
Abstract
Aims/hypothesis
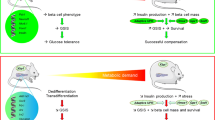
A strategy to enhance pancreatic islet functional beta cell mass (BCM) while restraining inflammation, through the manipulation of molecular and cellular targets, would provide a means to counteract the deteriorating glycaemic control associated with diabetes mellitus. The aims of the current study were to investigate the therapeutic potential of such a target, the islet-enriched and diabetes-linked transcription factor paired box 4 (PAX4), to restrain experimental autoimmune diabetes (EAD) in the RIP-B7.1 mouse model background and to characterise putative cellular mechanisms associated with preserved BCM.
Methods
Two groups of RIP-B7.1 mice were genetically engineered to: (1) conditionally express either PAX4 (BPTL) or its diabetes-linked mutant variant R129W (mutBPTL) using doxycycline (DOX); and (2) constitutively express luciferase in beta cells through the use of RIP. Mice were treated or not with DOX, and EAD was induced by immunisation with a murine preproinsulin II cDNA expression plasmid. The development of hyperglycaemia was monitored for up to 4 weeks following immunisation and alterations in the BCM were assessed weekly by non-invasive in vivo bioluminescence intensity (BLI). In parallel, BCM, islet cell proliferation and apoptosis were evaluated by immunocytochemistry. Alterations in PAX4- and PAX4R129W-mediated islet gene expression were investigated by microarray profiling. PAX4 preservation of endoplasmic reticulum (ER) homeostasis was assessed using thapsigargin, electron microscopy and intracellular calcium measurements.
Results
PAX4 overexpression blunted EAD, whereas the diabetes-linked mutant variant PAX4R129W did not convey protection. PAX4-expressing islets exhibited reduced insulitis and decreased beta cell apoptosis, correlating with diminished DNA damage and increased islet cell proliferation. Microarray profiling revealed that PAX4 but not PAX4R129W targeted expression of genes implicated in cell cycle and ER homeostasis. Consistent with the latter, islets overexpressing PAX4 were protected against thapsigargin-mediated ER-stress-related apoptosis. Luminal swelling associated with ER stress induced by thapsigargin was rescued in PAX4-overexpressing beta cells, correlating with preserved cytosolic calcium oscillations in response to glucose. In contrast, RNA interference mediated repression of PAX4-sensitised MIN6 cells to thapsigargin cell death.
Conclusions/interpretation
The coordinated regulation of distinct cellular pathways particularly related to ER homeostasis by PAX4 not achieved by the mutant variant PAX4R129W alleviates beta cell degeneration and protects against diabetes mellitus. The raw data for the RNA microarray described herein are accessible in the Gene Expression Omnibus database under accession number GSE62846.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The islet of Langerhans is the core unit of the endocrine pancreas, which regulates blood glucose homeostasis. Regulation is achieved by the release of insulin from beta cells in response to increasing levels of glucose and by the secretion of glucagon from alpha cells under fasting conditions. Imbalance in this circuitry leads to either hyperglycaemia, the hallmark of diabetes mellitus, or hypoglycaemia. Loss of beta cell function coupled to insulin resistance of target tissues, which usually associates with obesity and chronic low-grade inflammation, defines type 2 diabetes [1]; high-grade T lymphocyte inflammation mediating autoimmune beta cell destruction is characteristic of type 1 diabetes [2].
Emerging evidence suggests that alterations in endoplasmic reticulum (ER) function contribute to beta cell disarray in both type 1 and 2 diabetes [3]. In an attempt to restore ER function and prevent apoptosis, cells activate the unfolded protein response (UPR) [4]. The crucial role of the UPR in balancing beta cell death and survival is illustrated in the human Wolfram and Wolcott–Rallison syndromes, in which mutations in UPR genes result in unresolved ER stress, beta cell death and early-onset diabetes [5, 6]. Interestingly, MODY genes such as Pdx1 and Hnf1a regulate UPR-associated genes [7, 8]. These clinical conditions suggest that islet-enriched transcription factors involved in insulin biosynthesis and secretion also preserve the BCM by limiting ER stress.
Paired box (Pax) genes encode transcription factors critical for tissue development and cellular differentiation [9]. Paired box 4 (PAX4) is necessary for the generation of pancreatic islet cell progenitors and their differentiation towards beta cells [10, 11]. Pax4 gene mutations have been associated with type 1 and 2 diabetes as well as with ketosis-prone diabetes, suggesting a key role of PAX4 in mature islets [12, 13]. Accordingly, overexpression of PAX4 in adult beta cells was shown to block streptozotocin (STZ)-induced hyperglycaemia in mice whereas the diabetes-linked variant PAX4R129W was less efficient [14]. Despite differences in nitric oxide synthase 2 (NOS2) levels, both PAX4- and PAX4R129W-expressing islets exhibited similar levels of cytokine-induced NO production, indicating that the nuclear factor-κB (NF-κB) signalling pathway was fully activated and that additional anti-apoptotic pathways are involved in islet survival. Consistent with this premise, PAX4 islets expressed higher levels of B cell CLL/lymphoma 2 (BCL-2) [14]. Nonetheless, overexpression of BCL-2 in islets did not prevent autoimmune-mediated beta cell destruction and development of hyperglycaemia [15]. Thus, although these data highlight the protective function of PAX4 against a chemical acute stress, whether such an effect can also be conveyed in the context of a pathophysiological autoimmune attack and the molecular mechanism involved in this protection remain to be established.
Herein, we investigated whether PAX4 and PAX4R129W could promote beta cell health, preventing the development of hyperglycaemia in the RIP-B7.1 mouse model of experimental autoimmune diabetes (EAD), and sought to characterise the PAX4-regulated pathways implicated in islet survival and expansion.
Methods
Animals and bioluminescence imaging
Mouse experiments were approved by the local ethics committee and performed according to the Spanish law on animal use RD 53/2013. The rationale for using RIP-B7.1 (kindly supplied by B. O. Boehm) rather than NOD mice for the current study is provided in the electronic supplementary material (ESM) Methods. BPTL mice were derived as outlined in Fig. 1a, b and maintained on a C57bl/6 background. This mouse harbours four transgenes: (1) RIP-B7.1, a construct coding for the Cd80 gene under control of the rat insulin promoter (RIP); (2) the tetracycline response element (TRE)/cytomegalo mosaic virus (CMV) P ax4, a tetracycline-inducible CMV promoter driving Pax4 expression; (3) the RIP-reverse tetracycline trans-activator (rt T A), a construct allowing selective expression of Pax4 in beta cells exposed to DOX; and (4) the MIP-Luciferase (L uc), a construct that expresses luciferase under the mouse insulin promoter (MIP), allowing assessment of BCM using non-invasive in vivo imaging technology. Induction of Pax4 or Pax4R129W gene expression using doxycycline (DOX) was performed as previously described [14], while EAD was achieved by i.m. immunisation of 9-week-old BPTL animals with 50 μg of pC1/ppins plasmid DNA (1 μg/μl) encoding the murine preproinsulin II. Blood glucose levels were measured using a Precision Xceed glucometer (Abbott, Madrid, Spain). Bioluminescence imaging was performed using a Xenogen IVIS 50 imaging system as previously described [16].

Characterisation of the BPTL mice. (a) Schematic diagram showing the generation of the BPTL mouse. (b) Genotyping of BPTL mice by PCR using primer sets for Cd80, Pax4, Luc and rtTA. (c) The bioluminescent signal was correlated with age in BPTL male (white diamonds, n = 5) and female (black rectangles, n = 5) mice. (d) BCM calculated as a percentage of total islet cells and compared with 8-week-old animals was assessed in both sexes. On average, cells from at least 25 islets were counted per animal, with each group comprising at least 5–6 mice. (e) Blood glucose levels were measured for up to 35 days in BPTL mice (n = 3) immunised (black circles) or not (control, white squares). (f) Pancreatic sections of BPTL mice at the indicated time after immunisation were co-immunostained for glucagon (green) and insulin (red). Nuclei were stained with DAPI (blue). Scale bar, 25 μm. GCG, glucagon; INS, insulin
Islet isolation and treatment
Islets were isolated and cultured as previously described [14]. Total RNA was extracted using the RNeasy Micro Kit (Qiagen, Madrid, Spain) and quantitative (q)-PCR was performed as described previously [17]. Primer sequences can be obtained on request. For studies on thapsigargin-induced ER stress and apoptosis, islets from either Pax4/rtTA or Pax4R129W/rtTA transgenic mice were treated with 1 μg/l DOX for 96 h or left untreated [14]. Fluorescence of the Discosoma sp. red fluorescent protein (DsRed) correlating with Pax4 or Pax4R129W expression was monitored using an ImageXpress Micro System (Molecular Devices, Wokingham, UK). Islets were then treated or not with 1 μmol/l thapsigargin for 48 h and apoptosis was assessed by ELISA (Roche Diagnostics, Mannheim, Germany). Glucose-stimulated insulin secretion (GSIS) was performed as previously described [18].
Intracellular Ca2+ measurements
Isolated islets were incubated for 1 h at room temperature with 2 μmol/l Fura-2 (Qiagen). Fluorescence recordings were performed using an inverted epifluorescence microscope (Axiovert 200; Zeiss, Jena, Germany) equipped with 360 nm and 380 nm band-pass filters. Recordings were expressed as the ratio of fluorescence at 360 nm and 380 nm (F360/380). Images were taken every 3 s. Intracellular [Ca2+] changes in response to stimuli were analysed as previously described [19].
MIN6 cell culture and RNA interference
MIN6 cells were cultured as previously described [20] and transfected with either 50 μmol Pax4 small interfering (si)RNA (Sigma) or scramble siRNA using Oligofectamine (Life Technologies, Madrid, Spain). Cells were either processed for RNA or treated with thapsigargin (Sigma-Aldrich, Madrid, Spain) to assess apoptosis 48 h after transfection.
Immunohistochemistry and electron microscopy
Dissected pancreases were fixed in 4% paraformaldehyde and processed at the Histology Core Facility, Andalusian Center for Molecular Biology and Regenerative Medicine (CABIMER). A detailed immunocytochemistry protocol along with the list of antibodies used is provided in ESM Methods and ESM Table 1. Validation of antibodies was performed on appropriate control samples. The BCM and islet cell number were assessed as described elsewhere [21]. Apoptosis, proliferation and DNA damage were quantified by counting caspase-3-, Ki67- and p53BP1-positive cells, respectively, in at least 20 to 50 islets from three independent pancreatic sections from 3–4 mice per group. Insulitis scoring was performed as previously outlined [22]. For electron microscopy (EM), pancreatic islets were processed using a standard Spurr protocol [23]. Images were acquired with an electron-multiplying charge-coupled device (EMCCD) camera (TRS 2 k × 2 k).
RNA microarray
Labelled cRNA samples were prepared from pools of at least 100 islets isolated from either Pax4/rtTA or Pax4R121W/rtTA transgenic animals (8-week-old females) treated with DOX or not treated [14]. Three preparations of cRNA per group were then hybridised to the GeneChip Mouse Gene 1.0 ST Array chip (Affymetrix, Santa Clara, CA, USA) using the standard protocols of the Genomic Core Facility, CABIMER. Raw data are accessible in the Gene Expression Omnibus database under accession number GSE62846, while its analysis is described in ESM Methods.
Statistical analysis
Results are expressed as mean ± SEM. Statistical differences between two conditions were estimated using the unpaired Student’s t test. One-way ANOVA was used for comparison of more than two groups with Bonferroni post hoc test or non-parametric Mann–Whitney test (*p < 0.05 and **p < 0.01).
Results
PAX4 expression blunts EAD in immunised BPTL mice
We first monitored in vivo the bioluminescence intensity (BLI) emitted by beta cells of BPTL mice from 4 to 65 weeks of age (Fig. 1c). In 4-week-old mice, the bioluminescence signal was two fold higher in male mice than females. Consistent with a transient wave of beta cell apoptosis and decreased rate of islet growth around weaning [24], both sexes displayed a decline in BLI signal between the fourth and the ninth week (Fig. 1c). Thereafter, this signal did not significantly change with age (Fig. 1c). Morphometric evaluation revealed that the volume density of beta cells was similar in animals aged from 4 to 16 weeks (Fig. 1d). Thus, by 9 weeks of age the bioluminescent signal reflects the mass and function of beta cells, which we hereafter refer to as the functional beta cell mass (BCM). Similar to the RIP-B7.1 animal, immunised BPTL mice developed hyperglycaemia within 21 days (Fig. 1e) due to a gradual loss of beta cells (Fig. 1f), whereas non-immunised mice remained normoglycaemic (Fig. 1e, f).
We next determined whether PAX4 expression could prevent EAD. Five-week-old BPTL mice were treated with DOX for 4 weeks prior to immunisation. Compared with control mice, islets from treated mice revealed a ten fold increase in Pax4 expression (Fig. 2a), but no change in Cd80 transcript levels (Fig. 2b). DOX treatment did not alter the BCM or GSIS of PAX4-expressing islets (Fig. 2c, d). Nine-week-old untreated controls remained normoglycaemic and featured no variation in bioluminescent signal for up to 28 days (Fig. 2e). Immunised BPTL mice without DOX revealed a rapid decrease in BLI, reaching undetectable levels by day 28 post-immunisation, which coincided with sustained hyperglycaemia (Fig. 2f). Escalation in blood glucose level observed 21 days after immunisation correlated with a 60% decrease in BLI. DOX-treated and immunised BPTL mice maintained both normoglycaemia and bioluminescent signal (Fig. 2g). Protection was extended up to 63 days (Fig. 2h), at which point 65% of immunised and DOX-treated BPTL mice developed hyperglycaemia, probably because of the robust immune attack conveyed by CD80 overexpression. Immunised and DOX-treated RIP-B7.1 mice developed hyperglycaemia (ESM Fig. 1), excluding a protective effect mediated by the antibiotic through alteration in the gut microbiome [25, 26]. Non-DOX-treated BPTL mice suffered a 40% and 80% reduction in the functional BCM after 21 and 28 days of immunisation, respectively. Such changes were not observed in DOX-treated mice (Fig. 3a, b). By day 63, the latter animals retained approximately 50% of the functional BCM (Fig. 3b).

PAX4 prevents the development of hyperglycaemia in immunised BPTL mice. Islet (a) Pax4 and (b) Cd80 transcript levels in 5-week-old BPTL animals treated with DOX for 1 month or not treated (n = 4–6). Relative mRNA levels were normalised to the transcript levels of the housekeeping gene β-actin and/or Rps29. Data were calculated as fold change compared with non-DOX-treated mice. **p < 0.01. (c) BCM was calculated as a percentage of total islet cells and compared with the (−) DOX group. On average, cells from at least 25 islets were counted per animal (n = 4–6). (d) Insulin secretion by isolated BPTL islets treated or not with DOX was assessed in 30 min static incubations in response to glucose at 2.5 mmol/l (white bars) and 16.5 mmol/l (black bars). Insulin released was quantified by ELISA and expressed as a percentage of total cellular insulin content. n = 4–7, *p < 0.05. BLI (white diamonds) and blood glucose levels (black circles) was measured weekly for up to 4 weeks in (e) control untreated BPTL mice (n = 4), (f) immunised non-DOX-treated (n = 6) and (g) immunised DOX-treated BPTL animals (n = 6). (h) BLI and blood glucose level measurements were extended to 63 days in immunised and DOX-treated BPTL mice (n = 6). BLI results are presented as per cent change compared with day 0 (t = 0), while blood glucose levels are expressed as means ± SEM

PAX4 preserves the functional BCM in immunised BPTL mice. (a) Immunohistochemical analysis of glucagon (green) and insulin (red) in pancreases from BPTL mice treated or not with DOX and killed at days 0, 21, 28 and 63 post-immunisation. Nuclei are stained blue. Scale bar, 25 μm. (b) BCM in control non-immunised and non-DOX-treated (white bars), immunised and non-DOX-treated (grey bars) and immunised and DOX-treated (black bars) mice was calculated as a percentage of total islet cells and compared with the control group at each time point. On average, cells from at least 25 islets were counted per animal (n = 5–6). As similar results were obtained for days 28 and 35, the data were combined as a single time point. ND, not determined, as this group was killed at day 35 to comply with animal welfare guidelines. *p < 0.05 and **p < 0.01. GCG, glucagon; IMM, immunised; INS, insulin
PAX4 improves beta cell health and mitigates the autoimmune attack
Insulitis assessment at 28 days after immunisation revealed that 65% of islets of DOX-treated BPTL mice were insulitis free (grade 0), whereas 90% of islets derived from non-DOX-treated BPTL mice displayed severe insulitis (grades 2–4) (Fig. 4a). Even at 63 days after immunisation only 50% of islets from DOX-treated BPLT mice displayed mild insulitis (Fig. 4a). DOX treatment also diminished the percentage of cleaved CASPASE-3-positive islet cells in immunised animals (Fig. 4b and ESM Fig. 2a). DNA damage induced by NO and reactive oxygen species (ROS) contributes to beta cell death [27, 28]. Immunostaining for the double-strand DNA break marker p53BP1 revealed that PAX4 overexpression reduced DNA damage in islets from either immunised BPTL or STZ-treated mice (Fig. 4c). These changes were paralleled by increased cell proliferation in DOX-treated BPLT islets (Fig. 4d and ESM Fig. 2b).

BPTL mice overexpressing PAX4 display reduced insulitis, correlating with decreased islet apoptosis and DNA damage and increased proliferation. (a) Insulitis was scored as grade 0–4 according to the percentage of infiltrated islet area (0: 0%; 1: <10%; 2: >10% and <55%; 3: >55% and <75%; 4: >75%). n = 5–6, *p < 0.05 vs immunised day 0 and (−) DOX group; † p < 0.05 vs immunised day 28 and (−) DOX group. (b) Apoptosis was assessed by immunohistochemical analysis of cleaved caspase-3 in pancreatic islets from BPTL mice treated (black ovals) or not (white ovals) with DOX and killed at the time points. As similar results were obtained for days 28 and 35, the data were combined as a single time point. n = 5–6, *p < 0.05, (+) vs (−) DOX groups within each time point. (c) The average number of 53BP1 foci per beta cell was assessed from pancreatic islets of either 14-day-old immunised BPTL mice or 2-day-old STZ-treated PAX4 mice exposed (+) or not (−) to DOX. n = 5–6, *p < 0.05 vs untreated mice; † p < 0.05 vs DOX-untreated immunised or STZ-treated mice. (d) Cell proliferation was assessed by the combined immunohistochemical analysis of Ki67 in pancreatic islets from BPTL mice treated (black ovals) or not (white ovals) with DOX and killed at the time points. n = 5–6, *p < 0.05, (+) vs (−) DOX groups within the time point. IMM, immunised
PAX4 regulates genes important for beta cell proliferation and ER homeostasis
Transcriptome profiling was conducted on PAX4 and PAX4R129W islets to highlight PAX4 target genes involved in beta cell health, and those altered by the diabetes-linked mutant variant R129W. We initially demonstrated that DOX-treated and immunised mutBPTL mice developed hyperglycaemia (Fig. 5a) with an incidence of approximately 75% by day 28 compared with 100% in non-DOX-treated immunised animals (Fig. 5b). Transcriptome analysis revealed that 770 transcripts were upregulated and 449 were downregulated in PAX4 islets, whereas 1437 genes were upregulated and 1136 downregulated in PAX4R129W islets. Genes showing the largest changes are listed in ESM Table 2. Notwithstanding this list, we substantiated by qPCR our previous findings [14] that Mafa transcript levels were repressed whereas Nos2 levels were unchanged in PAX4 islets (ESM Fig. 3). However, Bcl2 levels were not increased in PAX4-overexpressing islets (ESM Fig. 3), arguing against a role of this factor in protecting from EAD [15].

ER homeostasis and cell cycle are key cellular pathways targeted by PAX4. (a) Blood glucose levels were measured for up to 28 days in control non-DOX-treated (control, white squares) and immunised DOX-treated (black circles) mutBPTL mice (n = 4). (b) Hyperglycaemia incidence was assessed in control non-treated (n = 5, white diamonds), immunised and non-DOX-treated (n = 6, white circles) and immunised and DOX-treated (n = 6, black circles) BPTL mice as well as in immunised and DOX-treated mutBPTL mice (n = 7, black squares). (c, d) Significantly enriched KEGG pathways (adjusted p value <0.05) altered by (c) PAX4 and (d) PAX4R129W. The dotted line shows the threshold of significance. (e, f) Heat maps displaying t statistic values of (e) cell cycle and (f) protein processing in the ER KEGG pathway genes modulated in either PAX4 or PAX4R129W islets vs control. Colours display the t statistic values for all genes within the corresponding KEGG pathway, estimated using the t statistic value from differential expression analysis. GPI, glycosylphosphatidylinositol
A functional enrichment analysis disclosed that the cell cycle and the protein processing in ER pathways were among the top upregulated Kyoto Encyclopaedia of Genes and Genomes (KEGG) pathways in islets overexpressing PAX4, but were among the most significantly downregulated pathways in islets overexpressing PAX4R129W (adjusted p values < 0.05) (Fig. 5c, d and ESM Tables 3 and 4). To contrast the expression levels of genes contributing to these pathways, we generated heat maps amenable to statistical analysis (raw p value < 0.05). Several genes associated with cell cycle were increased in PAX4 islets, while remaining unchanged or having lower levels in islets expressing PAX4R129W (Fig. 5e). Genes encoding proteins for peptide folding (Hspa5 [also known as Bip], Calr), ER–Golgi translocation (Lman1, Lman2, Sec23b and Plaa) and ER-associated protein degradation (ERAD) (Ufd1l, Derl3, Pdia4, Ssr3, Syvn1 and Dnaja2/Hsp40) were upregulated after overexpression of PAX4, but were downregulated after overexpression of PAX4R129W (Fig. 5f). The UPR-associated genes Mbtps2, Eif2ak3 and Mapk10 were upregulated in PAX4 islets and were marginally altered in PAX4R129W islets (Fig. 5f).
PAX4 targets ER integrity and calcium homeostasis, protecting cells against apoptosis
Given that PAX4 but not PAX4R129W targets genes that are involved in ER homeostasis, we investigated the contribution of ER to PAX4-mediated beta cell health. We initially assessed expression levels of calreticulin (Calr), a major Ca2+ binding protein of the ER lumen and of galectin-9 (Lgals9), a protein involved in immune modulation. Although Pax4 and Pax4R129W transcript levels were increased three fold, Calr and Lgals9 expression levels were only increased in PAX4 islets (Fig. 6a, b). As Ca2+ handling by the ER affects the cell sensitivity to apoptosis, we assessed whether PAX4 and PAX4R129W could protect against ER-stress-induced beta cell apoptosis. Islets treated in vitro with DOX exhibited DsRed fluorescence, confirming the expression of the transgenes (Fig. 6c). Thapsigargin exposure prompted a two fold increase in apoptosis in control mice (Fig. 6d). DOX reduced both basal and thapsigargin-induced apoptosis only in PAX4 islets (Fig. 6d). However, Pax4 siRNA-mediated repression in MIN6 cells caused a 60% decrease in Calr transcript levels, with a concomitant sensitisation to thapsigargin-induced apoptosis compared with siControl and thapsigargin-treated cells (Fig. 6e, f).

PAX4-regulated ER homeostasis prevents cell degeneration. Islet PAX4, galectin-9 (Lgals9) and calreticulin (Calr) transcript levels were assessed in (a) PAX4 or (b) PAX4R129W transgenic mice treated (black bars) or not treated (white bars) with DOX for 1 month. The relative mRNA levels were normalised to transcript levels of the housekeeping gene β-actin. Data were calculated as fold change compared with the values in control, non-DOX-treated animals. n = 3, **p < 0.01 and *p < 0.05. (c) Pax4 or Pax4R129W induction in DOX-treated islets was qualitatively assessed through DsRed fluorescence. Scale bar, 10 μm. (d) Control (white bars), PAX4-expressing (black bars) and PAX4R129W-expressing (grey bars) islets were then challenged with 1 μmol/l thapsigargin or not challenged and apoptosis was measured by ELISA. Results expressed as per cent change compared with control non-thapsigargin-treated islets. n = 5, *p < 0.05. (e) Pax4, Calr and cyclophylin expression levels were measured in MIN6 cells treated with siControl (white bars) and siPax4 (black bars). mRNA levels were normalised to the transcript levels of the housekeeping gene β-actin. Data were calculated as fold change compared with the values in siControl-treated cells. n = 4, *p < 0.05. (f) Apoptosis was evaluated in control MIN6 (white bars), and cells treated with siControl (grey bars) or siPax4 (black bars), untreated or treated with thapsigargin. Results are expressed as fold change compared with control non-thapsigargin-treated cells. n = 5, *p < 0.05
Perturbation in beta cell ER homeostasis has been linked to ER dilation [29, 30]. Accordingly, islets from non-DOX-treated Pax4 transgenic mice exposed in vitro to thapsigargin displayed distension and fragmentation of the rough ER (RER) in beta cells, an effect prevented by PAX4 overexpression (Fig. 7). ER calcium release in response to carbachol was then measured. Calcium release in PAX4-overexpressing beta cells from isolated islets was marginally higher than that of controls cells (Fig. 8a–d). As the higher ER-Ca2+-buffering capacity induced by PAX4 could mask the carbachol-stimulated calcium release, we measured cytosolic Ca2+ signals in response to glucose. Glucose-induced calcium oscillations were improved in isolated islets overexpressing PAX4 compared with control islets (Fig. 8e, f). The overall Ca2+ signal was also higher in PAX4-overexpressing cells (Fig. 8g–i). Thus, PAX4 overexpression leads to improve ER and Ca2+ homeostasis in the face of beta cell stress as encountered during the islet isolation procedure or an immune attack.

ER integrity is preserved in PAX4-overexpressing islets. Two sets (upper and lower) of beta cell micrographs from islets isolated from PAX4 transgenic mice, treated with DOX or not treated, and challenged with 1 μmol/l thapsigargin or not challenged. Arrows indicate the RER. Inset is enlarged below each image. Scale bar, 0.5 μm. N, nucleus; V, insulin vesicles

PAX4-overexpressing islets exhibit normal calcium oscillation in response to glucose. Carbachol-stimulated (100 μmol/l) intracellular release of Ca2+ was measured in intact islets (a) treated with DOX or (b) not treated. As indicated by the bars, islets were exposed to Ca2+-deficient medium containing 2 mmol/l EGTA prior to applying 100 μmol/l carbachol. (c) Analysis of the AUC for the time of the stimulus with carbachol from experiments shown in (a) and (b). (d) Analysis of the Ca2+ peak amplitude in response to carbachol challenge. (n = 5–6). Representative Ca2+ signals in response to 11 mmol/l glucose measured in intact islets (e) treated with DOX or (f) not treated. (g) Analysis of the AUC for the last 10 min of the glucose stimulus recording shown in (e) and (f). (h) Analysis of the amplitude of the first Ca2+ transient in response to the glucose challenge. (i) Analysis of the basal fluorescence during the first minute of the experiments. n = 9; **p ≤ 0.01 and ***p ≤ 0.001. ΔF; fluorescence increase
Discussion
Inflammation is a common denominator in types 1 and 2 diabetes, and leads to beta cell failure and death, predominantly by apoptosis. As yet, there is no ‘unifying hypothesis’ for the mechanisms triggering beta cell deterioration [31, 32]; deciphering the molecular roadmap regulated by factors such as PAX4—mutations in which are linked to both forms of diabetes—may help identify common pathways. Herein, we provide proof-of-concept that PAX4, but not PAX4R129W, preserves the BCM and delays the development of hyperglycaemia in the RIP-B7.1 mouse model of EAD. This highlights the mechanistic differences between the wild type and mutant variant.
Consistent with previous reports exploiting the RIP-B7.1 model, 90–100% of immunised non-DOX-treated RIP-B7.1 mice bearing either the Pax4 or Pax4R129W transgene developed hyperglycaemia within 3 weeks, correlating with insulitis and beta cell destruction [33]. DOX-mediated induction of Pax4 prevented hyperglycaemia development in BPTL animals up to 4 weeks after immunisation, whereas Pax4R129W induction partially reduced the hyperglycaemic incidence in mutBPTL mice. Thus, PAX4 blunts hyperglycaemia in an autoimmune context, in contrast to several other factors such as caspase-3-generated RAS p21 protein activator 1 (RasGAP) N-terminal fragment (fragment N), cytokine response modifier A (CRMA) or BCL-2 which, despite increasing BCM, could not prevent hyperglycaemia in animal models of type 1 diabetes [15, 22, 34]. Our data imply that in addition to inhibiting apoptosis, PAX4 is involved in additional regulatory pathways, possibly including immune modulation. Correspondingly, insulitis was reduced after PAX4 overexpression, an effect not attributable to a non-specific repression of the Cd80 transgene that facilitates the immune response. Our observations extend the analogous findings that inhibition of vascular endothelial growth factor receptor 2 (VEGFR-2) in NOD mice reversed hyperglycaemia by abrogating insulitis and restoring islet cell function [35]. Although the mechanism by which PAX4 acts at the interface of beta cells and the immune system to blunt insulitis and improve islet recovery remains undefined, our genetic analysis revealed that Lgals9 was specifically upregulated in PAX4 islets. This gene induces apoptosis of differentiated T helper 1 (Th1) cells [36], and its overexpression in NOD mice reduced insulitis and hyperglycaemia [37], prolonging the survival of grafts [38]. It is therefore plausible that, by enhancing Lgals9 expression, PAX4 may downregulate Th1 function, partially impeding insulitis and improving islet survival.
Transcriptome profiling revealed that cell-cycle-associated genes upregulated by PAX4 were downregulated by PAX4R129W, despite comparable expression levels of both transgenes. Specifically, the type 2 diabetes-associated cyclin-dependent kinase inhibitor 2A, which strongly inhibits the proliferative cyclin-dependent kinase 4 (CDK4) [39], was enriched in PAX4R129W islets whereas it was decreased in PAX4 islets. Reciprocally, cyclin D3, which promotes beta cell survival, was increased in PAX4- but not in PAX4R129W-overexpressing islets [40]. These findings provide some molecular insights into the pathogenic effect of the R129W mutation on beta cell plasticity.
Our data also reveal that PAX4, but not PAX4R129W, is a key regulator of ER function by a combined targeting of genes involved in UPR, Ca2+ homeostasis, ER–Golgi translocation, and ERAD. The functional importance of the transcriptional changes in these genes was validated by the capacity of PAX4 to prevent thapsigargin-induced ER ultrastructural abnormalities and apoptosis of beta cells, consistent with the finding that PAX4-binding sites are enriched within the promoter region of palmitate-modified ER stress response genes [41]. In contrast, repression of PAX4-sensitised MIN6 cells to thapsigargin cell death correlates with reduced CALR levels. Calreticulin is a Ca2+ chaperone of the ER, which contributes to the quality control of protein folding [42]. Overexpression of CALR in MIN6 cells enhances ER Ca2+ stores and prevents NO-induced apoptosis [43]. In this context, the production of NO and ROS induced by inflammatory cytokines in the diabetic environment promotes beta cell death by a variety of mechanisms, including the induction of irreversible double-strand DNA breaks [27, 44]. We found that PAX4 blunts DNA damage in two models of experimental diabetes, pointing to a general protective mechanism, possibly through preserved ER homeostasis, implicating CALR [43]. The finding that PAX4-overexpressing islets exhibited improved glucose-induced Ca2+ oscillations points to this premise. Loss of oscillatory capacity is associated with diminished islet glucose sensitivity and increased ER dysfunction [45]. Thus, amplified Ca2+ content in the CALR-enriched ER of PAX4-overexpressing beta cells may impact cytosolic calcium dynamics [43, 46], thereby preventing activation of downstream apoptotic pathways under stress conditions.
We conclude that PAX4 favours beta cell survival and regeneration in various deleterious inflammatory and high-grade inflammatory environments, such as autoimmunity, through the coordinated regulation of immune modulation, cell cycle, cell survival, ER homeostasis and DNA repair. While both PAX4 and PAX4R129W modulate these pathways, it is the wild-type transcription factor that conveys pro-survival properties by increasing the expression of selected adaptive genes.
Abbreviations
- BCL-2:
-
B cell CLL/lymphoma 2
- BCM:
-
Beta cell mass
- BLI:
-
Bioluminescence intensity
- BPTL:
-
RIP-B7.1/P ax4/rt T A/MIP-L uc
- CABIMER:
-
Andalusian Center for Molecular Biology and Regenerative Medicine
- CALR:
-
Calreticulin
- DOX:
-
Doxycycline
- DsRed:
-
Discosoma sp. red fluorescent protein
- EAD:
-
Experimental autoimmune diabetes
- EM:
-
Electron microscopy
- ER:
-
Endoplasmic reticulum
- ERAD:
-
ER-associated protein degradation
- GSIS:
-
Glucose-stimulated insulin secretion
- KEGG:
-
Kyoto Encyclopaedia of Genes and Genomes
- Luc:
-
Luciferase
- MIP:
-
Mouse insulin promoter
- PAX4:
-
Paired box 4
- RER:
-
Rough ER
- RIP:
-
Rat insulin II promoter
- rtTA:
-
Reverse tetracycline-controlled transactivator
- si:
-
Small interfering
- STZ:
-
Streptozotocin
- Th1:
-
T helper 1
- UPR:
-
Unfolded protein response
References
Donath MY (2014) Targeting inflammation in the treatment of type 2 diabetes: time to start. Nat Rev Drug Discov 13:465–476
Tisch R, Wang B (2008) Dysregulation of T cell peripheral tolerance in type 1 diabetes. Adv Immunol 100:125–149
Papa FR (2012) Endoplasmic reticulum stress, pancreatic beta-cell degeneration, and diabetes. Cold Spring Harb Perspect Med 2:a007666
Cao SS, Kaufman RJ (2014) Endoplasmic reticulum stress and oxidative stress in cell fate decision and human disease. Antioxid Redox Signal 21:396–413
Delepine M, Nicolino M, Barrett T, Golamaully M, Lathrop GM, Julier C (2000) EIF2AK3, encoding translation initiation factor 2-alpha kinase 3, is mutated in patients with Wolcott-Rallison syndrome. Nat Genet 25:406–409
Inoue H, Tanizawa Y, Wasson J et al (1998) A gene encoding a transmembrane protein is mutated in patients with diabetes mellitus and optic atrophy (Wolfram syndrome). Nat Genet 20:143–148
Kirkpatrick CL, Wiederkehr A, Baquie M et al (2011) Hepatic nuclear factor 1alpha (HNF1alpha) dysfunction down-regulates X-box-binding protein 1 (XBP1) and sensitizes beta-cells to endoplasmic reticulum stress. J Biol Chem 286:32300–32312
Sachdeva MM, Claiborn KC, Khoo C et al (2009) Pdx1 (MODY4) regulates pancreatic beta cell susceptibility to ER stress. Proc Natl Acad Sci U S A 106:19090–19095
Robson EJ, He SJ, Eccles MR (2006) A PANorama of PAX genes in cancer and development. Nat Rev Cancer 6:52–62
Greenwood AL, Li S, Jones K, Melton DA (2007) Notch signaling reveals developmental plasticity of Pax4(+) pancreatic endocrine progenitors and shunts them to a duct fate. Mech Dev 124:97–107
Sosa-Pineda B, Chowdhury K, Torres M, Oliver G, Gruss P (1997) The Pax4 gene is essential for differentiation of insulin-producing beta cells in the mammalian pancreas. Nature 386:399–402
Brun T, Gauthier BR (2008) A focus on the role of Pax4 in mature pancreatic islet beta-cell expansion and survival in health and disease. J Mol Endocrinol 40:37–45
Mauvais-Jarvis F, Smith SB, Le May C et al (2004) PAX4 gene variations predispose to ketosis-prone diabetes. Hum Mol Genet 13:3151–3159
Hu He KH, Lorenzo PI, Brun T et al (2011) In vivo conditional pax4 overexpression in mature islet beta cells prevents stress-induced hyperglycemia in mice. Diabetes 60:1705–1715
Allison J, Thomas H, Beck D et al (2000) Transgenic overexpression of human Bcl-2 in islet beta cells inhibits apoptosis but does not prevent autoimmune destruction. Int Immunol 12:9–17
Park SY, Wang X, Chen Z et al (2005) Optical imaging of pancreatic beta cells in living mice expressing a mouse insulin I promoter-firefly luciferase transgene. Genesis 43:80–86
Gauthier BR, Brun T, Sarret EJ et al (2004) Oligonucleotide microarray analysis reveals PDX1 as an essential regulator of mitochondrial metabolism in rat islets. J Biol Chem 279:31121–31130
Gauthier BR, Wiederkehr A, Baquie M et al (2009) PDX1 deficiency causes mitochondrial dysfunction and defective insulin secretion through TFAM suppression. Cell Metab 10:110–118
Gonzalez A, Merino B, Marroqui L et al (2013) Insulin hypersecretion in islets from diet-induced hyperinsulinemic obese female mice is associated with several functional adaptations in individual beta-cells. Endocrinology 154:3515–3524
Semache M, Ghislain J, Zarrouki B, Tremblay C, Poitout V (2014) Pancreatic and duodenal homeobox-1 nuclear localization is regulated by glucose in dispersed rat islets but not in insulin-secreting cell lines. Islets 6, e982376
Mellado-Gil J, Rosa TC, Demirci C et al (2011) Disruption of hepatocyte growth factor/c-Met signaling enhances pancreatic beta-cell death and accelerates the onset of diabetes. Diabetes 60:525–536
Bulat N, Jaccard E, Peltzer N et al (2011) RasGAP-derived fragment N increases the resistance of beta cells towards apoptosis in NOD mice and delays the progression from mild to overt diabetes. PLoS One 6, e22609
Spurr AR (1969) A low-viscosity epoxy resin embedding medium for electron microscopy. J Ultrastruct Res 26:31–43
Scaglia L, Cahill CJ, Finegood DT, Bonner-Weir S (1997) Apoptosis participates in the remodeling of the endocrine pancreas in the neonatal rat. Endocrinology 138:1736–1741
Alkanani AK, Hara N, Lien E et al (2014) Induction of diabetes in the RIP-B7.1 mouse model is critically dependent on TLR3 and MyD88 pathways and is associated with alterations in the intestinal microbiome. Diabetes 63:619–631
Angelakis E, Million M, Kankoe S et al (2014) Abnormal weight gain and gut microbiota modifications are side effects of long-term doxycycline and hydroxychloroquine treatment. Antimicrob Agents Chemother 58:3342–3347
Oleson BJ, Broniowska KA, Schreiber KH, Tarakanova VL, Corbett JA (2014) Nitric oxide induces ataxia telangiectasia mutated (ATM) protein-dependent gammaH2AX protein formation in pancreatic beta cells. J Biol Chem 289:11454–11464
Tornovsky-Babeay S, Dadon D, Ziv O et al (2014) Type 2 diabetes and congenital hyperinsulinism cause DNA double-strand breaks and p53 activity in beta cells. Cell Metab 19:109–121
Ravelli RB, Kalicharan RD, Avramut MC et al (2013) Destruction of tissue, cells and organelles in type 1 diabetic rats presented at macromolecular resolution. Sci Rep 3:1804
Marroqui L, Masini M, Merino B et al (2015) Pancreatic alpha cells are resistant to metabolic stress-induced apoptosis in type 2 diabetes. EBioMedicine 2:378–385
Cnop M, Welsh N, Jonas JC, Jorns A, Lenzen S, Eizirik DL (2005) Mechanisms of pancreatic beta-cell death in type 1 and type 2 diabetes: many differences, few similarities. Diabetes 54(Suppl 2):S97–S107
Donath MY, Halban PA (2004) Decreased beta-cell mass in diabetes: significance, mechanisms and therapeutic implications. Diabetologia 47:581–589
Karges W, Rajasalu T, Spyrantis A, Wieland A, Boehm B, Schirmbeck R (2007) The diabetogenic, insulin-specific CD8 T cell response primed in the experimental autoimmune diabetes model in RIP-B7.1 mice. Eur J Immunol 37:2097–2103
Millet I, Wong FS, Gurr W et al (2006) Targeted expression of the anti-apoptotic gene CrmA to NOD pancreatic islets protects from autoimmune diabetes. J Autoimmun 26:7–15
Villalta SA, Lang J, Kubeck S et al (2013) Inhibition of VEGFR-2 reverses type 1 diabetes in NOD mice by abrogating insulitis and restoring islet function. Diabetes 62:2870–2878
Zhu C, Anderson AC, Schubart A et al (2005) The Tim-3 ligand galectin-9 negatively regulates T helper type 1 immunity. Nat Immunol 6:1245–1252
Chou FC, Shieh SJ, Sytwu HK (2009) Attenuation of Th1 response through galectin-9 and T cell Ig mucin 3 interaction inhibits autoimmune diabetes in NOD mice. Eur J Immunol 39:2403–2411
Chou FC, Kuo CC, Wang YL et al (2013) Overexpression of galectin-9 in islets prolongs grafts survival via downregulation of Th1 responses. Cell Transplant 22:2135–2145
Scott LJ, Mohlke KL, Bonnycastle LL et al (2007) A genome-wide association study of type 2 diabetes in Finns detects multiple susceptibility variants. Science 316:1341–1345
Saavedra-Avila NA, Sengupta U, Sanchez B et al (2014) Cyclin D3 promotes pancreatic beta-cell fitness and viability in a cell cycle-independent manner and is targeted in autoimmune diabetes. Proc Natl Acad Sci U S A 111:E3405–E3414
Cnop M, Abdulkarim B, Bottu G et al (2014) RNA sequencing identifies dysregulation of the human pancreatic islet transcriptome by the saturated fatty acid palmitate. Diabetes 63:1978–1993
Wang WA, Groenendyk J, Michalak M (2012) Calreticulin signaling in health and disease. Int J Biochem Cell Biol 44:842–846
Oyadomari S, Takeda K, Takiguchi M et al (2001) Nitric oxide-induced apoptosis in pancreatic beta cells is mediated by the endoplasmic reticulum stress pathway. Proc Natl Acad Sci U S A 98:10845–10850
Cardozo AK, Ortis F, Storling J et al (2005) Cytokines downregulate the sarcoendoplasmic reticulum pump Ca2+ ATPase 2b and deplete endoplasmic reticulum Ca2+, leading to induction of endoplasmic reticulum stress in pancreatic beta-cells. Diabetes 54:452–461
Jahanshahi P, Wu R, Carter JD, Nunemaker CS (2009) Evidence of diminished glucose stimulation and endoplasmic reticulum function in nonoscillatory pancreatic islets. Endocrinology 150:607–615
Gilon P, Ravier MA, Jonas JC, Henquin JC (2002) Control mechanisms of the oscillations of insulin secretion in vitro and in vivo. Diabetes 51(Suppl 1):S144–S151
Acknowledgements
We thank F. Cortes (Department of Stem Cells, CABIMER, Seville, Spain) for kindly providing the 53BP1 antibody as well as his expert advice on DNA damage. We also thank J. Vallejo Ortega (Department of Stem Cells, CABIMER, Seville, Spain), E. Nadujar and M. Pérez (Genomic Core Facility, CABIMER, Seville, Spain) for excellent technical assistance.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Access to research materials
The raw data for the RNA microarray described herein are accessible in the Gene Expression Omnibus database under accession number GSE62846.
Funding
This work was funded by grants from the Consejeria de Salud, Fundacion Publica Andaluza Progreso y Salud, Junta de Andalucia (PI-0727-2010 to BRG and PI-0085-2013 to PIL), Consejeria de Economia, Innovacion y Ciencia (P10.CTS.6359 to BRG), Ministerio de Ciencia e Innovacion (BFU2013-42789-P to IQ) and the Ministerio de Economia y Competidividad, Instituto de Salud Carlos III co-funded by Fondos FEDER (PI10/00871 and PI13/00593 to BRG). NC-V is supported by a JDRF subsidy (17-2013-372 to BRG.). AM-M is a recipient of a Miguel Servet grant (CP14/00105) from the Instituto de Salud Carlos III co-funded by Fondos FEDER and EF-M is a recipient of a Juan de la Cierva Fellowship. PM is supported by Swiss National Science Foundation grant 310030-141162, and the European Union grant IMIDIA, C2008-T7. BOB is supported by grants from the Lee Kong Chian School of Medicine, Nanyang Technological University, Singapore, Republic of Singapore.
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Contribution statement
JMMG, CMJ-M and AM-M contributed equally to the conception and design of experiments as well as the acquisition and analysis of data. AIA-M, EF-M, IGHG, LL-N, NC-V, EB-T and IQ were involved in the acquisition and analysis of data. PIL, JS-L, JPF, AS, BOB and PM took part in the analysis and interpretation of data. BRG conceptualised and supervised all experiments and wrote the manuscript. All authors critically reviewed the manuscript for important intellectual content and approved the final version to be published. BRG is the guarantor of this work.
Additional information
José Manuel Mellado-Gil, Carmen María Jiménez-Moreno and Alejandro Martin-Montalvo contributed equally to this work
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM Methods
(PDF 63 kb)
ESM Fig. 1
(PDF 48 kb)
ESM Fig. 2
(PDF 440 kb)
ESM Fig. 3
(PDF 61 kb)
ESM Table 1
(PDF 50 kb)
ESM Table 2
(PDF 43 kb)
ESM Table 3
(PDF 62 kb)
ESM Table 4
(PDF 51 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Mellado-Gil, J.M., Jiménez-Moreno, C.M., Martin-Montalvo, A. et al. PAX4 preserves endoplasmic reticulum integrity preventing beta cell degeneration in a mouse model of type 1 diabetes mellitus. Diabetologia 59, 755–765 (2016). https://doi.org/10.1007/s00125-016-3864-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-016-3864-0