
Abstract
Aims/hypothesis
The clinical importance of fat deposition in the liver and pancreas is increasingly recognised. However, to what extent deposition of fat in these two depots is affected by intermediate variables is unknown. The aim of this work was to conduct a mediation analysis with a view to uncovering the metabolic traits that underlie the relationship between liver fat and intrapancreatic fat deposition (IPFD) and quantifying their effect.
Methods
All participants underwent MRI/magnetic resonance spectroscopy on the same 3.0 T scanner to determine liver fat and IPFD. IPFD of all participants was quantified manually by two independent raters in duplicate. A total of 16 metabolic traits (representing markers of glucose metabolism, incretins, lipid panel, liver enzymes, pancreatic hormones and their derivatives) were measured in blood. Mediation analysis was conducted, taking into account age, sex, ethnicity and BMI. Significance of mediation was tested by computing bias-corrected bootstrap CIs with 5000 repetitions.
Results
A total of 353 individuals were studied. Plasma glucose, HDL-cholesterol and triacylglycerol mediated 6.8%, 17.9% and 24.3%, respectively, of the association between liver fat and IPFD. Total cholesterol, LDL-cholesterol, alanine aminotransferase, aspartate aminotransferase, alkaline phosphatase, γ-glutamyl transpeptidase, insulin, glucagon, amylin, C-peptide, HbA1c, glucagon-like peptide-1 and gastric inhibitory peptide did not mediate the association between liver fat and IPFD.
Conclusions/interpretation
At least one-quarter of the association between liver fat and IPFD is mediated by specific blood biomarkers (triacylglycerol, HDL-cholesterol and glucose), after accounting for potential confounding by age, sex, ethnicity and BMI. This unveils the complexity of the association between the two fat depots and presents specific targets for intervention.
Graphical abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.

Introduction
While the dangers of excess body fat in general became well appreciated in the 20th century, the risks associated with excess ectopic fat deposition have been progressively brought to the fore in the 21st century. In particular, excess deposition of fat in the parenchymal cells of the liver (fatty liver disease) has emerged as the most common disorder of the liver in the western world and eastern world alike [1,2,3,4,5]. This fat deposition is a growing cause of cirrhosis, hepatocellular cancer and end-stage liver disease, which may require liver transplantation. Fatty liver disease is also a risk factor for CVD (regardless of traditional risk factors such as arterial hypertension) and chronic kidney disease [5]. Another parenchymal organ often bedevilled by excess fat deposition is the pancreas. Fatty pancreas disease is the most common disorder of the pancreas and is a harbinger of pancreatitis and pancreatic cancer [6,7,8]. Pancreatic cancer is one of the most lethal diseases, with the mortality rate being similar to incidence (seven and eight cases per 100,000 person-years, respectively) [9]. Pancreatitis has an incidence rate of 43 cases per 100,000 person-years and, while mortality from this disease is relatively low, can result in numerous new-onset metabolic sequelae such as post-pancreatitis diabetes mellitus, exocrine pancreatic dysfunction and osteopathy [9,10,11,12]. The burden of both pancreatic cancer and pancreatitis is projected to increase substantially by 2050 [13]. The liver and the pancreas share a common developmental origin and numerous studies over the past decade have shown a significant association between liver fat and intrapancreatic fat deposition (IPFD) [14]. While early studies on the topic should be interpreted with caution because of their use of ultrasound (which is semi-quantitative and operator-dependent) and small sample size [15,16,17], the association between liver fat and IPFD was conclusively shown in a large 2014 population-based study that employed chemical shift-encoded MRI (the gold standard for quantifying IPFD non-invasively) [18].
Now that scientific knowledge in the area is no longer in its rudimentary stage, it behoves researchers to develop a fine-grained understanding of the mechanisms that underlie the link between liver fat and IPFD. While BMI and sex are obvious confounders of the association between the two entities, the epistemological challenge is to establish exactly how these entities become intertwined [19]. Mediation analysis is positioned well to test hypotheses about the mechanisms that are at work as it determines the extent to which a potential causal variable influences an outcome, through an intermediate variable (also called ‘mediator’). Specifically, this path-analytic methodological framework enables partitioning of the influence of liver fat on IPFD into indirect (i.e. through the mediator of interest) and direct (i.e. through other mechanisms) components and allows the quantification of both (i.e. estimating the proportion mediated) [20]. As with any causal inference method, mediation analysis requires assumptions to be made about the causality of the effects in the mediation model. Specifically, it is assumed that changes in liver fat cause changes in metabolic traits and that changes in metabolic traits cause changes in intrapancreatic fat. The biological plausibility of these assumptions is strong in regard to liver enzymes, lipid panel, markers of glucose metabolism, incretins and pancreatic hormones. Fatty liver disease is widely regarded as the most common cause of elevated liver enzymes [2, 5, 21]. Liver enzymes were significantly associated with IPFD in our 2017 meta-analysis of biomarkers of IPFD (encompassing nearly 12,000 individuals from 17 observational studies) that informed the design of the present study [22]. Notably, γ-glutamyl transferase had the strongest association of all the biomarkers studied (though it was investigated in two studies only). Lipid metabolism is dysregulated in fatty liver disease as increased liver fat results in hepatic overproduction of VLDL particles and dysregulated clearance of lipoproteins from the circulation [1, 23]. Lipid panel was significantly associated with IPFD in the above-mentioned 2017 meta-analysis, with triacylglycerol being the most frequently investigated biomarker [22]. A Mendelian randomisation study demonstrated that liver fat causes type 2 diabetes (defined based on HbA1c and/or fasting plasma glucose levels) [24]. Hyperglycaemia in the non-diabetic range (defined based on HbA1c and/or fasting plasma glucose levels) was an independent predictor of increased IPFD after 5 years of follow-up in a longitudinal cohort study of individuals without diabetes [25]. Liver fat is almost universally associated with insulin resistance, with resulting changes in secretion of both pancreatic hormones and incretins [5, 26]. Pancreatic hormones were significantly associated with IPFD (with no heterogeneity between the studies that investigated insulin) in the above-mentioned 2017 meta-analysis [22].
The present study aimed to disentangle the relationships between the above metabolic traits and liver fat and IPFD, using mediation analysis.
Methods
Study design
This cross-sectional study, with the enrolment of four individual cohorts [27], was conducted at the University of Auckland (Auckland, New Zealand) and received ethical approval by the New Zealand Health and Disability Ethics Committee (13/STH/182, 16/STH/23, 17/NTA/172, 18/NTB/1). People aged >18 years residing in Auckland were recruited following written informed consent. They had no personal history of acute infectious or inflammatory disorders requiring medical treatment or evaluation in the preceding 6 months. Individuals were excluded if they had participated in a weight-loss programme, received dietetic support or education, undergone liver, pancreatic or bariatric surgery or organ transplantation, had chronic pancreatitis or any other pathology of the pancreas detectable on cross-sectional imaging, received any radiological or endoscopic intervention involving the liver or the pancreas, had malignancy, chronic liver disease or autoimmune disease, used systemic corticosteroids, had health issues that preclude undergoing MRI (e.g. end-stage renal failure, congestive heart failure, mental disorders), had metallic implants, heart pacemakers or other implantable electronic devices, or were pregnant or breastfeeding.
MRI measurements
Imaging protocol
A single 3.0 Tesla MAGNETOM Skyra scanner, VE 11A (Siemens, Erlangen, Germany) at the Centre for Advanced Magnetic Resonance Imaging at the University of Auckland (Auckland, New Zealand) was used to acquire abdominal images using the same protocol for all study participants. The protocol involved participants lying in the supine position, holding their breath for 11 s at end-expiration. Axial longitudinal relaxation time (T1)-weighted volumetric interpolated breath-hold examination Dixon sequence was applied with the following parameters: true form abdomen shim mode; field of view (FOV), 440 mm; basic resolution, 512; echo time (TE), 2.46 ms, 3.69 ms; repetition time (TR), 5.82 ms; flip angle, 9°; pixel bandwidth, 750 Hz; signal average, 1; slice thickness, 5 mm; field of view, 500 × 400 mm; matrix, 512 × 410; and partial Fourier and parallel imaging with a total acceleration factor of 2.8. Four types of images were generated: in-phase; out-of-phase; fat only; and water only.
Liver fat
Magnetic resonance spectroscopy (MRS) was used to determine liver fat. A single voxel (20 mm × 20 mm × 20 mm) was placed in the right lobe of the liver, away from the blood vessels and bile ducts and at least 10 mm away from the organ’s edge. Automated shimming was performed prior to signal acquisition to improve the homogeneity of B0, the main static magnetic field. Spectra were acquired using a free-breathing navigator-triggered spin-echo acquisition with the following parameters: TE, 33 ms; TR, 3000 ms; 50 averages. The acquisition time for each spectrum was 5 min. Both water-suppressed and non-water-suppressed spectra were taken, with the non-water-suppressed spectrum being the reference for liver fat quantification. Spectra were processed and analysed using the SIVIC software (University of California-San Francisco, USA). The fat fraction (%) was defined as the area under fat peak divided by area under fat and water peaks multiplied by 100 [28].
Intrapancreatic fat
IPFD was measured manually using a modified ‘MR-opsy’ technique, described in detail elsewhere [29]. Briefly, two candidate slices with clear visualisation of the pancreas were selected from a series of abdominal scans. Three regions of interest were placed in the head, body and tail regions of the pancreas to estimate IPFD. A thresholding range of 1–20% was applied to prevent the potential inclusion of non-parenchymal tissues (such as visceral fat, the main pancreatic duct, blood vessels) within the selected regions of interest [30]. IPFD was calculated, using ImageJ software (National Institutes of Health, USA), as the average pancreatic fat fraction of the two slices. Two raters measured IPFD independently for each participant and average values of two measurements were used for statistical analyses. Intra-class correlation was calculated to assess the inter-rater reliability. The inter-rater reliability was considered excellent if intra-class correlation was more than 0.90 [31].
Laboratory measurements
Venous blood samples were obtained from each participant at the time of their participation, after at least 8 h of fasting. These blood samples were centrifuged at 4000 g (4°C); plasma and serum were separated into aliquots and stored at −80°C until further use. Laboratory measurements were performed separately for each of the four individual cohorts, using the same laboratory methods. HbA1c was measured using a chromatography assay. Fasting plasma glucose was measured using a hexokinase colorimetric assay. Liver enzymes and lipid panel were analysed using standard methods in LabPlus, the tertiary referral medical laboratory of Auckland City Hospital (Auckland, New Zealand). Pancreatic hormones (insulin, glucagon, amylin [islet amyloid polypeptide]), their derivatives (C-peptide), and incretins (gastric inhibitory peptide [GIP], total glucagon-like peptide-1 [GLP-1]) were measured using the MILLIPLEX MAP human metabolic hormone magnetic bead panel based on the Luminex xMAP technology (Merck, Hesse, Germany) in line with the manufacturer’s instructions. Results were quantified based on fluorescent reporter signals recorded by the Luminex xPONENT software (MILLIPLEX Analyst 5.1). The intra-assay and inter-assay CV for all analytes was <10% and <15%, respectively.
Covariates
Anthropometric data (height and weight) of all participants were recorded to calculate BMI. All measurements were taken with participants wearing light clothing, and height and weight were measured in a standing position without shoes or headgear. Ethnicity was categorised as European White, Asian and Others.
Statistical analysis
Analyses were conducted using SPSS version 27.0 (IBM Corp., NY, USA) and SAS version 9.4 for Windows (SAS Institute, USA). Data were expressed as median and IQR or frequency. Data with skewed distribution were log-transformed when appropriate. A single-mediator model with single-level data for the mediation analysis was built to investigate whether the studied metabolic traits are mediators in the association between liver fat and IPFD [32]. Age, sex, ethnicity and BMI were treated as confounders. PROCESS macro for mediation analysis (https://processmacro.org/ version 3.4.1) was used to test statistical significance and magnitude of mediation in ordinary least-squares regression models, according to the method of Preacher and Hayes [33]. First, we calculated direct effect estimates of liver fat on IPFD (which included the exposure, confounders and mediator as independent variables). Then, we estimated the effect of liver fat on individual potential mediators. All the studied metabolic traits were considered as potential mediators. The indirect effect of potential mediators was then calculated by computing the product of the two regression coefficients of the potential mediators on liver fat and IPFD. The magnitude of indirect effect was calculated by dividing the coefficient of the indirect effect by the coefficient of the direct effect, according to the method of Preacher and Hayes [33].
Significance of mediation was tested by computing bias-corrected bootstrap CIs. Bootstrapping (a non-parametric resampling procedure) was used because it did not impose the assumption of normality of the sampling distribution. Bootstrapping involved repeated sampling from the dataset to estimate the indirect effect in each resampled dataset. By repeating this process 5000 times, an empirical approximation of the sampling distribution of the quantified indirect effect of the independent variable on the dependent variable through each potential mediator was built and used to yield CIs for the indirect effect [34]. A two-sided p value of less than 0.05 was deemed statistically significant. The mediation analysis was reported in line with the AGReMA (A Guideline for Reporting Mediation Analyses) guidelines [35].
Results
Characteristics of participants
A total of 410 participants met the inclusion criteria. Participants who had pathology of the pancreas on cross-sectional imaging (n=22), major health problems that precluded undergoing MRI (n=8), malignancy (n=3), autoimmune disease (n=3), an organ transplant (n=1), a heart pacemaker (n=1), or who used systemic corticosteroids (n=1) were excluded. In addition, participants were excluded if MRS was abandoned (n=15), the pancreas was not visible in its entirety on MRI (n=1), the participant developed claustrophobia while in the scanner (n=1), or i.v. cannulation failed (n=1). Three hundred and fifty-three individuals were analysed. Their median (IQR) age was 50.0 (37.0–60.0) years, and 151 (42.8%) of the study participants were men. Other baseline characteristics are presented in Table 1. The median (IQR) liver fat and IPFD was 5.7 (3.1–12.0)% and 8.7 (7.0–10.1)%, respectively. The intra-class correlation of IPFD measurement was 0.967 (95% CI 0.960, 0.973) (Fig. 1).

Concordance between intrapancreatic fat measurements by two independent raters
Lipid panel as mediators
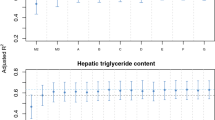
Triacylglycerol, total cholesterol, HDL-cholesterol and LDL-cholesterol were considered as potential mediators in the association between liver fat and IPFD (Fig. 2). While the exposure–mediator effect was statistically significant for triacylglycerol and HDL-cholesterol, the mediator–outcome effect was statistically significant for triacylglycerol, HDL-cholesterol and LDL-cholesterol (Table 2). Based on the statistical significance of the indirect effect estimates, triacylglycerol and HDL-cholesterol were mediators whereas total cholesterol and LDL-cholesterol were not (Fig. 3). Triacylglycerol and HDL-cholesterol mediated 24.3% and 17.9%, respectively, of the association between liver fat and IPFD (Table 3).

Scheme of the study design and key findings. Single-headed arrows represent regression paths, circles represent independent and dependent variables, ovals represent covariates and rectangles represent potential mediators. Standardised coefficients are shown for statistically significant indirect effects (solid lines) only. Dashed lines represent non-significant indirect effects; dotted lines represent covariates. ALP, alkaline phosphatase; ALT, alanine aminotransferase; AST, aspartate aminotransferase; GGT, γ-glutamyl transpeptidase

Indirect effects (with 95% CIs) from the bootstrap samples. ALP, alkaline phosphatase; ALT, alanine aminotransferase; AST, aspartate aminotransferase; GGT, γ-glutamyl transpeptidase
Markers of glucose metabolism as mediators
Fasting plasma glucose and HbA1c were considered as potential mediators in the association between liver fat and IPFD (Fig. 2). Both the exposure–mediator effect and the mediator–outcome effect were statistically significant for glucose but neither were significant for HbA1c (Table 2). Based on the statistical significance of the indirect effect estimates, glucose was a mediator whereas HbA1c was not (Fig. 3). Glucose mediated 6.8% of the association between liver fat and IPFD (Table 3).
Incretins as mediators
GLP-1 and GIP were considered as potential mediators in the association between liver fat and IPFD (Fig. 2). While the exposure–mediator effect was not statistically significant for either GLP-1 or GIP, the mediator–outcome effect was statistically significant for GIP but not GLP-1 (Table 2). Based on the statistical significance of the indirect effect estimates, neither incretin was a mediator (Fig. 3).
Pancreatic hormones and derivatives as mediators
Insulin, amylin, C-peptide and glucagon were considered as potential mediators in the association between liver fat and IPFD (Fig. 2). Neither the exposure–mediator effect nor the mediator–outcome effect was statistically significant for any of the studied pancreatic hormones (Table 2). Based on the statistical significance of the indirect effect estimates, none of the pancreatic hormones were mediators (Fig. 3).
Liver enzymes as mediators
Alanine aminotransferase, aspartate aminotransferase, alkaline phosphatase and γ-glutamyl transpeptidase were considered as potential mediators in the association between liver fat and IPFD (Fig. 2). While the exposure–mediator effect was statistically significant for all the studied liver enzymes, the mediator–outcome effect was not statistically significant for any of them (Table 2). Based on the statistical significance of the indirect effect estimates, none of the liver enzymes were mediators (Fig. 3).
Discussion
This study represents one of the largest MRI cohorts of people with measured IPFD in the literature. The 2008 ‘twin cycle’ hypothesis theorised the interconnectedness of IPFD and liver fat as a key factor influencing the development of type 2 diabetes as well as its remission [36]. The predictions of this hypothesis were subsequently confirmed in interventional studies of a low-energy diet: the Counterpoint, Counterbalance and DiRECT trials [37,38,39]. Further, a 2022 data-driven cluster analysis partitioned individuals without diabetes based on their abdominal fat distribution and then studied the longitudinal association of membership in the resulting clusters with incident type 2 diabetes [40]. The study showed that individuals with excess liver fat and those with excess IPFD (but not those with excess skeletal muscle fat deposition) are at remarkably similar (fourfold and 3.4-fold higher, respectively) risk of type 2 diabetes, further supporting the link between liver fat and IPFD [40]. The use of the path-analytic methodological framework and the large sample size of the present observational study enabled us to provide robust complementary evidence that supports the interconnectedness of IPFD and liver fat. We demonstrated, for the first time, that the influence of liver fat on IPFD can be partitioned into direct and indirect components using mediation analysis. Out of the 16 metabolic traits studied, three traits (triacylglycerol, HDL-cholesterol and glucose) met the established formal criteria for mediators. This held true irrespective of age, sex, ethnicity and BMI. Further, we were able to quantify the magnitude of the indirect effect that these circulating mediators have on the association between liver fat and intrapancreatic fat, ranging from 24.3% for triacylglycerol to 6.8% for glucose.
These three metabolic traits were previously identified as reasonably accurate biomarkers of IPFD in our 2017 meta-analysis of small observational studies [22]. Among the lipid metabolism-related metabolic traits identified, triacylglycerol and HDL-cholesterol had weighted correlation coefficients of 0.38 (95% CI 0.31, 0.46) and −0.33 (95% CI −0.35, −0.31), respectively, with IPFD [22]. They were investigated in seven studies (with 10.5% heterogeneity) and five studies (with 0% heterogeneity), respectively. When individuals with and without fatty pancreas were compared, triacylglycerol had a large positive standardised mean effect size (d+ = 0.49) whereas HDL-cholesterol had a medium negative standardised mean effect size (d+ = −0.32) [22]. In addition, glucose had a weighted r of 0.30 (95% CI 0.26, 0.33) with IPFD and was investigated in seven studies (with 0% heterogeneity) [22]. When individuals with and without fatty pancreas were compared, glucose had a medium positive standardised mean effect size (d+ = 0.36) [22]. However, the above correlations between IPFD and the three metabolic traits were investigated predominantly in individuals with obesity and/or diabetes but rarely in healthy non-obese individuals. The weighted mean correlation coefficients of IPFD were higher with HbA1c than with glucose (r=0.39 vs r=0.30) in that meta-analysis. However, while the studies on glucose had 0% heterogeneity, those on HbA1c had 77.8% heterogeneity. In addition, only six out of 17 studies included in the 2017 meta-analysis used MRI [6]. Most importantly, correlation is one of the simplest statistical analyses and it cannot provide insights into the extent to which potential causal variables influence IPFD.
The present study adds to the existing literature by delineating potentially causal mechanisms that are involved in deposition of fat in the pancreas. Of the three metabolic traits identified as mediators in the present study, triacylglycerol had the largest mediation effect on the association between liver fat and IPFD. Liver fat increased triacylglycerol by 18% of an SD. One SD increase in triacylglycerol changed IPFD by a factor of 0.61. The indirect effect of triacylglycerol on the association between liver fat and IPFD accounted for 24.3% of the direct effect. HDL-cholesterol had the second-largest mediation effect on the association between liver fat and IPFD. Liver fat decreased HDL-cholesterol by 10% of an SD. One SD decrease in HDL-cholesterol changed IPFD by a factor of 0.83. The indirect effect of HDL-cholesterol on the association of liver fat with IPFD accounted for 17.9% of the direct effect. Last, glucose had the smallest mediation effect on the association between liver fat and IPFD. Liver fat increased glucose by 3% of an SD. One SD increase in fasting plasma glucose changed IPFD by a factor of 1.14. The indirect effect of glucose on the association of liver fat and IPFD accounted for 6.8% of the direct effect. Based on the above findings, triacylglycerol, HDL-cholesterol and glucose can be conceptualised as the conduits through which deposition of fat in the liver affects the deposition of fat in the pancreas.
Several limitations are to be acknowledged. First, although the path-analytic methodological framework employed in the present study has been used previously [32, 33], it has not been applied to body composition and metabolic traits. IPFD encompasses multiple subcategories with potentially disparate causal factors that may not be part of the same mechanisms [6]. In addition, one could argue which direction cause is occurring in cross-sectional studies or whether a variable is a presumed causal consequence of the exposure (which would help to clearly distinguish between a mediator and a confounder). Genome-wide and Mendelian randomisation studies may help to provide further causal evidence. Second, although the mediation analysis enabled us to gain important mechanistic insights into the pathogenesis of IPFD, IPFD and liver fat were measured at a single time point. Prospective longitudinal cohort studies are warranted to demonstrate conclusively the causal relationship between liver fat and IPFD. Third, a single-mediator model was used in the present study. It is possible that multiple mediators affect one another, and that these mediators may act as confounders of the effects of other mediators [34]. Given that triacylglycerol and HDL-cholesterol may not be causally independent, caution is necessary when interpreting the estimates of their effects in the present study. Fourth, histological confirmation of fat in the pancreas (and liver) was not available. This is because biopsy of the pancreas is highly invasive and cannot be ethically performed in a large study. However, the present study used 3.0 T MRI, the gold standard for non-invasive quantification of fat in the pancreas. Further, a single scanner/image acquisition protocol was used for all study participants, ensuring consistency of the measurements. Fifth, all the metabolic traits were investigated in the fasted state only. It is conceivable that the indirect effects of postprandial plasma glucose [41, 42], lipid profile [43, 44] and incretins [42, 45, 46] on the association between liver fat and IPFD may differ from those in fasted state. Further mediation analyses may consider exploring metabolic traits in a postprandial state. Last, habitual dietary intake is known to affect both liver fat and IPFD [47,48,49,50] but was not investigated in the present study. The role of dietary intake in the association between liver fat and IPFD remains to be studied in the future.
In conclusion, out of the 16 metabolic traits (lipid panel, liver enzymes, pancreatic hormones, markers of glucose metabolism and incretins) investigated in the present study, three traits, namely triacylglycerol, HDL-cholesterol and glucose, were established to be mediators in the association between liver fat and IPFD. This calls for retiring the simplistic paradigm of a direct linear association between liver fat and IPFD. Targeting the circulating levels of triacylglycerol, HDL-cholesterol and/or glucose holds promise for reducing IPFD.
Data availability
The datasets generated and analysed during the present study are available from the corresponding author upon reasonable request.
Abbreviations
- GIP:
-
Gastric inhibitory peptide
- GLP-1:
-
Glucagon-like peptide-1
- IPFD:
-
Intrapancreatic fat deposition
- MRS:
-
Magnetic resonance spectroscopy
- TE:
-
Echo time
- TR:
-
Repetition time
References
Powell EE, Wong VW, Rinella M (2021) Non-alcoholic fatty liver disease. Lancet 397(10290):2212–2224. https://doi.org/10.1016/S0140-6736(20)32511-3
Stefan N, Häring HU, Cusi K (2019) Non-alcoholic fatty liver disease: Causes, diagnosis, cardiometabolic consequences, and treatment strategies. Lancet Diabetes Endocrinol 7(4):313–324. https://doi.org/10.1016/S2213-8587(18)30154-2
Geier A, Tiniakos D, Denk H, Trauner M (2021) From the origin of NASH to the future of metabolic fatty liver disease. Gut 70(8):1570–1579. https://doi.org/10.1136/gutjnl-2020-323202
Younossi ZM (2019) Non-alcoholic fatty liver disease – A global public health perspective. J Hepatol 70(3):531–544. https://doi.org/10.1016/j.jhep.2018.10.033
Cotter TG, Rinella M (2020) Nonalcoholic fatty liver disease 2020: The state of the disease. Gastroenterology 158(7):1851–1864. https://doi.org/10.1053/j.gastro.2020.01.052
Petrov MS, Taylor R (2022) Intra-pancreatic fat deposition: Bringing hidden fat to the fore. Nat Rev Gastroenterol Hepatol 19(3):153–168. https://doi.org/10.1038/s41575-021-00551-0
Petrov MS, Yadav D (2019) Global epidemiology and holistic prevention of pancreatitis. Nat Rev Gastroenterol Hepatol 16(3):175–184. https://doi.org/10.1038/s41575-018-0087-5
Sreedhar UL, DeSouza SV, Park B, Petrov MS (2020) A systematic review of intra-pancreatic fat deposition and pancreatic carcinogenesis. J Gastrointest Surg 24:2560–2569. https://doi.org/10.1007/s11605-019-04417-4
Xiao AY, Tan ML, Wu LM et al (2016) Global incidence and mortality of pancreatic diseases: a systematic review, meta-analysis, and meta-regression of population-based cohort studies. Lancet Gastroenterol Hepatol 1:45–55. https://doi.org/10.1016/S2468-1253(16)30004-8
Petrov MS (2021) Post-pancreatitis diabetes mellitus: prime time for secondary disease. Eur J Endocrinol 184:R137–R149. https://doi.org/10.1530/EJE-20-0468
Petrov MS, Basina M (2021) Diagnosing and classifying diabetes in diseases of the exocrine pancreas. Eur J Endocrinol 184:R151–R163. https://doi.org/10.1530/EJE-20-0974
Petrov MS (2021) Post-pancreatitis diabetes mellitus and excess intra-pancreatic fat deposition as harbingers of pancreatic cancer. World J Gastroenterol 27:1936–1942. https://doi.org/10.3748/wjg.v27.i17.1936
Cho J, Petrov MS (2020) Pancreatitis, pancreatic cancer, and their metabolic sequelae: projected burden to 2050. Clin Transl Gastroenterol 11:e00251. https://doi.org/10.14309/ctg.0000000000000251
Singh RG, Yoon HD, Wu LM, Lu J, Plank LD, Petrov MS (2017) Ectopic fat accumulation in the pancreas and its clinical relevance: A systematic review, meta-analysis, and meta-regression. Metabolism 69(4):1–13. https://doi.org/10.1016/j.metabol.2016.12.012
Lee JS, Kim SH, Jun DW et al (2009) Clinical implications of fatty pancreas: Correlations between fatty pancreas and metabolic syndrome. World J Gastroenterol 15(15):1869–1875. https://doi.org/10.3748/wjg.15.1869
Choi CW, Kim GH, Kang DH et al (2010) Associated factors for a hyperechogenic pancreas on endoscopic ultrasound. World J Gastroenterol 16(34):4329–4334. https://doi.org/10.3748/wjg.v16.i34.4329
Sepe PS, Ohri A, Sanaka S et al (2011) A prospective evaluation of fatty pancreas by using EUS. Gastrointest Endosc 73(5):987–993. https://doi.org/10.1016/j.gie.2011.01.015
Wong VW, Wong GL, Yeung DK et al (2014) Fatty pancreas, insulin resistance, and β-cell function: A population study using fat-water magnetic resonance imaging. Am J Gastroenterol 109(4):589–597. https://doi.org/10.1038/ajg.2014.1
Adams LA, Anstee QM, Tilg H, Targher G (2017) Non-alcoholic fatty liver disease and its relationship with cardiovascular disease and other extrahepatic diseases. Gut 66(6):1138–1153. https://doi.org/10.1136/gutjnl-2017-313884
Lee H, Herbert RD, McAuley JH (2019) Mediation analysis. JAMA 321(7):697–698. https://doi.org/10.1001/jama.2018.21973
Ekstedt M, Franzén LE, Mathiesen UL et al (2006) Long-term follow-up of patients with NAFLD and elevated liver enzymes. Hepatology 44(4):865–873. https://doi.org/10.1002/hep.21327
Singh RG, Yoon HD, Poppitt SD, Plank LD, Petrov MS (2017) Ectopic fat accumulation in the pancreas and its biomarkers: A systematic review and meta-analysis. Diabetes Metab Res Rev 33(8):e2918. https://doi.org/10.1002/dmrr.2918
Deprince A, Haas JT, Staels B (2020) Dysregulated lipid metabolism links NAFLD to cardiovascular disease. Mol Metab 42:101092. https://doi.org/10.1016/j.molmet.2020.101092
Martin S, Sorokin EP, Thomas EL et al (2022) Estimating the effect of liver and pancreas volume and fat content on risk of diabetes: A Mendelian randomization study. Diabetes Care 45(2):460–468. https://doi.org/10.2337/dc21-1262
Yamazaki H, Tauchi S, Kimachi M et al (2018) Independent association between prediabetes and future pancreatic fat accumulation: A 5-year Japanese cohort study. J Gastroenterol 53(7):873–882. https://doi.org/10.1007/s00535-017-1422-2
Svegliati-Baroni G, Patrício B, Lioci G, Macedo MP, Gastaldelli A (2020) Gut-pancreas-liver axis as a target for treatment of NAFLD/NASH. Int J Mol Sci 21(16):5820. https://doi.org/10.3390/ijms21165820
Skudder-Hill L, Sequeira IR, Cho J, Ko J, Poppitt SD, Petrov MS (2022) Fat distribution within the pancreas according to diabetes status and insulin traits. Diabetes 71(6):1182–1192. https://doi.org/10.2337/db21-0976
Singh RG, Nguyen NN, DeSouza SV, Pendharkar SA, Petrov MS (2019) Comprehensive analysis of body composition and insulin traits associated with intra-pancreatic fat deposition in healthy individuals and people with new-onset prediabetes/diabetes after acute pancreatitis. Diabetes Obes Metab 21(2):417–423. https://doi.org/10.1111/dom.13523
Singh RG, Cervantes A, Kim JU et al (2019) Intrapancreatic fat deposition and visceral fat volume are associated with the presence of diabetes after acute pancreatitis. Am J Physiol Gastrointest Liver Physiol 316(6):G806–G815. https://doi.org/10.1152/ajpgi.00385.2018
Al-Mrabeh A, Hollingsworth KG, Steven S, Tiniakos D, Taylor R (2017) Quantification of intrapancreatic fat in type 2 diabetes by MRI. PLoS One 12(4):1–19. https://doi.org/10.1371/journal.pone.0174660
DeSouza SV, Priya S, Cho J, Singh RG, Petrov MS (2019) Pancreas shrinkage following recurrent acute pancreatitis: An MRI study. Eur Radiol 29(7):3746–3756. https://doi.org/10.1007/s00330-019-06126-7
Hayes AF (2009) Beyond Baron and Kenny: Statistical mediation analysis in the new millennium. Commun Monogr 76(4):408–420. https://doi.org/10.1080/03637750903310360
Preacher KJ, Hayes AF (2004) SPSS and SAS procedures for estimating indirect effects in simple mediation models. Behav Res Methods Instrum Comput 36(4):717–731. https://doi.org/10.1002/jcp.28952
Preacher KJ, Hayes AF (2008) Asymptotic and resampling strategies for assessing and comparing indirect effects in multiple mediator models. Behav Res Methods 40(3):879–891. https://doi.org/10.3758/brm.40.3.879
Lee H, Cashin AG, Lamb SE et al (2021) A guideline for reporting mediation analyses of randomized trials and observational studies: The AGReMA statement. JAMA 326(11):1045–1056. https://doi.org/10.1001/jama.2021.14075
Taylor R (2008) Pathogenesis of type 2 diabetes: Tracing the reverse route from cure to cause. Diabetologia 51(10):1781–1789. https://doi.org/10.1007/s00125-008-1116-7
Lim EL, Hollingsworth KG, Aribisala BS, Chen MJ, Mathers JC, Taylor R (2011) Reversal of type 2 diabetes: Normalisation of beta cell function in association with decreased pancreas and liver triacylglycerol. Diabetologia 54(10):2506–2514. https://doi.org/10.1007/s00125-011-2204-7
Steven S, Hollingsworth KG, Al-Mrabeh A et al (2016) Very low-calorie diet and 6 months of weight stability in type 2 diabetes: Pathophysiological changes in responders and nonresponders. Diabetes Care 39(5):808–815. https://doi.org/10.2337/dc15-1942
Al-Mrabeh A, Zhyzhneuskaya SV, Peters C et al (2020) Hepatic lipoprotein export and remission of human type 2 diabetes after weight loss. Cell Metab 31(2):233–249. https://doi.org/10.1016/j.cmet.2019.11.018
Yamazaki H, Tauchi S, Machann J et al (2022) Fat distribution patterns and future type 2 diabetes. Diabetes 71(9):1937–1945. https://doi.org/10.2337/db22-0315
Vitale M, Giacco R, Laiola M et al (2021) Acute and chronic improvement in postprandial glucose metabolism by a diet resembling the traditional Mediterranean dietary pattern: Can SCFAs play a role? Clin Nutr 40(2):428–437. https://doi.org/10.1016/j.clnu.2020.05.025
Bharmal SH, Cho J, Alarcon Ramos GC, Ko J, Cameron-Smith D, Petrov MS (2021) Acute nutritional ketosis and its implication for plasma glucose and glucoregulatory peptides in adults with prediabetes: A crossover placebo-controlled randomized trial. J Nutr 151(4):921–929. https://doi.org/10.1093/jn/nxaa417
Monfort-Pires M, Delgado-Lista J, Gomez-Delgado F, Lopez-Miranda J, Perez-Martinez P, Ferreira SR (2016) Impact of the content of fatty acids of oral fat tolerance tests on postprandial triglyceridemia: Systematic review and meta-analysis. Nutrients 8(9):580. https://doi.org/10.3390/nu8090580
Liu Y, Bharmal SH, Kimita W, Petrov MS (2022) Effect of acute ketosis on lipid profile in prediabetes: findings from a cross-over randomized controlled trial. Cardiovasc Diabetol 21(1):138. https://doi.org/10.1186/s12933-022-01571-z
Asmar A, Cramon PK, Asmar M et al (2020) Increased oral sodium chloride intake in humans amplifies selectively postprandial GLP-1 but not GIP, CCK, and gastrin in plasma. Physiol Rep 8(15):e14519. https://doi.org/10.14814/phy2.14519
Pendharkar SA, Singh RG, Cervantes A, DeSouza SV, Bharmal SH, Petrov MS (2019) Gut hormone responses to mixed meal test in new-onset prediabetes/diabetes after acute pancreatitis. Horm Metab Res 51(3):191–199. https://doi.org/10.1055/a-0802-9569
Anania C, Massimo Perla F, Olivero F, Pacifico L, Chiesa C (2018) Mediterranean diet and nonalcoholic fatty liver disease. World J Gastroenterol 24(19):2083–2094. https://doi.org/10.3748/wjg.v24.i19.2083
Ko J, Skudder-Hill L, Tarrant C, Kimita W, Bharmal SH, Petrov MS (2021) Intra-pancreatic fat deposition as a modifier of the relationship between habitual dietary fat intake and insulin resistance. Clin Nutr 40(7):4730–4737. https://doi.org/10.1016/j.clnu.2021.06.017
Matsuda A, Makino N, Tozawa T et al (2014) Pancreatic fat accumulation, fibrosis, and acinar cell injury in the Zucker diabetic fatty rat fed a chronic high-fat diet. Pancreas 43(5):735–743. https://doi.org/10.1097/MPA.0000000000000129
He K, Li Y, Guo X, Zhong L, Tang S (2020) Food groups and the likelihood of non-alcoholic fatty liver disease: A systematic review and meta-analysis. Br J Nutr 124(1):1–13. https://doi.org/10.1017/S0007114520000914
Acknowledgements
The study was part of the COSMOS programme (www.cosmos.auckland.ac.nz).
Authors’ relationships and activities
The authors declare that there are no relationships or activities that might bias, or be perceived to bias, their work.
Contribution statement
MSP conceived the study. JK, IRS, SDP and JC recruited participants. JK and LSH collected the imaging data. JK and JC conducted the mediation analysis. JK wrote the initial draft. IRS, LSH, JC, SDP and MSP critically reviewed and revised the manuscript for important intellectual content. All authors read and approved the final version of the paper. MSP is the guarantor of this work.
Funding
Open Access funding enabled and organized by CAUL and its Member Institutions. This study was funded by the Royal Society of New Zealand (Rutherford Discovery Fellowship to MSP) and the New Zealand National Science Challenge High Value Nutrition Program, Ministry for Business, Innovation and Employment (grant 3710040).
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ko, J., Sequeira, I.R., Skudder-Hill, L. et al. Metabolic traits affecting the relationship between liver fat and intrapancreatic fat: a mediation analysis. Diabetologia 66, 190–200 (2023). https://doi.org/10.1007/s00125-022-05793-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-022-05793-4