
Abstract
Aims/hypothesis
Data analyses from Swedish individuals with newly diagnosed diabetes have suggested that diabetes could be classified into five subtypes that differ with respect to the progression of dysglycaemia and the incidence of diabetes consequences. We assessed this classification in a multiethnic cohort of participants with established and newly diagnosed diabetes, randomly allocated to insulin glargine vs standard care.
Methods
In total, 7017 participants from the Outcome Reduction with Initial Glargine Intervention (ORIGIN) trial were assigned to the five predefined diabetes subtypes (namely, severe auto-immune diabetes, severe insulin-deficient diabetes, severe insulin-resistant diabetes, mild obesity-related diabetes, mild age-related diabetes) based on the age at diabetes diagnosis, BMI, HbA1c, fasting C-peptide levels and the presence of glutamate decarboxylase antibodies at baseline. Differences between diabetes subtypes in cardiovascular and renal outcomes were investigated using Cox regression models for a median follow-up of 6.2 years. We also compared the effect of glargine vs standard care on hyperglycaemia, defined by having a mean post-randomisation HbA1c ≥6.5%, between subtypes.
Results
The five diabetes subtypes were replicated in the ORIGIN trial and exhibited similar baseline characteristics in Europeans and Latin Americans, compared with the initially described clusters in the Swedish cohort. We confirmed differences in renal outcomes, with a higher incidence of events in the severe insulin-resistant diabetes subtype compared with the mild age-related diabetes subtype (i.e., chronic kidney disease stage 3A: HR 1.49 [95% CI 1.31, 1.71]; stage 3B: HR 2.25 [1.82, 2.78]; macroalbuminuria: HR 1.56 [1.22, 1.99]). No differences were observed in the incidence of retinopathy and cardiovascular diseases after adjusting for multiple hypothesis testing. Diabetes subtypes also differed in glycaemic response to glargine, with a particular benefit of receiving glargine (vs standard care) in the severe insulin-deficient diabetes subtype compared with the mild age-related diabetes subtype, with a decreased occurrence of hyperglycaemia by 13% (OR 1.36 [1.30, 1.41] on glargine; OR 1.49 [1.43, 1.57] on standard care; p for interaction subtype × intervention = 0.001).
Conclusions/interpretation
Cluster analysis enabled the characterisation of five subtypes of diabetes in a multiethnic cohort. Both the incidence of renal outcomes and the response to insulin varied between diabetes subtypes. These findings reinforce the clinical utility of applying precision medicine to predict comorbidities and treatment responses in individuals with diabetes.
Trial registration
ORIGIN trial, ClinicalTrials.gov NCT00069784.
Graphical abstract

Similar content being viewed by others
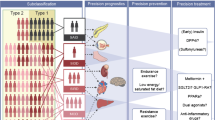



Introduction
The doubling of the prevalence of diabetes over the past 25 years and the high risk it confers of serious health-related consequences has made diabetes a major public health challenge [1]. Despite progress in the discovery and development of pharmacological treatments, one in four people with diabetes will develop macrovascular complications and half will develop microvascular complications [2]. Given the heterogeneity in clinical presentation and disease progression, the identification of diabetes subgroups, beyond the usual classification into type 1 and type 2 diabetes, may improve risk stratification for complications and optimise treatment strategies [3].
The characterisation of five distinct diabetes subgroups using sex-stratified, data-driven cluster analyses in the All New Diabetics in Scania (ANDIS) cohort marked an important milestone [4]. Using six clinico–biological variables routinely collected at diabetes diagnosis, such as age at diagnosis, BMI, HbA1c level, GAD antibodies, fasting glucose, and HOMA2 estimates of beta cell function and insulin resistance based on C-peptide levels, Ahlqvist et al categorised adults with newly diagnosed diabetes into five mutually exclusive clusters, distinguished by different underlying pathophysiology—i.e., severe auto-immune diabetes (SAID), severe insulin-deficient diabetes (SIDD), severe insulin-resistant diabetes (SIRD), mild obesity-related diabetes (MOD) or mild age-related diabetes (MARD). These clusters exhibited distinct baseline phenotypic characteristics and diabetic complication trajectories. For instance, participants in the most insulin-resistant cluster had a 2–3-fold higher risk of developing chronic kidney disease and macroalbuminuria whereas those in the most insulin-deficient cluster had a 1.5-fold higher risk of developing retinopathy. Independent analyses demonstrated that Iraqi immigrants were more often assigned to the SIDD cluster compared with the native Swedes, and had higher risk of coronary artery diseases but lower risk of chronic kidney diseases [5]. The distribution of clusters and baseline patient characteristics was replicated in two cross-sectional populations in China and the USA, respectively [6]. In a Japanese cohort, participants with SAID or SIDD had the highest incidence of retinopathy and those with SIRD had the highest incidence of chronic kidney disease over approximately 15 years of follow-up [7]. In an Asian Indian population, the SIDD cluster had the highest hazards for developing retinopathy, while the combined insulin-resistant and -deficient diabetes cluster had the highest hazards for kidney disease [8]. These observations were also consistently made in the German Diabetes Study [9], and in the ADOPT, RECORD [10], DEVOTE, LEADER and SUSTAIN-6 trials [11], in which the clustering approach was similarly applied.
This refined diabetes classification system thus seems to be a powerful tool that is consistent with a precision medicine approach to diabetes care [3], although evidence is required to ensure its applicability in clinical practice. For instance, this clustering approach has not been evaluated in populations with diverse ethnic groups within which factors related to diabetes progression and complications may vary [2]. Moreover, the effects of different glucose-lowering therapeutic strategies have not been fully evaluated between clusters.
The Outcome Reduction with Initial Glargine Intervention (ORIGIN) trial was a randomised controlled trial that enrolled participants from multiple ancestries with dysglycaemia. Participants were randomly allocated to insulin glargine vs standard care groups and monitored for cardiovascular and renal events for a median follow-up of 6.2 years. Herein, we replicated the clustering method described by Ahlqvist et al to categorise ORIGIN participants with newly diagnosed or established diabetes into the five aforementioned subgroups. We investigated whether either incident selected diabetes consequences (cardiovascular events, chronic kidney diseases and/or severe retinopathy) or participant glycaemic response to insulin differed between clusters.
Methods
Study population
The design of the ORIGIN trial has been described previously [12]. Between 2003 and 2005, a total of 578 clinical sites in 40 countries enrolled 12,537 participants aged ≥50 years with established or newly detected diabetes (with HbA1c levels ≤75 mmol/mol [9%]), impaired glucose tolerance (IGT) or impaired fasting glucose (IFG) levels, and additional cardiovascular risk factors. Following random allocation of participants to two therapies using a factorial design (either one daily injection of insulin glargine [Sanofi Aventis Deutschland, Frankfurt am Main, Germany] with the dose titrated to achieve a fasting plasma glucose level ≤5.3 mmol/l [95 mg/dl] or standard care; and either omega 3 fatty acid supplement or placebo), participants were monitored for a median duration of 6.2 years for cardiovascular events and other health outcomes. The ORIGIN trial complies with the Declaration of Helsinki and was approved by the Ethical Committee at each site and each participant provided written informed consent. A subset of 8494 participants also consented to provide blood samples at baseline for further analyses (including C-peptide and GAD antibodies). A total of 7017 participants with established diabetes or newly diagnosed diabetes and available baseline data (i.e., age at diabetes diagnosis, BMI, HbA1c, fasting C-peptide levels and GAD antibodies) were included in these analyses.
Study measurements
Age at diabetes diagnosis and measured BMI were collected at the recruitment visit of the ORIGIN trial. HbA1c levels were assayed at local laboratories at baseline, annually and at the end of the treatment period for all participants [13]. GAD antibodies were assayed at the Clinical Research Laboratory and Biobank at the Population Health Research Institute in Hamilton, Canada, using the commercially available Kronus anti-GAD ELISA kit (Star, ID, USA). The results were considered negative if the concentration was <5 U/ml [14]. C-peptide was measured in fasting samples using a multiplex assay including 237 cardiometabolic biomarkers (Luminex, Myriad RBM, Austin, TX, USA), as previously reported [15, 16]. All assays were performed blinded to the treatment allocation, and without knowledge of any clinical participant information.
Study outcomes
ORIGIN participants were asked about serious health outcomes every 4 months. Major cardiovascular events were defined as death from cardiovascular causes, nonfatal myocardial infarction, nonfatal stroke or a composite of them [12]. Eye disease information was collected for retinopathy requiring retinal photocoagulation or vitrectomy. Serum creatinine was measured in local laboratories at baseline, after 2 years and upon study completion. First-voided urine samples were collected at the research sites following the same schedule and forwarded to the Clinical Research Laboratory and Biobank at the Population Health Research Institute in Hamilton, Canada for the measurement of urine albumin and urine creatinine concentrations. Urine albumin was measured using a turbidimetric method (with the Beckman MA reagent) and urine creatinine was measured using a modified Jaffé reaction (with the Beckman CR-S reagent) on a Beckman UniCel DxC 600 Instrument (Beckman Coulter, Fullerton, CA, USA) [17]. The eGFR was calculated based on the Modification of Diet in Renal Disease (MDRD) formula, and used to define kidney function as normal (stage 1, eGFR >90 ml min−1 1.73 m−2), mildly impaired (stage 2, eGFR 60–90 ml min−1 1.73 m−2) or moderately impaired (stage 3A, 45≤eGFR<60 ml min−1 1.73 m−2; and stage 3B, 30≤eGFR<45 ml min−1 1.73 m−2). An albumin/creatinine ratio equal to or greater than 25 mg/mmol in men and ≥35 mg/mmol in women defined macroalbuminuria [18]. Hyperglycaemia was defined as a mean post-randomisation HbA1c ≥47.5 mmol/mol (6.5%). The mean HbA1c was calculated using the mean value of all available annual measurements, excluding the baseline value, and including the 1 year value, up to and including the 5 years value [13]. All clinical outcomes were adjudicated by a committee that was blinded to the treatment allocation.
Statistical analyses
We applied the clustering methodology provided by Ahlqvist et al on ORIGIN participants with established or newly diagnosed diabetes. The SAID cluster included participants with positive GAD antibodies. We used the sex-specific nearest centroid approach, adapted for the use of C-peptide only (rather than HOMA2-B and HOMA2-IR as initially described), to assign the remaining participants into the four predefined clusters, SIDD, SIRD, MOD and MARD. Variables including age at diabetes diagnosis, BMI, and HbA1c and C-peptide levels for each participant were scaled and centred. Participants were then assigned to one of the four clusters based on the cluster they were most similar to, estimated as the nearest Euclidean distance to the cluster centre, derived from ANDIS coordinates (see electronic supplementary material [ESM] Table 1 for cluster centre coordinates). Data are presented as the mean (SD), median (IQR) or proportion (%). Cox regression models were used to test the predictive values of baseline clustering on the incidence of diabetes-related consequences over 6.2 years of follow-up. The proportional hazards assumption was first verified, and then Cox regression models were performed including each of the diabetes-related consequences (i.e., coronary event, stroke, retinopathy, mildly impaired kidney function, moderately impaired kidney function, macroalbuminuria) as dependent variables, and the categorical cluster variable as the independent variable of interest, with the MARD cluster used as reference. Models were adjusted for age at diabetes diagnosis, sex, self-reported ethnicity, diabetes duration at recruitment, allocation of glargine, allocation of fish oil and usage of other glucose-lowering medications.
To test whether the effect of glargine on diabetes-related consequences differed by cluster, we performed Cox regression models, including each of the diabetes-related consequences as dependent variables, and glargine allocation, cluster and the interaction between glargine and cluster as independent variables, in addition to the aforementioned adjustment covariates. Post hoc pairwise comparisons of the effects across clusters were also performed. To determine whether the effect of glargine on hyperglycaemia differed by cluster, a logistic regression model was performed with the dependent variable being hyperglycaemia and the independent variables being glargine allocation, cluster and interaction between the two. Glargine allocation was compared with standard care, and the MARD cluster was used as reference. This model was adjusted for age at diabetes diagnosis, sex, self-reported ethnicity, diabetes duration at recruitment, allocation of fish oil, usage of other glucose-lowering medications and the inverse probability of selection weighting to minimise the bias related to post-randomisation subgroup analyses. All statistical analyses were conducted using R (version 3.6.0) [19]. Two-tailed p values <0.05 were considered statistically significant, with adjustments for multiple hypothesis testing applied, as appropriate.
Results
A total of 6492 ORIGIN participants with established diabetes and 525 ORIGIN participants with newly detected diabetes were assigned to one of the five clusters. The individual distribution and baseline characteristics of participants are presented in Table 1 and Fig. 1. The MARD cluster of older participants (median [IQR] age at diagnosis, 63 [58–69] years) with modest metabolic abnormalities (median HbA1c level, 43.2 [38.8–47.8] mmol/mol; 6.1% [5.7–6.5%]; median BMI, 27.4 [25.2–29.7] kg/m2; and median C-peptide level, 0.40 [0.29–0.53] nmol/l) included 38% of the ORIGIN participants. Twenty-three per cent of participants were assigned to the MOD cluster and were consistently characterised by the highest BMI (33.9 [31.2–37.3] kg/m2), as well as a median HbA1c level of 47.1 mmol/mol (42.1–52.2 mmol/mol; 6.5% [6.0–6.9%]) and a median C-peptide level of 0.53 nmol/l (0.40–0.70 nmol/l). Another 23% of participants were assigned to the SIDD cluster and were characterised by low C-peptide levels (0.43 [0.28–0.57] nmol/l) and high HbA1c levels (60.7 [57.1–65.5] mmol/mol; 7.7% [7.4–8.1%]). A total of 13% of participants were categorised into the SIRD cluster and exhibited the highest C-peptide levels (1.03 [0.83–1.30] nmol/l). A minority of participants (3%) had positive GAD antibodies and were assigned to the SAID cluster. Those participants had a median age at diagnosis of 59 years (52–66 years), a median BMI of 29.2 kg/m2 (26.4–32.0 kg/m2), a median HbA1c level of 47.6 mmol/mol (41.0–55.6 mmol/mol; 6.5% [5.9–299 7.2%]) and overt insulin deficiency (C-peptide level, 0.43 [0.32–0.61] nmol/l). Given the multiethnic ancestries of ORIGIN participants, we further characterised the population by performing descriptive analyses separately in the two major ethnic groups (87% of the overall sample), namely Europeans (n = 3661) and Latin Americans (n = 2428), after clusters were established in the overall sample. The participant distributions showed that a larger proportion of individuals of Latin American ancestries were assigned to the SIDD cluster compared with the individuals of European ancestries (28% vs 18%, respectively) (Fig. 1). The baseline characteristics of participants from each cluster were consistent across ethnic groups (ESM Table 2).

Distribution and characteristics of the five diabetes subtypes in ORIGIN. (a) Distribution. (b–e) Characteristics of the overall population. (f–i) Characteristics separately in European and Latin American populations. Boxplots represent the median and 2nd and 3rd quartiles; error bars represent the 95% CI. The key in (a) applies to all figure parts. Eur, Europeans; Lat, Latin Americans
We next performed Cox regression analyses to determine whether the incidence of diabetes-related consequences differed between clusters. HRs of incident cardiovascular diseases, retinopathy and nephropathy over the 6.2 years of follow-up are presented for each cluster in Fig. 2 (see ESM Table 3 for number of participants and person-years, and ESM Table 4 for pairwise comparisons). Participants in the SIRD cluster had a 49% increase in the incidence of moderately impaired kidney function (stage 3A; HR 1.49 [95% CI 1.31, 1.71], p = 4.1×10−9), a 125% increase in the incidence of stage 3B kidney impairment (HR 2.25 [1.82, 2.78], p = 5.6×10−14) and a 56% increase in the incidence of macroalbuminuria (HR 1.56 [1.22, 1.99], p = 0.0003), compared with the participants in the MARD cluster. Individuals in the SIDD cluster also exhibited a 36% increase in the incidence of macroalbuminuria compared with the individuals in the MARD cluster (HR 1.36 [1.11, 1.66], p = 0.003). Incident retinopathy was nominally increased in the SIDD cluster (HR 2.04 [1.16, 3.58], p = 0.01), as were incident coronary events in the SIRD cluster (HR 1.64 [1.15, 2.33], p = 0.01), compared with the MARD cluster.

Adjusted HRs of cardiovascular, renal and retinal outcomes in ORIGIN. For ORIGIN, HRs were adjusted for sex, age at diagnosis of diabetes, diabetes duration at recruitment, glargine allocation, fish oil allocation, self-reported ethnicity and usage of other glucose-lowering medications. The MARD cluster was used as referent group (REF) for all the analyses. Error bars represent 95% CI. CKD, chronic kidney disease
Then, to determine whether the effect of glargine on hyperglycaemia differed between clusters, logistic regression was performed by including the interaction term between glargine allocation and clusters. We found a significant interaction between glargine allocation and clusters on hyperglycaemia in the SAID, SIDD and MOD clusters (Fig. 3). Indeed, random assignment to glargine (vs standard care) significantly decreased hyperglycaemia by 19% in the SAID cluster (OR 1.10 [1.01, 1.19] on glargine, OR 1.29 [1.18, 1.42] on standard care, vs MARD cluster; p for interaction = 0.008), by 13% in the SIDD cluster (OR 1.36 [1.30, 1.41] on glargine, OR 1.49 [1.43, 1.57] on standard care, vs MARD cluster; p for interaction = 0.001) and by 9% in the MOD cluster (OR 1.07 [1.02, 1.12] on glargine, OR 1.16 [1.10, 1.22] on standard care, vs MARD cluster; p for interaction = 0.006). No significant interaction was found in the SIRD cluster (vs MARD cluster). However, we did not observe a different effect of glargine allocation on incidence of diabetes-related consequences between the clusters (all p for interaction >0.05; ESM Fig. 1).

ORs for hyperglycaemia with glargine allocation vs standard care, by clusters in the ORIGIN cohort. Hyperglycaemia was defined as a mean post-randomisation HbA1c ≥6.5%. The MARD cluster was used as referent group for all analyses. Error bars represent 95% CI
Discussion
When clustering ORIGIN participants based on five clinico–biological measures related to diabetes, we found consistent observations with the report from the ANDIS cohort and the Scania Diabetes Registry (SDR) with respect to participant distribution across clusters, baseline phenotypic characteristics and the incidence of diabetes-related consequences. In particular, insulin resistance has been consistently associated with impaired renal function, since participants assigned to the SIRD cluster showed an accelerated decline of eGFR and macroalbuminuria in ORIGIN, here and in previous reports [7, 9,10,11]. We nominally replicated the association between the increased risk of retinopathy and the SIDD cluster observed in the ANDIS cohort, although only severe retinopathy was recorded in ORIGIN participants, which reduced our number of events and limited our power to detect a significant effect after multiple hypothesis correction. Consistent with the ANDIS cohort, no significant association of clusters with incident coronary events and stroke was observed in ORIGIN participants. This latter finding was in contrast to the reports from the DEVOTE, LEADER and SUSTAIN-6 trials [11], which showed an increased incidence in cardiovascular events in the cluster characterised by the highest HbA1c and lowest BMI, compared with the age-related diabetes cluster. However, the populations in these trials included participants with longer duration of diabetes than participants in the ORIGIN and the ANDIS cohorts. Finally, consistent with the German Diabetes Study [9], the predictive value of diabetes-related consequences derived from cluster analysis was preserved, regardless of the duration of diabetes at the time when the clustering was applied [9].
There are no current clinical recommendations based on the five diabetes clusters, but our findings, combined with those of others, suggest that individuals with SIRD would benefit from the early detection of kidney disease, and, thus, from the prevention of major renal outcomes. While based on a lower level of evidence, cautious screening for retinopathy may also be warranted for individuals with SIDD. Indeed, evaluation of cluster-specific complications may guide personalised medical management of diabetes subgroups. However, it has not been fully established whether diabetes clustering is useful beyond risk stratification for complications. In particular, how such clustering may dictate responses to different glucose-lowering therapies and help identify individuals who may benefit from a specific drug class for hyperglycaemia and for diabetes-related comorbidities has not been fully determined, and this latter assumes a homogenous response to a given treatment within a cluster. Our study is the first to show a particular benefit on hyperglycaemia with the early use of insulin in the SIDD cluster. Clustering applied in the RECORD and ADOPT trials, respectively, demonstrated more beneficial effects of thiazolidinediones allocation in participants assigned to the SIRD cluster, and of sulfonylureas in participants assigned to the MARD cluster [10]. Although diabetes is caused by a combination of disrupted pathophysiological processes rather than a single defect [20], assigning individuals to one of the five mutually exclusive clusters to which they were the closest helped to distinguish individuals based on their dominant biological defect, and allowed us to decipher who was more likely to develop specific diabetes consequences or to have a better glycaemic response to a given glucose-lowering therapeutic strategy.
The strengths of our study included the use of data from a large randomised trial that enrolled participants from multiple ancestries, which enabled us to replicate the diabetes clustering across ethnicities, and to describe the progression of diabetes-related consequences and the response to glargine therapy along clusters. As we positioned our analyses as a replication of the clustering algorithm developed by Ahlqvist et al, we did not perform a de novo clustering analysis, but instead used the cluster coordinates determined in the ANDIS study, as was similarly and previously performed in other populations with diabetes [6,7,8,9,10,11]. The first limitation was that our clustering method did not include HOMA indices as there were no fasting plasma glucose measurements available concomitantly to the C-peptide measurements in ORIGIN participants. Although our results reinforce that the clustering method can provide accurate patient risk stratification without using HOMA-B and HOMA-IR and using fasting C-peptide level alone as a surrogate for both HOMA indices, this might explain the increased proportion of ORIGIN participants in the SIDD cluster compared with the proportion observed by Ahlqvist et al [4]. Nonetheless, these observations provide realistic implications for the clinical utility of diabetes clustering in patients, since the use of HOMA indices is not recommended in daily practice. A second limitation was that our results were observed for individuals enrolled in a clinical trial, and, thus, were potentially not representative of the general population due to the trial inclusion criteria. The clinical trial design, in particular the inclusion criteria, could also explain differences between cluster baseline characteristics reported in ORIGIN compared with the ones previously reported in other studies, in respect of age at diabetes diagnosis and levels of HbA1c at baseline [6,7,8,9,10,11]. However, trial data have advantages over real-world data as they are collected through a protocol follow-up and enable a systematic assessment of disease progression and therapy introduction. The third limitation was that we were unable to assess other diabetes-related outcomes, such as incidence of neuropathies, which has been described to be highest in the SIDD cluster [9], or incidence of non-alcoholic fatty liver diseases, reported to be highest in the SIRD cluster [9]. Finally, our study was designed to assign individuals to one of the five mutually exclusive clusters, and did not have the purpose to compare the predictive performance of these models with probabilistic individualised prediction models combining the same clinical features as continuous measures [21]. Moreover, whether the predictive performance of models using simple continuous measures would be superior to models using data-driven clusters is still conceptually debated.
In total, our study shows that individuals with newly diagnosed or established diabetes, regardless of their ethnicity, can be allocated to five specific clusters that show distinct metabolic alterations and different risk patterns for the development of diabetes-related comorbidities, and responses to insulin glargine allocation. Indeed, we identified the highest incidence of kidney diseases in the SIRD cluster, and a particular benefit on hyperglycaemia with insulin glargine allocation in the SIDD cluster. Our findings provide evidence that clustering for the management of diabetes would improve patient risk stratification for diabetes-related consequences, and further optimise screening for such complications and prevention. However, further investigations are required to guide cluster-specific treatment decisions, and potentially improve the clustering approach by adding additional relevant biomarkers and genetic information.
Data availability
No additional data are available.
Abbreviations
- ADOPT:
-
A Diabetes Outcome Progression Trial
- ANDIS:
-
All New Diabetics in Scania
- DEVOTE:
-
Trial Comparing Cardiovascular Safety of Insulin Degludec Versus Insulin Glargine in Subjects With Type 2 Diabetes at High Risk of Cardiovascular Events
- LEADER:
-
Liraglutide Effect and Action in Diabetes: Evaluation of Cardiovascular Outcome Results
- MARD:
-
Mild age-related diabetes
- MOD:
-
Mild obesity-related diabetes
- ORIGIN:
-
Outcome Reduction with Initial Glargine Intervention
- RECORD:
-
Rosiglitazone Evaluated for Cardiac Outcomes and Regulation of Glycaemia in Diabetes
- SAID:
-
Severe auto-immune diabetes
- SIDD:
-
Severe insulin-deficient diabetes
- SIRD:
-
Severe insulin-resistant diabetes
- SUSTAIN-6:
-
Trial to Evaluate Cardiovascular and Other Long-term Outcomes With Semaglutide in Subjects With Type 2 Diabetes
References
World Health Organization (2021) Diabetes. Available from http://www.who.int/news-room/fact-sheets/detail/diabetes. Accessed 3 May 2021
Harding JL, Pavkov ME, Magliano DJ, Shaw JE, Gregg EW (2019) Global trends in diabetes complications: a review of current evidence. Diabetologia 62(1):3–16. https://doi.org/10.1007/s00125-018-4711-2
Ahlqvist E, Tuomi T, Groop L (2019) Clusters provide a better holistic view of type 2 diabetes than simple clinical features. Lancet Diabetes Endocrinol 7(9):668–669. https://doi.org/10.1016/S2213-8587(19)30257-8
Ahlqvist E, Storm P, Käräjämäki A et al (2018) Novel subgroups of adult-onset diabetes and their association with outcomes: a data-driven cluster analysis of six variables. Lancet Diabetes Endocrinol 6(5):361–369. https://doi.org/10.1016/S2213-8587(18)30051-2
Bennet L, Nilsson C, Mansour-Aly D, Christensson A, Groop L, Ahlqvist E (2020) Adult-onset diabetes in Middle Eastern immigrants to Sweden: Novel subgroups and diabetic complications-The All New Diabetes in Scania cohort diabetic complications and ethnicity. Diabetes Metab Res Rev e3419. https://doi.org/10.1002/dmrr.3419
Zou X, Zhou X, Zhu Z, Ji L (2019) Novel subgroups of patients with adult-onset diabetes in Chinese and US populations. Lancet Diabetes Endocrinol 7(1):9–11. https://doi.org/10.1016/S2213-8587(18)30316-4
Tanabe H, Saito H, Kudo A et al (2020) Factors Associated with Risk of Diabetic Complications in Novel Cluster-Based Diabetes Subgroups: A Japanese Retrospective Cohort Study. J Clin Med 9(7):2083. https://doi.org/10.3390/jcm9072083
Anjana RM, Baskar V, Nair ATN et al (2020) Novel subgroups of type 2 diabetes and their association with microvascular outcomes in an Asian Indian population: a data-driven cluster analysis: the INSPIRED study. BMJ Open Diabetes Res Care 8(1):e001506. https://doi.org/10.1136/bmjdrc-2020-001506
Zaharia OP, Strassburger K, Strom A et al (2019) Risk of diabetes-associated diseases in subgroups of patients with recent-onset diabetes: a 5-year follow-up study. Lancet Diabetes Endocrinol 7(9):684–694. https://doi.org/10.1016/S2213-8587(19)30187-1
Dennis JM, Shields BM, Henley WE, Jones AG, Hattersley AT (2019) Disease progression and treatment response in data-driven subgroups of type 2 diabetes compared with models based on simple clinical features: an analysis using clinical trial data. Lancet Diabetes Endocrinol 7(6):442–451. https://doi.org/10.1016/S2213-8587(19)30087-7
Kahkoska AR, Geybels MS, Klein KR et al (2020) Validation of distinct type 2 diabetes clusters and their association with diabetes complications in the DEVOTE, LEADER and SUSTAIN-6 cardiovascular outcomes trials. Diabetes Obes Metab 22(9):1537–1547. https://doi.org/10.1111/dom.14063
ORIGIN Trial Investigators, Gerstein HC, Bosch J et al (2012) Basal insulin and cardiovascular and other outcomes in dysglycemia. N Engl J Med 367(4):319–328. https://doi.org/10.1056/NEJMoa1203858
ORIGIN Trial Investigators (2013) Characteristics associated with maintenance of mean A1C<6.5% in people with dysglycemia in the ORIGIN trial. Diabetes Care 36(10):2915–2922. https://doi.org/10.2337/dc12-2238
Birkeland KI, Grill V, Wium C et al (2019) The association of basal insulin treatment versus standard care with outcomes in anti-GAD positive and negative subjects: A post-hoc analysis of the ORIGIN trial. Diabetes Obes Metab 21(2):429–433. https://doi.org/10.1111/dom.13528
Gerstein HC, Paré G, McQueen MJ et al (2015) Identifying Novel Biomarkers for Cardiovascular Events or Death in People With Dysglycemia. Circulation 132(24):2297–2304. https://doi.org/10.1161/CIRCULATIONAHA.115.015744
Gerstein HC, Paré G, McQueen MJ, Lee SF, Hess S, ORIGIN Trial Investigators (2017) Validation of the ORIGIN Cardiovascular Biomarker Panel and the Value of Adding Troponin I in Dysglycemic People. J Clin Endocrinol Metab 102(7):2251–2257. https://doi.org/10.1210/jc.2017-00273
ORIGIN trial investigators, Gilbert RE, Mann JFE et al (2014) Basal insulin glargine and microvascular outcomes in dysglycaemic individuals: results of the Outcome Reduction with an Initial Glargine Intervention (ORIGIN) trial. Diabetologia 57(7):1325–1331. https://doi.org/10.1007/s00125-014-3238-4
Mattix HJ, Hsu C, Shaykevich S, Curhan G (2002) Use of the albumin/creatinine ratio to detect microalbuminuria: implications of sex and race. J Am Soc Nephrol JASN 13(4):1034–1039. https://doi.org/10.1681/ASN.V1341034
R Core Team (2013) R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria
Udler MS, Kim J, von Grotthuss M et al (2018) Type 2 diabetes genetic loci informed by multi-trait associations point to disease mechanisms and subtypes: A soft clustering analysis. PLoS Med 15(9):e1002654. https://doi.org/10.1371/journal.pmed.1002654
Dennis JM (2020) Precision Medicine in Type 2 Diabetes: Using Individualized Prediction Models to Optimize Selection of Treatment. Diabetes 69(10):2075–2085. https://doi.org/10.2337/dbi20-0002
Acknowledgements
We are thankful to all the participants for contributing to this project.
Authors’ relationships and activities
MP declares that there are no relationships or activities that might bias, or be perceived to bias, her work. SH is an employee of Sanofi. GP has received consulting fees from Sanofi, Bristol Myers Squibb, Lexicomp and Amgen, and support for research through his institution from Sanofi. HG has received consulting fees from Sanofi, Novo Nordisk, Lilly, AstraZeneca, Boehringer Ingelheim and GlaxoSmithKline, and support for research or continuing education through his institution from Sanofi, Lilly, Takeda, Novo Nordisk, Boehringer Ingelheim and AstraZeneca.
Funding
The ORIGIN trial and biomarker project were supported by Sanofi and the Canadian Institutes of Health Research (CIHR). The biomarker project was led by ORIGIN investigators at the Population Health Research Institute (Hamilton, Canada) with the active collaboration of Sanofi scientists. Sanofi directly compensated Myriad RBM, Inc. for measurement of the biomarker panel and the Population Health Research Institute for scientific, methodological and statistical work. This project was also supported by the Innovative Medicines Initiative 2 Joint Undertaking under grant agreement no. 115974 (BEAt-DKD). This Joint Undertaking receives support from the European Union’s Horizon 2020 Research and Innovation Programme, the European Federation of Pharmaceutical Industries and Associations, and JDRF. Genetic analysis of ORIGIN participants was supported by CIHR (award 125794 to GP). MP is supported by the E.J. Moran Campbell Internal Career Research Award from McMaster University. MFG is supported by the Swedish Heart and Lung Foundation (20160872), the Swedish Research Council (2018-02837; 2014-03352; EXODIAB 2009-1039) and the Swedish Foundation for Strategic Research (LUDC-IRC 15-0067). GP is supported by the Canada Research Chair in Genetic and Molecular Epidemiology and the CISCO Professorship in Integrated Health Biosystems. The study funder was not involved in the design of the study; the collection, analysis, and interpretation of data; writing the report; and did not impose any restrictions regarding the publication of the report.
Author information
Authors and Affiliations
Contributions
SH and LG proposed validating the type 2 diabetes clusters in the ORIGIN cohort. MP, GP and HG designed the study, planned the analyses, interpreted the results and wrote the manuscript. MP and OA performed the statistical and bioinformatics analyses. SH suggested including C-peptide in the biomarker panel and coordinated the biomarker screen at Myriad RBM, Inc. (Austin, TX, USA). MFG contributed to the interpretation of the data. All authors contributed to the critical reading and revision of the manuscript. All authors have approved the submitted version of this manuscript. HG is the guarantor of this work.
Corresponding author
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
ESM
(PDF 185 kb)
Rights and permissions
About this article

Cite this article
Pigeyre, M., Hess, S., Gomez, M.F. et al. Validation of the classification for type 2 diabetes into five subgroups: a report from the ORIGIN trial. Diabetologia 65, 206–215 (2022). https://doi.org/10.1007/s00125-021-05567-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-021-05567-4