7.4697
Kratofil RM, Kubes P, Deniset JF (2017) Monocyte conversion during inflammation and injury. Arterioscler Thromb Vasc Biol 37(1):35–42. https://doi.org/10.1161/ATVBAHA.116.308198
Serbina VN, Pamer GE (2006) Monocyte emigration from bone marrow during bacterial infection requires signals mediated by chemokine receptor CCR2. Nat Immunol 7:311–317. https://doi.org/10.1038/ni1309
Sunderkötter C, Nikolic T, Dillon JM et al (2004) Subpopulations of mouse blood monocytes differ in maturation stage and inflammatory response. J Immunol 172:4410–4417. https://doi.org/10.4049/jimmunol.172.7.4410
Tsou C-L, Peters W, Si Y et al Monocyte chemoattractant protein-1 (MCP-1/CCL2) in diabetic retinopathy: latest evidence and clinical considerations. J Clin Investig 117:902–909. https://doi.org/10.1172/JCI29919
Kuziel AW, Morgan SJ, Dawson TC et al (1997) Severe reduction in leukocyte adhesion and monocyte extravasation in mice deficient in CC chemokine receptor 2. Proc Natl Acad Sci USA 94:12053–12058. https://doi.org/10.1073/pnas.94.22.12053
Monickaraj F, Oruganti SR, McGuire P, Das A (2021) A potential novel therapeutic target in diabetic retinopathy: a chemokine receptor (CCR2/CCR5) inhibitor reduces retinal vascular leakage in an animal model. Graefe’s Arch Clin Exp Ophthalmol 259(1):93–100. https://doi.org/10.1007/s00417-020-04884-5
Orban T, Leinonen H, Getter T et al (2018) A combination of G protein-coupled receptor modulators protects photoreceptors from degeneration. J Pharmacol Exp Ther 364(2):207–220. https://doi.org/10.1124/jpet.117.245167
Saadane A, Lessieur EM, Du Y, Liu H, Kern TS (2020) Successful induction of diabetes in mice demonstrates no gender difference in development of early diabetic retinopathy. PloS One 15:e0238727. https://doi.org/10.1371/journal.pone.0238727
Saadane A, Mast N, Charvet CD et al (2014) Retinal and nonocular abnormalities in Cyp27a1(-/-)Cyp46a1(-/-) mice with dysfunctional metabolism of cholesterol. Am J Pathol 184:2403–2419. https://doi.org/10.1016/j.ajpath.2014.05.024
Lee CA, Li G, Patel MD et al (2013) Diabetes-induced impairment in visual function in mice: contributions of p38 MAPK, RAGE, leukocytes, and aldose reductase. Investig Ophthalmol Vis Sci 93:135–143
Liu H, Tang J, Lee CA, Kern TS (2015) Metanx and early stages of diabetic retinopathy. Investig Ophthalmol Vis Sci 56:647–653. https://doi.org/10.1167/iovs.14-15220
Prusky GT, Alam NM, Beekman S, Douglas RM (2004) Rapid quantification of adult and developing mouse spatial vision using a virtual optomotor system. Investig Ophthalmol Vis Sci 45:4611–4616. https://doi.org/10.1167/iovs.04-0541
Du Y, Cramer M, Lee CA et al (2015) Adrenergic and serotonin receptors affect retinal superoxide generation in diabetic mice: relationship to capillary degeneration and permeability. FASEB J 29(5):2194–2204. https://doi.org/10.1096/fj.14-269431
Veenstra A, Liu H, Lee CA, Du Y, Tang J, Kern TS (2015) Diabetic retinopathy: retina-specific methods for maintenance of diabetic rodents and evaluation of vascular histopathology and molecular abnormalities. Curr Protoc Mouse Biol 5(3):247–270. https://doi.org/10.1002/9780470942390.mo140190
Su X, Sorenson CM, Sheibani N (2003) Isolation and characterization of murine retinal endothelial cells. Mol Vis 9:171–178
León B, Martínez del Hoyo G, Parrillas V et al (2004) Dendritic cell differentiation potential of mouse monocytes: monocytes represent immediate precursors of CD8- and CD8+ splenic dendritic cells. Blood 103(7):2668–2676. https://doi.org/10.1182/blood-2003-01-0286
Asnaghi V, Gerhardinger C, Hoehn T, Adeboje A, Lorenzi M (2003) A role for the polyol pathway in the early neuroretinal apoptosis and glial changes induced by diabetes in the rat. Diabetes 52(2):506–511. https://doi.org/10.2337/diabetes.52.2.506
Feit-Leichman RA, Kinouchi R, Takeda M et al (2005) Vascular damage in a mouse model of diabetic retinopathy: relation to neuronal and glial changes. Investig Ophthalmol Vis Sci 46:4281–4287. https://doi.org/10.1167/iovs.04-1361
Zheng L, Howell SJ, Hatala DA, Huang K, Kern TS (2007) Salicylate-based anti-inflammatory drugs inhibit the early lesion of diabetic retinopathy. Diabetes 56(2):337–345. https://doi.org/10.2337/db06-0789
Sasaki M, Ozawa Y, Kurihara T et al (2010) Neurodegenerative influence of oxidative stress in the retina of a murine model of diabetes. Diabetologia 53(5):971–979. https://doi.org/10.1007/s00125-009-1655-6
Al-Shabrawey M, Bartoli M, El-Remessy A et al (2008) Role of NADPH oxidase and STAT3 in statin-mediated protection against diabetic retinopathy. Investig Ophthalmol Vis Sci 49:3231–3238. https://doi.org/10.1167/iovs.08-1754
Al-Shabrawey M, Rojas M, Sanders T et al (2008) Role of NADPH oxidase in retinal vascular inflammation. Investig Ophthalmol Vis Sci 49:3239–3244. https://doi.org/10.1167/iovs.08-1755
Berkowitz BA, Bredell BX, Davis C, Samardzija M, Grimm C, Roberts R (2015) Measuring in vivo free radical production by the outer retina. Investig Ophthalmol Vis Sci 56(13):7931–7938. https://doi.org/10.1167/iovs.15-18420
Du Y, Veenstra A, Palczewski K, Kern TS (2013) Photoreceptor cells are major contributors to diabetes-induced oxidative stress and local inflammation in the retina. Proc Natl Acad Sci USA 110:16586–16591. https://doi.org/10.1073/pnas.1314575110
Kanwar M, Chan PS, Kern TS, Kowluru RA (2007) Oxidative damage in the retinal mitochondria of diabetic mice: possible protection by superoxide dismutase. Investig Ophthalmol Vis Sci 48(8):3805–3811. https://doi.org/10.1167/iovs.06-1280
Kowluru RA (2001) Diabetes-induced elevations in retinal oxidative stress, protein kinase C and nitric oxide are interrelated. Acta Diabetol 38(4):179–185. https://doi.org/10.1007/s592-001-8076-6
Barber AJ, Antonetti DA, Kern TS et al (2005) The Ins2Akita mouse as a model of early retinal complications in diabetes. Investig Ophthalmol Vis Sci 46(6):2210–2218. https://doi.org/10.1167/iovs.04-1340
Zhang JZ, Xi X, Gao L, Kern TS (2007) Captopril inhibits capillary degeneration in the early stages of diabetic retinopathy. Curr Eye Res 32:883–889. https://doi.org/10.1080/02713680701584123
Chibber R, Ben-Mahmud BM, Chibber S, Kohner EM (2007) Leukocytes in diabetic retinopathy. Curr Diabetes Rev 3(1):3–14. https://doi.org/10.2174/157339907779802139
Tang J, Lee CA, Du Y et al (2013) MyD88-dependent pathways in leukocytes affect the retina in diabetes. PloS One 8:e68871. https://doi.org/10.1371/journal.pone.0068871
Bajpai G, Bredemeyer A, Li W et al (2019) Tissue resident CCR2- and CCR2+ cardiac macrophages differentially orchestrate monocyte recruitment and fate specification following myocardial injury. Circ Res 124(2):263–278. https://doi.org/10.1161/CIRCRESAHA.118.314028
Evans TA, Barkauskas DS, Myers JT et al (2014) High-resolution intravital imaging reveals that blood-derived macrophages but not resident microglia facilitate secondary axonal dieback in traumatic spinal cord injury. Exp Neurol 254:109–120. https://doi.org/10.1016/j.expneurol.2014.01.013
Wattananit S, Tornero D, Graubardt N et al (2016) Monocyte-derived macrophages contribute to spontaneous long-term functional recovery after stroke in mice. J Neurosci 36(15):4182–4195. https://doi.org/10.1523/JNEUROSCI.4317-15.2016
Gautier LE, Jakubzick C, Randolph JG (2009) Regulation of the migration and survival of monocyte subsets by chemokine receptors and its relevance to atherosclerosis. Arterioscler Thromb Vasc Biol 29:1412–1418. https://doi.org/10.1161/ATVBAHA.108.180505
Tacke F, Randolph JG (2006) Migratory fate and differentiation of blood monocyte subsets. Immunobiology 211:609–618. https://doi.org/10.1016/j.imbio.2006.05.025
Feng C, Wang X, Liu T, Zhang M, Xu G, Ni Y (2017) Expression of CCL2 and its receptor in activation and migration of microglia and monocytes induced by photoreceptor apoptosis. Mol Vis 23:765–777
Roubeix C, Dominguez E, Raoul W et al (2019) Mo-derived perivascular macrophage recruitment protects against endothelial cell death in retinal vein occlusion. J Neuroinflammation 16:1547. https://doi.org/10.1186/s12974-019-1547-8
Taghavi Y, Hassanshahi G, Kounis GN, Koniari I, Khorramdelazad H (2019) Monocyte chemoattractant protein-1 (MCP-1/CCL2) in diabetic retinopathy: latest evidence and clinical considerations. J Cell Commun Signal 13:451–462. https://doi.org/10.1007/s12079-018-00500-8
Dong N, Li X, Xiao L, Yu W, Wang B, Chu L (2012) Upregulation of retinal neuronal MCP-1 in the rodent model of diabetic retinopathy and its function in vitro. Investig Ophthalmol Vis Sci 53:7567–7575. https://doi.org/10.1167/iovs.12-9446
Maus UA, Waelsch K, Kuziel WA et al (2003) Monocytes are potent facilitators of alveolar neutrophil emigration during lung inflammation: role of the CCL2-CCR2 axis. J Immunol 170(6):3273–3278. https://doi.org/10.4049/jimmunol.170.6.3273
McLeod DS, Lefer DJ, Merges C, Lutty GA (1995) Enhanced expression of intercellular adhesion molecule-1 and P-selectin in the diabetic human retina and choroid. Am J Pathol 147:642–653
Berkowitz BA, Gradianu M, Bissig D, Kern TS, Roberts R (2009) Retinal ion regulation in a mouse model of diabetic retinopathy: natural history and the effect of Cu/Zn superoxide dismutase overexpression. Investig Ophthalmol Vis Sci 50:2351–2358. https://doi.org/10.1167/iovs.08-2918
Kowluru RA, Tang J, Kern TS (2001) Abnormalities of retinal metabolism in diabetes and experimental galactosemia. VII. Effect of long-term administration of antioxidants on the development of retinopathy. Diabetes 50(8):1938–1942. https://doi.org/10.2337/diabetes.50.8.1938
Talahalli R, Zarini S, Tang J et al (2013) Leukocytes regulate retinal capillary degeneration in the diabetic mouse via generation of leukotrienes. J Leukoc Biol 93:135–143. https://doi.org/10.1189/jlb.0112025
Tang J, Kern TS (2011) Inflammation in diabetic retinopathy. Prog Retin Eye Res 30(5):343–358. https://doi.org/10.1016/j.preteyeres.2011.05.002
Tian P, Ge H, Liu H et al (2013) Leukocytes from diabetic patients kill retinal endothelial cells: effects of berberine. Mol Vis 19:2092–2105
Joussen AM, Poulaki V, Qin W et al (2002) Retinal vascular endothelial growth factor induces intercellular adhesion molecule-1 and endothelial nitric oxide synthase expression and initiates early diabetic retinal leukocyte adhesion in vivo. Am J Pathol 160(2):501–509. https://doi.org/10.1016/S0002-9440(10)64869-9
Vincent JA, Mohr S (2007) Inhibition of caspase-1/interleukin-1beta signaling prevents degeneration of retinal capillaries in diabetes and galactosemia. Diabetes 56(1):224–230. https://doi.org/10.2337/db06-0427
Zheng L, Kern T (2009) Role of nitric oxide, superoxide, peroxynitrite and poly(ADP-ribose) polymerase in diabetic retinopathy. Front Biosci 14:3974–3987. https://doi.org/10.2741/3505
Liu H, Lessieur EM, Saadane A, Lindstrom SI, Taylor PR, Kern TS (2019) Neutrophil elastase contributes to the pathological vascular permeability characteristic of diabetic retinopathy. Diabetologia 62(12):2365–2374. https://doi.org/10.1007/s00125-019-04998-4
Kantari C, Pederzoli-Ribeil M, Witko-Sarsat V (2008) The role of neutrophils and monocytes in innate immunity. Contrib Microbiol 15:118–146. https://doi.org/10.1159/000136335
Maus U, von Grote K, Kuziel WA et al (2002) The role of CC chemokine receptor 2 in alveolar monocyte and neutrophil immigration in intact mice. Am J Respir Crit Care Med 166(3):268–273. https://doi.org/10.1164/rccm.2112012
Soehnlein O, Lindbom L, Weber C (2009) Mechanisms underlying neutrophil-mediated monocyte recruitment. Blood 114(21):4613–4623. https://doi.org/10.1182/blood-2009-06-221630
Soehnlein O, Zernecke A, Eriksson EE et al (2008) Neutrophil secretion products pave the way for inflammatory monocytes. Blood 112(4):1461–1471. https://doi.org/10.1182/blood-2008-02-139634
Acknowledgements
We thank J. Atwood, the flow core facility manager (Flow cytometry core institute for immunology at University California Irvine, Irvine, USA), for helping us with flow cytometry data analysis and thank Chieh Allen Lee (Case Western Reserve University, Cleveland, USA) and Jianying Kiser (University California Irvine, Irvine, USA) for management of the diabetic animal colony.
Authors’ relationship and activities
The authors declare that there are no relationships or activities that might bias, or be perceived to bias, their work.
Contribution statement
AS designed the experiments, acquired the data, analysed the data and wrote the manuscript. AAV, MSM, JT, YD, FAE and EML acquired the data, were involved in the analysis and interpretation of the data and reviewed the manuscript. EP was involved in analysis and interpretation of data, and reviewed/edited the manuscript. TSK designed the experiments, acquired the data, analysed the data and reviewed/edited the manuscript. All the authors approved the final version of the manuscript to be published. TSK is the guarantor of this work.
Funding
This work was supported by NIH grants EY022938 and R24 EY024864, and BX003604 from the Department of Veterans Affairs. TSK was the recipient of a Research Career Scientist award from the Department of Veterans Affairs. The authors acknowledge departmental support from an RPB (Research to Prevent Blindness) unrestricted grant (University of California, Irvine).
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
ESM
(PDF 938 kb)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Saadane, A., Veenstra, A.A., Minns, M.S. et al. CCR2-positive monocytes contribute to the pathogenesis of early diabetic retinopathy in mice. Diabetologia 66, 590–602 (2023). https://doi.org/10.1007/s00125-022-05860-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-022-05860-w