
Abstract
Pregnancy is a key mammalian reproductive event in which growth and differentiation of the fetus imposes extra metabolic and hormonal demands on the mother. Its successful outcome depends on major changes in maternal blood circulation, metabolism and endocrine function. One example is the endocrine pancreas, where beta cells undergo a number of changes in pregnancy that result in enhanced functional beta cell mass in order to compensate for the rising metabolic needs for maternal insulin. During the last 5 years, a series of studies have increased our understanding of the molecular events involved in this functional adaptation. In the mouse, a prominent functional change during pregnancy is the capacity of some beta cells to produce serotonin. In this review we will discuss the mechanism and potential effects of pregnancy-related serotonin production in beta cells, considering functional consequences at the local intra-islet and systemic level.
Similar content being viewed by others


Introduction
Pregnancy and lactation are key reproductive events that distinguish mammals from other vertebrates. To regulate these processes, the endocrine networks of the pregnant mother undergo drastic changes in order to accommodate the high metabolic demand of fetal growth. In one such network, pancreatic beta cells dynamically upregulate the capacity for insulin secretion to meet elevated physiological demands [1]. The importance of this adaptation is illustrated by gestational diabetes, a condition that develops when beta cells fail to meet these demands and that has adverse health effects both on fetus and mother [2].
Two components enhance the secretory capacity of the pancreatic beta cell population during pregnancy. First, beta cell number increases because some of the already existing beta cells in the pre-pregnant state replicate [3]. Second, as a functional community, beta cells work harder in pregnancy, delivering more insulin into the circulation per unit of time. This is achieved by a combination of factors such as cellular hypertrophy [1], increased proinsulin biosynthesis [4], larger insulin stores [4] and a greater elevation in insulin secretory response after glucose stimulation [5].
The present review addresses two aspects of an endocrine network in which the plasticity of mouse beta cells is involved. First, placental lactogens (PLs), a series of peptides closely related to growth hormone (GH) and prolactin (PRL) [6], have potent and diverse effects on beta cells [1]. These effects are mediated via prolactin receptors (PRLRs), which are abundantly expressed on mouse beta cells [7]. Second, among the most upregulated genes of PL-stimulated mouse beta cells are the genes encoding the two non-allelic isoforms of tryptophan hydroxylase (TPH), the rate-limiting enzyme for serotonin biosynthesis. The upregulated synthesis and secretion of serotonin during pregnancy may contribute to an endocrine network of reproductive metabolism and this needs to be studied in further detail. But there are many questions and few final answers. What could the physiological role of this beta cell serotonin be? Could the amine act as a local regulator of beta cell function? Would the secreted serotonin—together with insulin—represent a feedback hormonal signal from PL-stimulated beta cells to the placenta and, if so, could beta cell serotonin be relevant for reproductive fitness?
PLs, PRLRs and pregnancy-related changes in rodent beta cells
Placental hormones drive many of the metabolic changes that occur during pregnancy. An important example is the PL family, which has a different evolutionary origin in various mammalian species [8]. As illustrated in Fig. 1, species-specific gene amplification events in two different loci contribute to a variety of different peptides. A solitary Gh gene is found in rodents, while the Prl gene is amplified to 26 different paralogous genes in mice and 20 genes in rats. In contrast, humans and other primates have a single PRL gene but GH is copied several times, giving rise to the chorionic somatomammotropin gene family.

The genomic organisation of the PRL and GH gene loci in mice and humans. The human PRL family has only one member, PRL, while in mice there are many local gene duplications resulting in a gene cluster on chromosome 13. The figure shows 23 genes of the Prl family that are located in one large gene cluster; the remaining three duplicate genes are outside this cluster (not shown). In contrast, the murine Gh gene family contains only one member, while in the human genome, the orthologous gene is duplicated several times, giving rise to the chorionic somatomammotropin gene cluster on chromosome 17
PLs act on PRLRs, which together with GH receptors belong to the class 1 cytokine receptor family. Characteristic features of the family members include possession of one transmembrane domain and a signal transduction machinery that depends on activation of Janus kinase 2 (JAK2) and phosphorylation of signal transducer and activator of transcription 5 (STAT5) [7]. Multiple Prlr transcript variants are known to exist; the encoded protein variants differ in their cytoplasmic tail length, signal transduction pathways used and tissue specificity of expression [9]. While liver predominantly expresses the short isoform, pancreatic beta cells primarily utilise the long variant of the receptor. Of all tissues examined, expression of PRLRs on rodent beta cells is among the highest and PLs are known to be an important signal for the altered regulation of glucose homeostasis during pregnancy [10, 11]. However, a number of interspecies differences exist and these may contribute to the wide variation in reproductive and growth patterns seen between different mammalian families. The striking interspecies difference in gene duplication events of the Prl and Gh gene loci (Fig. 1) may explain why human GH is more PRL-like than mouse GH in terms of pregnancy-induced changes in beta cells [12]. Mice and rats also exhibit some subtle differences in the expression profile of Ghr and Prlr, particularly in response to pregnancy. Moldrup et al [13] observed a low Prlr mRNA signal in the liver of male rats, and these authors measured pregnancy-induced increase in Ghr signal, in both the liver and pancreatic islet samples. In the liver of male mice, however, expression of the short PRLR isoform was detected [14, 15]. Moreover, in pancreatic islets of pregnant mice, Prlr expression was upregulated but no change occurred in Ghr expression [16, 17]. We will further comment on the differences between human and mouse islets in a separate section.
To better understand the molecular basis of PL on pancreatic beta cells, several research groups have analysed mRNA in murine islets of Langerhans during pregnancy [16–19] and differentially expressed genes have been classified into various clusters. A first interesting regulated gene cluster concentrates on the direct biological effects of the PRLR itself. Confirming earlier reports, it was found that the mRNA signal encoding PRLR is significantly upregulated during pregnancy [16], indicating the presence of a positive feedback loop by which initial stimulation of PRLR by PL leads to reinforcement of the signalling axis. On the other hand, the feed-forward signal is counterbalanced by upregulation of two transcripts encoding suppressors of cytokine signalling, CISH [20] and SOCS2 [21]. In addition, in a mouse model of PRLR deficiency we have shown that expression of this receptor is required for the induction of many of the pregnancy-related mRNA changes in mouse islets of Langerhans [14]. We observed that the largest functional cluster of upregulated genes during pregnancy encodes cell cycle proteins and that their expression peaks at around day 9.5 of pregnancy (P9.5) [17]. Examples of these upregulated genes include Mki67, Top2a and genes encoding different cyclins. The expression of two genes that were earlier mentioned in the literature, Foxm1 and Men1 [22, 23], was, however, found not to be significantly altered in these screens. This may be explained by the too-low sensitivity of the screen or by the possibility that many regulatory events of replication occur at the post RNA level.
Serotonin production in mouse beta cells during pregnancy
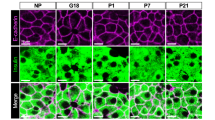
Transcriptome analysis of islets isolated from pregnant mice identified very strong upregulation of the two paralogous genes encoding tryptophan hydroxylase, Tph1 (Fig. 2a) and Tph2 [16, 18, 19, 24]. These isoforms catalyse the same chemical reaction: the NADPH-dependent hydroxylation of carbon 5 of tryptophan to form 5-hydroxytryptophan, which is the substrate of the second step of serotonin biosynthesis mediated by the enzyme aromatic amino acid decarboxylase (also known as DOPA decarboxylase or DDC) [25]. As Ddc is already expressed in islets from non-pregnant female mice and in those from male mice, the upregulation of the tryptophan hydroxylase reaction introduces the complete pathway of serotonin biosynthesis in islets. Previous literature mentions the tissue-specific use of the two paralogous genes: Tph1 in enterochromaffin cells, mammary and pineal glands [26, 27] and Tph2 in the raphe nuclei of the brain [28]. A new tissue pattern was found in islets where both Tph1 and Tph2 are induced during pregnancy [16, 18, 19, 24], fitting the idea that endocrine beta cells share many characteristics with neuronal cells [29]. Moreover, in one study [24] it was verified that the spectacular increase in Tph expression occurred specifically in pancreatic islets and not in other tissues. Are both paralogous genes important for serotonin production in beta cells? Time course analysis of pregnant mice indicated that in isolated islets Tph1 expression correlates better than the expression of Tph2 to serotonin content [18, 24]. Furthermore, the immunoreactivity of islets from pregnant whole-body Tph1-knockout mice was reduced below the level of detection (Fig. 2b) [24].

Tph1 mRNA is highly induced in the islets of pregnant mice. (a) The microarray expression profile of Tph1 mRNA in 28 mouse tissues, MIN6 cells and embryonic stem (ES) cells, presented as mean ± SD. (b) Heterogeneous pattern of pancreatic islet serotonin immunostaining at day 12.5 of pregnancy (P12.5); the immunoreactive signal is lost in islets from Tph1-knockout (Tph1KO) mice. Magnification ×40. Figure adapted with permission from Schraenen et al [24]. (c) Fluorescent staining of serotonin and insulin in MIN6 cells that were cultured with PL. Scale bar, 10 μm. Figure adapted with permission from Schraenen et al [24]. (d) Syngeneic islets from male donors transplanted under the kidney capsule of female acceptor mice stained for serotonin at day 12.5 of pregnancy. Scale bar, 50 μm. Figure adapted from Goyvaerts et al [14]
The upregulation of Tph1 and Tph2 is dependent on activation of PRLRs [18, 24]. This has been demonstrated in in vitro models wherein the increase in Tph1/2 mRNA can be mimicked in a monolayer culture of islets of non-pregnant mice by stimulating them with nanomolar concentrations of ovine PL. The same phenomenon can be induced in the mouse beta cell line MIN6 (Fig. 2c).
Among the different possible signalling mechanisms of the long PRLR isoform, the JAK2–STAT5 pathway seems most important (Fig. 3). PRL signalling is triggered by PRL binding to preformed PRLR homodimers [7, 24, 30]. JAK2 is constitutively associated with the PRLR and is phosphorylated after PL binding. Phosphorylated JAK2 recruits and phosphorylates STAT5 [31], which dimerises and migrates to the nucleus where it acts as a transcription factor. STAT5 recognises its target genes via an interferon γ-activated motif, which was recently identified in the Tph1 gene promoter [30].

Mechanism and consequences of serotonin production in mouse pancreatic beta cells during pregnancy. Binding of PL/PRL to the PRLR activates the JAK2–STAT5 signalling cascade. Phosphorylated STAT5 dimerises and translocates to the nucleus where it targets specific promoter sequences of Tph1/Tph2 genes resulting in mRNA transcription, enzyme production and serotonin biosynthesis in a subset of beta cells. Secreted serotonin can act locally on other beta cells through HTR2B and HTR3 receptors or it might regulate islet blood flow. In addition, beta cell serotonin could have a systemic effect when it enters the blood circulation. DDC, DOPA decarboxylase
Beta cell heterogeneity
Early studies of glucose-induced insulin release [32] and glucose-stimulated insulin biosynthesis [33] indicated that important phenotypic differences exist between adult beta cells, so that a common signal for all cells elicits a heterogeneous cellular response. A related phenomenon was found when assessing PL-induced serotonin immunoreactivity in individual beta cells, both in vivo during pregnancy (Fig. 2b) and in vitro—either with isolated islets or with the mouse beta cell line MIN6 (Fig. 2c) [24]. Although the percentage of serotonin-producing cells was dependent on the PL concentration, it never exceeded 50%. A similar cellular heterogeneity was observed in an islet transplantation model (Fig. 2d) wherein female mice transplanted with islets from male or female mice were studied during pregnancy [14]. Why only part of the beta cell population is triggered to produce serotonin after stimulation with PL is not known. It is conceivable that the serotonin-immunoreactive beta cells are enriched in the beta cell subpopulations that do or do not proliferate during pregnancy, but no quantitative data are yet available in the literature. There are no anatomical details available indicating whether serotonin-producing beta cells are closer to nerve endings or arterioles of the islet microcirculation. What surprises most is that this type of heterogeneity is also present in a cultured cell line (Fig. 2c) indicating that stochastic models of chromatin conformation could apply to the Tph1 promoter region [34].
Local effects of islet serotonin
In this section and the next we will review studies that examined the local and distal effects of serotonin that could contribute to the outcome of pregnancy in mice. For serotonin to exert local actions, serotonin-responsive beta cells must express serotonin receptors at sufficient levels to become biologically stimulated or inhibited (Fig. 3). In the mouse genome, 14 different serotonin receptor genes are known [35]. Serotonin receptors are classified into seven different families (serotonin receptor 1–7 [HTR1–7]), some of which contain different subtypes. All but one of these receptor types are G-protein-coupled receptors (GPCRs), the exception being serotonin 3 receptors (HTR3A and HTR3B), which are ligand-gated cation channels. The different GPCR subtypes couple to different signalling cascades. As a consequence, serotonin has a wide variety of functions in different organs in the body [26].
Which of these serotonin receptors could play a functional role during pregnancy in mouse islets? Relatively few studies are available at present and unfortunately the results obtained are inconsistent. Kim et al reported that mRNA encoding serotonin 2B receptors (Htr2b) was upregulated between day 6 and 18 of pregnancy [18]. The authors assessed beta cell proliferation using BrdU staining in Htr2b −/− mice and observed fewer proliferating beta cells when compared with Htr2b +/+ mice. In addition, the Htr2b −/− mice had impaired glucose tolerance at day 13 of pregnancy when compared with wild-type mice. Towards the end of pregnancy, the same authors found that there was an increase in Htr1d mRNA and they correlated this event to normalisation of beta cell mass after delivery [18]. Their interpretation was that pregnancy-related beta cell proliferation in the mouse is upregulated by HTR2B and downregulated by HTR1D on beta cells, making serotonin a paracrine regulator of beta cell proliferation. This interesting hypothesis fitted with data in other organs, such as the regenerating liver, where platelet-derived serotonin acts as a mitogen [36]. However, the findings of Kim et al [18] were not confirmed by other groups. For instance, Layden et al [19] screened for Gpcr mRNA signals in pancreatic islets, detected mRNA expression of 343 different GPCRs in mouse islets and found 216 GPCRs above the detection limit in islets from pregnant mice. However, these authors did not find pregnancy-related changes in Htr2b mRNA in islets and reported the Htr1d signal to be undetectable [19]. In our laboratory, we performed quantitative RT-PCR analysis on mRNA of islets from pregnant and non-pregnant mice: signals for both Htr1d and Htr2b mRNA were below the detection limit [24]. Furthermore, our analyses of beta cell proliferation in a mouse model of whole-body Tph1 deficiency still detected normal levels of beta cell proliferation during pregnancy, while the signal of serotonin immunoreactivity in the islets disappeared (Fig. 2b) [24]. The basis for the discrepant results between different laboratories is not clear, so currently there is no agreement as to whether or not serotonin regulates beta cell proliferation in the mouse.
Could paracrine or autocrine serotonin regulate insulin release? This question was examined using a mouse model of Htr3a deficiency; this gene is constitutively expressed in islets of non-pregnant and pregnant wild-type mice [37]. The homozygous Htr3a −/− mouse strain had normal glucose tolerance except during pregnancy; this abnormality was accompanied by reduced compensatory upregulation of glucose-induced insulin secretion during pregnancy. The proposed underlying mechanism is a lack of HTR3A channels, which in wild-type animals allow serotonin-mediated influx of cations, depolarising the resting membrane potential and lowering the threshold for glucose-induced insulin secretion [37]. However, other studies have already shown that insulin secretion is increased due to a PRL-induced increase in glucokinase expression [38]. More research is needed to investigate how these two pathways regulate insulin secretion together during pregnancy.
Another possible local effect is that islet blood flow is regulated by pregnancy-induced serotonin production. This merits further investigation as serotonin is known to have potent vasoconstricting effects [39] and because pancreatic islets are known to have a very high local blood flow that may underlie part of the functional heterogeneity between islets [40]. Previous studies of blood flow in islets of pregnant rats demonstrated that islet blood flow was increased at day 15 of pregnancy but decreased at day 18 of pregnancy [30]. Regarding the heterogeneous pattern of the serotonin-producing cells throughout the islet, it could be that local production of serotonin at strategically interesting locations in the islet regulates arterioles in islets of pregnant mice.
Effects of maternal serotonin on reproductive outcome
In addition to the potential local effects of serotonin, it is conceivable that beta cell serotonin is secreted into the bloodstream in which it would contribute to the free circulating or platelet serotonin pool and exert effects on other organs. In this context it is interesting to consider a bidirectional endocrine islet–placenta axis of communication in which PL-stimulated beta cells use the serotonin signal to communicate with the different placental/fetal units of a pregnant mouse. Former studies have shown that maternal serotonin is important for normal reproduction and fetal development [27, 41, 42]. Deletion of the Htr2b gene leads to some embryonic lethality before day 11.5 of pregnancy [41]. Surviving embryos display impaired heart development, including enlarged hearts and pericardial blood leakage, and surviving Htr2b −/− offspring display a progressive cardiomyopathy in which ventricular mass decreases due to a reduction in both number and size of the cardiomyocytes [43].
Consistent with these findings, Côté et al studied Tph1-deficient mice and found that the mutant mice displayed abnormal cardiac activity, resulting in progressive heart failure [44]. Another interesting finding was that Tph1 +/− embryos born from Tph1 −/− mothers had severe brain abnormalities, while Tph1 −/− embryos from Tph1 +/− mothers were normal [42] indicating that the fetal phenotype is correlated with the maternal rather than the fetal genotype. The interpretation was that maternal serotonin contributes to embryonic brain development before a fetus starts to synthesise its own brain serotonin. The maternal genotype was also found to correlate with the severity of cardiomyopathy of Tph1 mutant mice [45]. These studies did not identify the maternal tissue responsible for the serotonin needed for normal embryonic development. It remains to be investigated whether the pregnancy-induced production of serotonin in beta cells could be relevant. One possibility could be that beta cell signals contribute to serotonin production in the fetal part of the placenta from day 10.5 of pregnancy, which has been suggested to be important for brain development [46].
Of mice and men
Figure 1 indicates that primates and rodents evolve differently to produce placental paralogues of the PRL–GH family. This poses the question as to whether or not PL-induced changes observed in islets from pregnant mice also occur in pregnant humans. Early studies with purified rat beta cells and human islets indicated that profound interspecies differences exist for very important membrane proteins such as GLUT2 [47]. In a genome-wide search for differences between human and rodent islets, PRLR was among the mRNA transcripts that were much more highly expressed in rodents than in humans [48]. An important difference between human and murine islets during pregnancy is that in humans there is little or no beta cell proliferation [49]. Moreover, PRL cannot induce human beta cell proliferation [15]. This unresponsiveness has been explained by a combination of low PRLR expression and low mitogenic effect of STAT5 signalling [50]. Literature concerning human autopsy material of patients that died during pregnancy is scarce. In the study by Kim et al [18] serotonin-immunoreactive staining of the human endocrine pancreas was reported, but these observations have not yet been confirmed by others. Taken together, these studies indicate that it may not be possible to extrapolate the pregnancy-induced serotonin production in mouse islets to human pregnancy.
Conclusion and perspectives
In summary, a series of recent studies indicate that during pregnancy in the mouse, PLs stimulate islet PRLRs to trigger a strong upregulation of both isoforms of TPH, which activate serotonin synthesis and secretion by some (but not all) pancreatic beta cells. There are preliminary data that suggest islet serotonin may mediate some of the seminal changes (proliferation, higher insulin secretory response) that occur in the islets of pregnant mice, but these findings still need to be confirmed. The availability of a beta cell-specific Tph1-knockout mouse model would be a welcome addition to the research field. There are compelling reasons, other than the physiology of pregnancy, for clearly understanding the functional consequences of serotonin production by mouse beta cells, as serotonin production is artificially induced in many commonly used transgenic mouse models. The underlying reason is that the human GH minigene is used as a technical device to drive expression of the desired transgene [12].
Another point of interest is the mechanism of heterogeneity: why is the intense production of serotonin only triggered in a subset of the beta cell population; does the anatomical position in the islet matter and could this be related to changes in pancreatic blood flow?
Also desirable is a better understanding of the important species differences between mammals. Rodent models are often used in diabetes research, with the assumption that results can be extrapolated to humans. But many differences between human and rodent beta cells have been found over the past decades and it seems likely that pregnancy-induced serotonin production in beta cells is one of these points of difference. Figure 1 explains that the paralogues of the Prl and Gh families have expanded in a fundamentally different manner in rodents on the one hand and humans on the other. This difference makes human GH much more ‘lactogenic’ than mouse GH. Furthermore, the expression level of GH receptors and PRLRs is much lower in human islets than in mouse islets [48], leading to the prediction that serotonin is not induced in beta cells during human pregnancy. Even between mouse and rat beta cells there may be important differences in islet Ghr and Prlr mRNA expression [13]. Because serotonin reuptake inhibitors are commonly used in patients with depression, a condition that is sometimes associated with pregnancy, it is of great interest to study the influence of serotonin on human beta cells in greater detail.
Abbreviations
- GH:
-
Growth hormone
- GPCR:
-
G-protein-coupled receptor
- HTR:
-
Serotonin receptor
- JAK2:
-
Janus kinase 2
- PL:
-
Placental lactogen
- PRL:
-
Prolactin
- PRLR:
-
Prolactin receptor
- STAT5:
-
Signal transducer and activator of transcription 5
- TPH:
-
Tryptophan hydroxylase
References
Sorenson RL, Brelje TC (1997) Adaptation of islets of Langerhans to pregnancy: beta-cell growth, enhanced insulin secretion and the role of lactogenic hormones. Horm Metab Res 29:301–307
Barbour LA, McCurdy CE, Hernandez TL, Kirwan JP, Catalano PM, Friedman JE (2007) Cellular mechanisms for insulin resistance in normal pregnancy and gestational diabetes. Diabetes Care 30(Suppl. 2): S112–S119
Rieck S, Kaestner KH (2010) Expansion of β-cell mass in response to pregnancy. Trends Endocrinol Metab 21:151–158
Bone AJ, Howell SL (1977) Alterations in regulation of insulin biosynthesis in pregnancy and starvation studied in isolated rat islets of Langerhans. Biochem J 166:501–507
Green IC, Perrin D, Howell SL (1978) Insulin release in isolated islets of Langerhans of pregnant rats. Relationship between glucose metabolism and cyclic AMP. Horm Metab Res 10:32–35
Soares MJ, Konno T, Alam SM (2007) The prolactin family: effectors of pregnancy-dependent adaptations. Trends Endocrinol Metab 18:114–121
Bole-Feysot C, Goffin V, Edery M, Binart N, Kelly PA (1998) Prolactin (PRL) and its receptor: actions, signal transduction pathways and phenotypes observed in PRL receptor knockout mice. Endocr Rev 19:225–268
Soares MJ (2004) The prolactin and growth hormone families: pregnancy-specific hormones/cytokines at the maternal-fetal interface. Reprod Biol Endocrinol 2:51
Binart N, Bachelot A, Bouilly J (2010) Impact of prolactin receptor isoforms on reproduction. Trends Endocrinol Metab 21:362–368
Sorenson RL, Brelje TC (2009) Prolactin receptors are critical to the adaptation of islets to pregnancy. Endocrinology 150:1566–1569
Huang C, Snider F, Cross JC (2009) Prolactin receptor is required for normal glucose homeostasis and modulation of β-cell mass during pregnancy. Endocrinology 150:1618–1626
Brouwers B, de Faudeur G, Osipovich AB et al (2014) Impaired islet function in commonly used transgenic mouse lines due to human growth hormone minigene expression. Cell Metab 20:979–990
Moldrup A, Petersen ED, Nielsen JH (1993) Effects of sex and pregnancy hormones on growth hormone and prolactin receptor gene expression in insulin-producing cells. Endocrinology 133:1165–1172
Goyvaerts L, Lemaire K, Arijs I et al (2015) Prolactin receptors and placental lactogen drive male mouse pancreatic islets to pregnancy-related mRNA changes. PLoS One 10:e0121868
Davis JA, Linzer DI (1989) Expression of multiple forms of the prolactin receptor in mouse liver. Mol Endocrinol 3:674–680
Rieck S, White P, Schug J et al (2009) The transcriptional response of the islet to pregnancy in mice. Mol Endocrinol 23:1702–1712
Schraenen A, de Faudeur G, Thorrez L et al (2010) mRNA expression analysis of cell cycle genes in islets of pregnant mice. Diabetologia 53:2579–2588
Kim H, Toyofuku Y, Lynn FC et al (2010) Serotonin regulates pancreatic beta cell mass during pregnancy. Nat Med 16:804–808
Layden BT, Durai V, Newman MV et al (2010) Regulation of pancreatic islet gene expression in mouse islets by pregnancy. J Endocrinol 207:265–279
Yoshimura A, Ohkubo T, Kiguchi T et al (1995) A novel cytokine-inducible gene CIS encodes an SH2-containing protein that binds to tyrosine-phosphorylated interleukin 3 and erythropoietin receptors. EMBO J 14:2816–2826
Rico-Bautista E, Flores-Morales A, Fernandez-Perez L (2006) Suppressor of cytokine signaling (SOCS) 2, a protein with multiple functions. Cytokine Growth Factor Rev 17:431–439
Zhang H, Zhang J, Pope CF et al (2010) Gestational diabetes mellitus resulting from impaired beta-cell compensation in the absence of FoxM1, a novel downstream effector of placental lactogen. Diabetes 59:143–152
Karnik SK, Chen H, McLean GW et al (2007) Menin controls growth of pancreatic beta-cells in pregnant mice and promotes gestational diabetes mellitus. Science 318:806–809
Schraenen A, Lemaire K, de Faudeur G et al (2010) Placental lactogens induce serotonin biosynthesis in a subset of mouse beta cells during pregnancy. Diabetologia 53:2589–2599
Tyce GM (1990) Origin and metabolism of serotonin. J Cardiovasc Pharmacol 16(Suppl. 3): S1–S7
Berger M, Gray JA, Roth BL (2009) The expanded biology of serotonin. Annu Rev Med 60:355–366
Matsuda M, Imaoka T, Vomachka AJ et al (2004) Serotonin regulates mammary gland development via an autocrine-paracrine loop. Dev Cell 6:193–203
Walther DJ, Peter JU, Bashammakh S et al (2003) Synthesis of serotonin by a second tryptophan hydroxylase isoform. Science 299:76
Thiel G, Schuit F (2008) No REST for healthy beta cells. Diabetologia 51:1343–1346
Iida H, Ogihara T, Min MK et al (2015) Expression mechanism of tryptophan hydroxylase 1 in mouse islets during pregnancy. J Mol Endocrinol 55:41–53
Horvath CM (2000) STAT proteins and transcriptional responses to extracellular signals. Trends Biochem Sci 25:496–502
Salomon D, Meda P (1986) Heterogeneity and contact-dependent regulation of hormone secretion by individual B cells. Exp Cell Res 162:507–520
Schuit FC, In’t Veld PA, Pipeleers DG (1988) Glucose stimulates proinsulin biosynthesis by a dose-dependent recruitment of pancreatic beta cells. Proc Natl Acad Sci U S A 85:3865–3869
Kaern M, Elston TC, Blake WJ, Collins JJ (2005) Stochasticity in gene expression: from theories to phenotypes. Nat Rev Genet 6:451–464
Hannon J, Hoyer D (2008) Molecular biology of 5-HT receptors. Behav Brain Res 195:198–213
Lesurtel M, Graf R, Aleil B et al (2006) Platelet-derived serotonin mediates liver regeneration. Science 312:104–107
Ohara-Imaizumi M, Kim H, Yoshida M et al (2013) Serotonin regulates glucose-stimulated insulin secretion from pancreatic beta cells during pregnancy. Proc Natl Acad Sci U S A 110:19420–19425
Weinhaus AJ, Stout LE, Bhagroo NV, Brelje TC, Sorenson RL (2007) Regulation of glucokinase in pancreatic islets by prolactin: a mechanism for increasing glucose-stimulated insulin secretion during pregnancy. J Endocrinol 193:367–381
Rapport MM, Green AA, Page IH (1948) Partial purification of the vasoconstrictor in beef serum. J Biol Chem 174:735–741
Lau J, Svensson J, Grapensparr L, Johansson A, Carlsson PO (2012) Superior beta cell proliferation, function and gene expression in a subpopulation of rat islets identified by high blood perfusion. Diabetologia 55:1390–1399
Nebigil CG, Choi DS, Dierich A et al (2000) Serotonin 2B receptor is required for heart development. Proc Natl Acad Sci U S A 97:9508–9513
Côté F, Fligny C, Bayard E et al (2007) Maternal serotonin is crucial for murine embryonic development. Proc Natl Acad Sci U S A 104:329–334
Nebigil CG, Hickel P, Messaddeq N et al (2001) Ablation of serotonin 5-HT(2B) receptors in mice leads to abnormal cardiac structure and function. Circulation 103:2973–2979
Côté F, Thevenot E, Fligny C et al (2003) Disruption of the nonneuronal tph1 gene demonstrates the importance of peripheral serotonin in cardiac function. Proc Natl Acad Sci U S A 100:13525–13530
Fligny C, Fromes Y, Bonnin P et al (2008) Maternal serotonin influences cardiac function in adult offspring. FASEB J 22:2340–2349
Bonnin A, Goeden N, Chen K et al (2011) A transient placental source of serotonin for the fetal forebrain. Nature 472:347–350
De Vos A, Heimberg H, Quartier E et al (1995) Human and rat beta cells differ in glucose transporter but not in glucokinase gene expression. J Clin Invest 96:2489–2495
Benner C, van der Meulen T, Caceres E, Tigyi K, Donaldson CJ, Huising MO (2014) The transcriptional landscape of mouse beta cells compared to human beta cells reveals notable species differences in long non-coding RNA and protein-coding gene expression. BMC Genomics 15:620
Butler AE, Cao-Minh L, Galasso R et al (2010) Adaptive changes in pancreatic beta cell fractional area and beta cell turnover in human pregnancy. Diabetologia 53:2167–2176
Chen H, Kleinberger JW, Takane KK et al (2015) Augmented Stat5 signaling bypasses multiple impediments to lactogen-mediated proliferation in human β-cells. Diabetes 64:3784–3797
Acknowledgements
We thank K. Lemaire (Cellular and Molecular Medicine, KU Leuven, Belgium) for critical reading of this manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
Research from the Gene Expression Laboratory mentioned in this review was supported by grants from the JDRF (grants 1-2010-393), the KU Leuven (GOA/2009/10) and the Fonds voor Wetenschappelijk Onderzoek Vlaanderen (FWO grant G0B2516N and 12C2912N).
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Contribution statement
All authors were responsible for drafting the article and revising it critically for important intellectual content. All authors approved the version to be published.
Rights and permissions
About this article

Cite this article
Goyvaerts, L., Schraenen, A. & Schuit, F. Serotonin competence of mouse beta cells during pregnancy. Diabetologia 59, 1356–1363 (2016). https://doi.org/10.1007/s00125-016-3951-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-016-3951-2