
Abstract
Aims/hypothesis
Tankyrase (TNKS) is a ubiquitously expressed molecular scaffold that is implicated in diverse processes. The catalytic activity of TNKS modifies substrate proteins through poly-ADP-ribosylation (PARsylation) and is responsive to cellular energetic state. Global deficiency of the TNKS protein in mice accelerates glucose utilisation and raises plasma adiponectin levels. The aim of this study was to investigate whether the PARsylation activity of TNKS in adipocytes plays a role in systemic glucose homeostasis.
Methods
To inhibit TNKS-mediated PARsylation, we fed mice with a diet containing the TNKS-specific inhibitor G007-LK. To genetically inactivate TNKS catalysis in adipocytes while preserving its function as a molecular scaffold, we used an adipocyte-selective Cre transgene to delete TNKS exons that encoded the catalytic domain at the C-terminus. Tissue-specific insulin sensitivity in mice was investigated using hyperinsulinaemic–euglycaemic clamps. To model adipose–liver crosstalk ex vivo, we applied adipocyte-conditioned media to hepatocytes and assessed the effect on gluconeogenesis.
Results
The TNKS inhibitor G007-LK improved glucose tolerance and insulin sensitivity and promptly increased plasma adiponectin levels. In female mice, but not in male mice, adipocyte-selective genetic inactivation of TNKS catalysis improved hepatic insulin sensitivity and post-transcriptionally increased plasma adiponectin levels. Both pharmacological and genetic TNKS inhibition in female mouse-derived adipocytes induced a change in secreted factors to decrease gluconeogenesis in primary hepatocytes.
Conclusions/interpretation
Systemic glucose homeostasis is regulated by the PARsylation activity of TNKS in adipocytes. This regulation is mediated in part by adipocyte-secreted factors that modulate hepatic glucose production. Pharmacological TNKS inhibition could potentially be used to improve glucose tolerance.
Similar content being viewed by others

Introduction
Tankyrase (TNKS) is a ubiquitously expressed modular protein that functions both as a molecular scaffold and as an enzyme [1]. The ankyrin repeats near the TNKS N-terminus provide a multivalent scaffold that binds to diverse proteins containing the sequence motif RxxPDG [2]. Established TNKS partners include insulin-responsive aminopeptidase (IRAP, a vesicular membrane protein [2]) and axin (a key component of the β-catenin degradasome [3]). The catalytic domain in the TNKS C-terminus can poly-ADP-ribosylate (PARsylate) TNKS itself as well as many of its binding partners [3, 4]. In this post-translational modification, NAD+ is consumed as a co-substrate to label acceptor proteins with poly-ADP-ribose (PAR). In insulin-secreting cells, glucose and other nutrients can augment NAD+ production to stimulate TNKS autoPARsylation [5]. The downstream effects of TNKS-mediated PARsylation are not fully understood. In the case of TNKS itself and axin, PARsylation acts to destabilise both proteins by recruiting a PAR-directed E3 ubiquitin ligase [6]. Certain TNKS partners do not undergo PARsylation [7]; they presumably use TNKS as a scaffold to interact with other RxxPDG-containing partners.
TNKS resides in multiple subcellular locations and regulates diverse processes ranging from telomere homeostasis to mitotic progression [1, 8, 9]. In non-cycling cells, TNKS localises primarily to sorting stations for vesicular cargos and can either promote or repress the exocytosis of specific cargos [10, 11]. In 3T3-L1 adipocytes, TNKS co-fractionates with microsomes as a peripheral membrane protein and co-localises with GLUT4 vesicles near the Golgi [4, 12]. Given its Golgi localisation and its impact on the exocytosis of various cargos, TNKS seems poised to modulate the secretory pathway of adipocytes.
The PARsylation activity of TNKS, by destabilising axin, can augment the canonical Wnt–β-catenin pathway. In the absence of Wnt ligands, this pathway is inhibited since an axin-based degradasome (destruction complex) destabilises β-catenin through N-terminal phosphorylation, triggering ubiquitination and proteasomal degradation [13]. Wnt ligands inhibit the degradasome, allowing dephosphorylation and stabilisation of β-catenin to modulate the expression of target genes [13]. Many PARsylation inhibitors with various degrees of specificity for TNKS have been identified by virtue of blocking Wnt signalling [3, 14–16] but none has been investigated in vivo for potential effects on glucose homeostasis.
We have previously shown that global deficiency of the TNKS protein in mice led to greater energy expenditure, higher muscle insulin sensitivity and higher plasma levels of the insulin-sensitising hormone adiponectin [17]. In the current study we aimed to specifically implicate the PARsylation activity of TNKS in glucose homeostasis in mice. Since many endocrine organs are responsive to factors secreted by adipocytes [18], and secretory pathways are subject to regulation by TNKS [10, 11], we focused on establishing TNKS as a regulator of adipokine secretion that has an impact on systemic metabolism.
Methods
Generation and care of mouse models
A neomycin-resistant targeting construct was transfected into ES cells (129/SvEv) by InGenious Laboratory (Stony Brook, NY, USA) to flox exons 25–26 of the TNKS gene. After confirming homologous recombination and establishing germ-line transmission, the neomycin cassette was removed by mating with mice expressing FLP recombinase. Mice carrying the resulting floxed TNKS allele (TNKS +/f) were mated with mice expressing Cre recombinase from the adenovirus EIIa promoter (Jackson No. 003724; Jackson Laboratory, Bar Harbor, ME, USA) to generate the ΔPARP allele (Fig. 2a). The floxed allele and the ΔPARP allele were identifiable as a 387-bp and a 336-bp band, respectively, by PCR using the three primers indicated in Fig. 2a (sequences listed in electronic supplementary material [ESM] Table 1) and a genotyping protocol [17]. For adipose-selective inactivation of TNKS, TNKS f/f mice were mated with aP2 (also known as Fabp4)-Cre Salk [19] or adiponectin (Adipoq)-Cre transgenic mice (Jackson No. 010803). Adiponectin knockout mice were from the Jackson Laboratory. Up to four mice per cage were maintained in a controlled environment (23 ± 1°C, lights on from 06:00 to 18:00 hours) with free access to water and either normal chow (Diet 5001, LabDiet Inc., St. Louis, MO, USA) or a high-fat diet (HFD, TD.96132; Harlan, Indianapolis, IN, USA), which provided 13.5% and 40% of energy intake from fat, respectively. The same diets were reformulated by Research Diets (New Brunswick, NJ, USA) to contain G007-LK [20] at 0.1% wt/wt (chow) or 0.015% wt/wt (HFD). At a daily food consumption of 2.5 g by a 25 g (chow) or a 40 g (HFD) mouse, these formulations provided a daily G007-LK dose of 100 and 10 mg/kg, respectively. All procedures were approved by the Institutional Animal Care and Use Committee at the University of California, San Diego.
Mouse phenotyping
After nine or ten generations of backcrossing with C57BL/6N mice (Harlan), TNKS f/f mice were bred with aP2-Cre Salk TNKS f/f mice to generate littermates for phenotyping. Body composition was measured in an EchoMRI-100 scanner (EchoMRI, Houston, TX, USA) without fasting or sedation. Hyperinsulinaemic–euglycaemic clamps were conducted as described [17] after mice had been fasted for 6 h. Insulin was infused at 4 mU kg−1 min−1 and blood glucose was clamped at ∼6.7 mmol/l. To assess insulin-stimulated Akt phosphorylation in vivo, 8-month-old mice fasted for 6 h were anaesthetised. A piece of the liver, one gastrocnemius and one perigonadal fat pad were resected as basal samples. After injecting insulin (0.25 U/kg) into the inferior vena cava, the remaining liver, gastrocnemius and perigonadal fat pad were resected at 3, 7 and 10 min, respectively. Extraction of tissue proteins and quantification of triacylglycerol were as described [17]. Relative abundance of plasma adiponectin and retinol-binding protein 4 (RBP4) was determined by immunoblotting and densitometry as described [17]. Methods related to protein and mRNA analyses, the generation of adipocyte-conditioned media and ex vivo gluconeogenesis assay are detailed in ESM Methods.
Statistical analyses
All data represent mean ± SEM. Statistical significance was calculated using Student’s unpaired two-tailed t test (p < 0.05).
Results
Pharmacological TNKS inhibition enhances glucose tolerance and insulin sensitivity
To investigate the role of TNKS catalytic activity in metabolism, we treated wild-type male mice with G007-LK, a PARsylation inhibitor specific for TNKS and the closely related TNKS2 [21]. To deliver this compound orally at a daily dose of 10 mg/kg to a typical 40 g mouse that consumed 2.5 g of HFD each day, we impregnated the diet with 0.015% (wt/wt) of G007-LK. This formulation effectively attenuated TNKS activity in vivo as assessed by upregulation of axin1 and downregulation of active (unphosphorylated) β-catenin in adipose tissue (Fig. 1a). Compared with HFD alone, treatment with G007-LK-containing HFD for 6 months did not affect body weight (Fig. 1b), food intake (ESM Fig. 1a), plasma albumin levels (an index of liver synthetic function, ESM Fig. 1b) or intestinal mucosal histology (ESM Fig. 1c), all arguing against overt toxicity. G007-LK treatment did not affect overall adiposity as determined by body composition analysis (Fig. 1c) despite there being a modest reduction in individual adipocyte size (Fig. 1e). Importantly, G007-LK treatment lowered fasting glucose levels (Fig. 1g), improved glucose tolerance (Fig. 1h) and potentiated the hypoglycaemic effect of insulin (Fig. 1i).

Pharmacological effects of the TNKS inhibitor G007-LK on male C57BL/6 mice. (a–k) Mice were started on an HFD at the age of 3 months. G007-LK was added to the diet (0.015% wt/wt) for half of the cohort at the age of 6 months (black bars). The other half of the cohort was continued on plain HFD as controls (Con., white bars). (a) Immunoblots of epididymal adipose tissue extracts and quantification of the indicated proteins (at 12 months, n = 10 per group). ABC, active (nonphosphorylated) β-catenin. (b) Body weight during G007-LK treatment (n = 10 per group). (c) Fat mass as a percentage of body weight (at 11 months). (d) Weight of various organs (12 months, n = 8 or 9 per group). (e) Areal size of epididymal adipocytes (at 12 months). (f–i) After mice had been fasted for 6 h, plasma insulin (f) and blood glucose (g) in the fasting state, and glycaemic excursion after i.p. injection of glucose (1 g/kg) (h) or insulin (0.65 U/kg) (i) were measured (at 8–9 months, n = 10 per group). (j, k) After the indicated number of days of G007-LK treatment, fasting plasma was immunoblotted to quantify RBP4 (j) and adiponectin (k) as described in Methods (n = 10 per group). The immunoblots and quantification of day 60 samples are shown. AU, arbitrary units. (l) Mice (at 4 months, n = 5 per group) on a chow diet were started on G007-LK treatment (0.1% wt/wt in chow) at 16:00 hours, using drug-free chow as control. Plasma was collected 16 h and 22 h later without fasting of mice. Adiponectin levels determined by immunoblotting were normalised to pre-treatment (16:00 hours) average of the two groups. For all line graphs: black squares, G007-LK-treated mice; white rectangles, control mice. *p < 0.05 and **p < 0.01 for G007-LK vs control
Since glucose homeostasis is modulated by adipokines, we investigated the effect of G007-LK on plasma RBP4 (a pro-diabetic adipokine [22]) and adiponectin. Immunoblotting of plasma showed that during G007-LK treatment RBP4 levels became progressively lower than in controls (Fig. 1j) whereas adiponectin levels persistently exceeded those in controls (Fig. 1k). Since G007-LK did not increase adipose tissue stores of adiponectin mRNA (ESM Fig. 1d) or protein (Fig. 1a), its effect on plasma adiponectin was likely post-transcriptional. Also supporting a post-transcriptional mechanism was the finding that overnight G007-LK treatment at a higher dose (100 mg/kg per day) robustly increased plasma adiponectin (Fig. 1l).
Adipose-selective deletion of the TNKS catalytic domain
The effect of G007-LK on glucose metabolism (Fig. 1g–i) could reflect inhibition of PARsylation in any of the many tissues that express TNKS or TNKS2. However, the rapidity of the effect on plasma adiponectin (Fig. 1l) pointed to adipose tissue as a key target. To confirm this, we set out to genetically inactivate TNKS selectively in adipocytes. First, we used homologous recombination to flox the TNKS gene as depicted in Fig. 2a. Cre-mediated recombination of the floxed allele TNKS f, by shifting the open-reading frame, was predicted to delete the entire PARP domain (aa 1179–1320 encoded by exons 25–27) and shorten the protein product by 6 kDa. This was confirmed using mice homozygous for the recombinant allele ΔPARP (Fig. 2b). As expected, the recombination deleted the C-terminal epitopes of TNKS (Fig. 2b). This deletion did not disrupt the scaffolding function of native TNKS since the resulting fragment TNKSΔPARP was pulled down by GST-IRAPaa78-101 containing the TNKS-binding motif RxxPDG (Fig. 2b).

Adipose-selective deletion of the PARP domain of TNKS. (a) A Cre-Lox strategy that deletes the PARP domain from TNKS to generate the ΔPARP allele (not drawn to scale). Also indicated are the positions of PCR primers for genotyping. (b) Adipose tissue extracts from TNKS f/f (lane 1) and TNKS ΔPARP/ΔPARP mice (lane 2) were incubated with GST-IRAPaa78-108 [5] to pull down TNKS. The precipitates were immunoblotted for TNKS using the T1S antibody followed by the H-350 antibody. Epitopes recognised by these antibodies are shown in (a). (c) Adipose tissue extracts from an aP2-Cre TNKS f/f mouse (lane 2), three adiponectin-Cre TNKS f/f mice (lanes 4, 6 and 8), and their respective Cre-naive TNKS f/f littermates were immunoblotted using the TNKS antibody H-350. (d) Extracts of three organs and intraperitoneal macrophages of aP2-Cre TNKS f/f mice (CITA) and wild-type (WT) littermates (TNKS f/f) were immunoblotted for TNKS as in (c). (e, f) Extracts of perigonadal fat (e) and liver (f) of CITA mice (black bars, n = 11) and WT littermates (white bars, n = 15) (8–9 months old) were immunoblotted for axin1 and active (unphosphorylated) β-catenin (ABC) and quantified by densitometry. *p < 0.05 for CITA vs WT
To selectively truncate TNKS in adipocytes, we expressed Cre recombinase in TNKS f/f mice using either the aP2 Salk or the adiponectin promoter [23, 24]. Fig. 2c shows that only aP2-Cre induced efficient TNKS truncation in adipose tissue. Since no truncation was detected in the pancreas, heart, liver or peritoneal macrophages (Fig. 2d), aP2-Cre TNKS f/f mice were referred to as CITA (catalytically inactive TNKS in adipocytes) in subsequent phenotyping. To corroborate the tissue specificity of this conditional model, we showed (Fig. 2e, f) that the CITA adipose tissue, but not liver, exhibited upregulation of axin1 and downregulation of active (unphosphorylated) β-catenin, the expected consequences of TNKS inactivation.
Female CITA mice have greater glucose tolerance and insulin sensitivity
Female CITA mice fed a chow diet exhibited lower overall adiposity (Fig. 3b) and smaller adipocyte size (Fig. 3d) than wild-type littermates despite upregulation of adipogenic markers including lipogenic genes (Fig. 3e). Plasma resistin and leptin levels did not differ between the CITA and wild-type mice (Fig. 3f, g) whereas plasma adiponectin was significantly increased in CITA mice (Fig. 3h). This increase was a post-transcriptional effect since neither adiponectin mRNA (Fig. 3e) nor protein (Fig. 3h) in the CITA adipose tissue was increased. Importantly, female CITA mice exhibited greater glucose tolerance (Fig. 3i) and insulin sensitivity than their wild-type littermates (Fig. 3j). This glucose phenotype and increased adiponectin in plasma was not discernible in male CITA mice (ESM Fig. 2) or ovariectomised female CITA mice (ESM Fig. 3), a sexual dimorphism that prompted us to further phenotype only female mice.

Adipose TNKS inactivation increases glucose tolerance and adiponectin levels in chow-fed female mice. (a, b) Body weight (a) and percentage fat mass (b) of CITA mice and wild-type (WT, TNKS f/f ) littermates (at 8–9 months). (c) The weight of various organs in CITA (n = 11) and WT (n = 8) mice (at 8–9 months). (d) Sections of perigonadal fat were stained with haematoxylin and eosin (scale bar, 100 μm) and areal size of adipocytes was quantified using NIH ImageJ (available from http://rsb.info.nih.gov/ij/download.html). (e) Gene expression analysis of perigonadal fat in CITA (n = 11) and WT (n = 15) mice at 8–9 months. (f, g) Plasma resistin (f) and leptin levels (g) (4–5 months) were determined as described in Methods. (h) Plasma (at 6 months) and adipose tissue (from the same gel as shown in Fig. 2e) were immunoblotted for adiponectin using immunoglobulin and HSP90 as loading controls (CITA, n = 11; WT, n = 15). One arbitrary unit (AU) of plasma adiponectin corresponded to 24 μg/ml. (i, j) Blood glucose excursion of CITA (n = 11) and WT (n = 15) mice (at 4–5 months) injected intraperitoneally with glucose (2 g/kg) (i) or insulin (0.35 U/kg) (j) after a 6 h fast. White rectangles and bars, WT; black squares and bars, CITA. *p < 0.05, **p < 0.01 and † p = 0.2 for CITA vs WT
Liver of female CITA mice on a chow diet is more insulin sensitive
To specify the tissue(s) that mediated the glucose phenotype of female CITA mice on a chow diet, we performed hyperinsulinaemic–euglycaemic clamp studies. We found comparable rates of hepatic glucose production between CITA mice and controls in the basal state (23.1 ± 0.8 vs 23.6 ± 0.8 mg kg−1 min−1, p = 0.72). During insulin infusion, CITA mice tolerated a higher glucose infusion rate than wild-type mice (Fig. 4a) and showed a greater suppression of hepatic glucose production (Fig. 4b) and plasma NEFA levels (Fig. 4c), indicative of increased insulin sensitivity in liver and fat, respectively. This was in contrast to the muscle, where the insulin-stimulated glucose disposal rate indicated comparable insulin sensitivity between the two genotypes (Fig. 4d). To corroborate the above tissue specificity, we harvested various tissues before and after in vivo insulin stimulation to compare insulin signalling. Figure 4e shows that insulin-stimulated Akt phosphorylation in the liver and fat, but not muscle, of the CITA mice was more robust than that in control mice.

Adipose TNKS inactivation reduces hepatic glucose output in chow-fed female mice. (a–d) Hyperinsulinaemic–euglycaemic clamp studies were carried out in 6-month-old CITA and wild-type (WT) mice to quantify glucose infusion rate during the clamp (a), insulin-induced suppression of hepatic glucose production (HGP) (b), insulin-induced suppression of plasma NEFA (c) and insulin-stimulated glucose disposal rate (GDR) (d). (e) Basal and insulin-stimulated Akt phosphorylation in the indicated tissues of 8-month-old mice was determined as described in Methods (n = 8 for WT, n = 11 for CITA). The bar graph represents the ratio of stimulated over basal phospho-Akt levels, each normalised to corresponding total Akt levels. (f, g) Gene expression (f) and triacylglycerol (g) analyses of liver of the same mice as in Fig. 3e (n = 15 for WT, n = 11 for CITA). White bars, WT; black bars, CITA. *p < 0.05 and **p < 0.01 for CITA vs WT
The CITA liver exhibited downregulation of the gluconeogenic marker Pdk4 and the inflammatory markers F4/80 (also known as Adgre1) and Mcp-1 (Fig. 4f). It also contained less triacylglycerol than control liver (Fig. 4g), which presumably reflected decreased lipogenesis and increased lipolysis, since the CITA liver showed downregulation of lipogenic genes (Acc, Acs and Acl [Acly]), upregulation of the lipolytic gene Atgl (Pnpla2), and no changes in markers of triacylglycerol export (Apob and Mtp) (Fig. 4f). Collectively, these data indicate that the CITA liver, despite being free from TNKS inactivation (Fig. 2d, f), was indirectly affected by TNKS inactivation in adipose tissue.
Liver of female CITA mice on an HFD is more insulin sensitive
To find out if the metabolic phenotype of lean CITA mice is conserved in the obese state, we fed female mice an HFD. This diet did not alter TNKS abundance (ESM Fig. 4a) or prevent the CITA mutation from blocking Wnt signalling in fat (Fig. 5a) and raising plasma adiponectin levels (Fig. 5e). The liver of HFD-fed CITA mice, like its counterpart from lean mice, showed downregulation of lipogenic genes, inflammatory markers and Pdk4 (ESM Fig. 5a). Importantly, HFD-fed CITA mice also exhibited greater glucose tolerance (Fig. 5f) and insulin sensitivity (Fig. 5g). During hyperinsulinaemic–euglycaemic clamps, the hepatic glucose production in CITA mice tended to be lower than that in wild-type mice in the basal state (20.3 ± 1.5 vs 25.2 ± 1.3 mg kg−1 min−1, p = 0.13) and was significantly more sensitive to suppression by insulin (Fig. 5i). This was in contrast to the findings in fat and muscle, where the two genotypes exhibited comparable insulin sensitivity based on suppression of plasma NEFA levels (Fig. 5j) and stimulation of glucose disposal rate (Fig. 5k). Collectively these data indicate that in diet-induced obese female mice, liver insulin sensitivity was enhanced by inhibition of TNKS-mediated PARsylation in adipose tissue. This phenotype was not attributable to differences in the severity of obesity since body weight and overall adiposity were comparable between the two genotypes (Fig. 5b, c).

Adipose TNKS inactivation reduces hepatic glucose output in female mice on an HFD. Female CITA mice and wild-type (WT) littermates were started on an HFD at 2 months. (a–e) Extracts of perigonadal fat were immunoblotted for the indicated proteins at 9 months (a), and body weight (b) and percentage fat mass (c) were measured at 7 months. The weights of the indicated organs were measured at 11 months (d) (n = 9 for CITA; n = 5 for WT). Plasma adiponectin levels were measured at 7 months (e). One arbitrary unit (AU) corresponds to 38 μg/ml adiponectin. (f, g) Blood glucose excursion of 5-month-old mice injected intraperitoneally with glucose (2 g/kg) (f) or insulin (0.35 U/kg) (g) after a 6 h fast (n = 11 for WT; n = 7 for CITA). (h–k) Hyperinsulinaemic–euglycaemic clamp studies in 9-month-old mice as described in Methods. (h) Glucose infusion rate during the clamp. (i) Insulin-induced suppression of hepatic glucose production (HGP). (j) Insulin-induced suppression of plasma NEFA. (k) Insulin-stimulated glucose disposal rate (GDR). White bars and rectangles, WT; black squares and bars, CITA. ABC, active β-catenin. *p < 0.05, **p < 0.01 and † p = 0.18 for CITA vs WT
Adipose TNKS activity modulates secreted factors to regulate hepatocyte gluconeogenesis
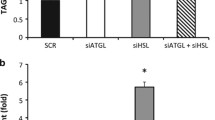
Given the indirect hepatic effect of adipose TNKS inactivation, we hypothesised that this effect was mediated by adipocyte-derived factors whose secretion was regulated by TNKS-mediated PARsylation. To test this hypothesis, we compared ex vivo gluconeogenesis in hepatocytes pre-treated with conditioned media (CM) generated by CITA or wild-type (TNKS f/f ) adipocytes. Figure 6a (bar 3 vs bar 4) shows decreased gluconeogenesis in hepatocytes pre-treated with CM from female CITA vs wild-type mouse adipocytes. This anti-gluconeogenic effect was not observed in CM from male CITA mouse adipocytes (Fig. 6a, bar 1 vs bar 2), a sexual dimorphism that mirrored the female specificity of CITA phenotype in vivo. To corroborate the effect of genetic TNKS inactivation, we also treated female wild-type mouse adipocytes with G007-LK or vehicle (DMSO), and applied the resulting CM to hepatocytes. Fig. 6b (bar 1 vs bar 2) shows that CM from G007-LK-treated adipocytes imparted an anti-gluconeogenic effect on hepatocytes. This effect was not due to G007-LK carryover from adipocytes to hepatocytes, since direct G007-LK treatment of hepatocytes did not affect gluconeogenesis (Fig. 6c). Furthermore, we treated 3T3-L1 adipocytes with IWR-1, a structurally distinct TNKS inhibitor [14], and rinsed off the inhibitor before CM collection. Figure 6d shows that CM from adipocytes pre-treated with IWR-1 also exhibited an anti-gluconeogenic effect. Collectively, these ex vivo studies indicate that upon genetic or pharmacological TNKS inactivation, adipocytes used secreted factors to communicate with hepatocytes to reduce gluconeogenesis.

TNKS inactivation in adipocytes, through soluble factors besides adiponectin, suppresses hepatocyte gluconeogenesis ex vivo. Primary hepatocytes were treated overnight with adipocyte-conditioned media (a, b, d) or adipocyte-naive media (c). After washing off the treatment, hepatocytes were incubated with [14C]pyruvate, and the release of [14C]glucose into the culture media was quantified as described in Methods. (a) Perigonadal adipocytes were isolated from mice (at 4–6 months) of the indicated sex. White bars, TNKS f/f; black bars, CITA. (b) During overnight collection of conditioned media (CM), adipocytes were treated with G007-LK (0.5 μmol/l, black bars) or DMSO (white bars). CM was diluted 1:1 with Medium E before applying to hepatocytes as described in Methods. (c) Hepatocytes were treated directly with G007-LK (0.25 μmol/l) or DMSO in adipocyte-naive DMEM (diluted 1:1 with Medium E) as described in Methods. (d) Differentiated (day 8) 3T3-L1 adipocytes were pre-treated with IWR-1 (10 μmol/l) or DMSO for 4 days as described in Methods. After removing IWR-1, CM was collected overnight and applied to hepatocytes as described in Methods. Data are representative of three or four independent studies, each conducted in 4–6 replicates. *p < 0.05 and **p < 0.01 for indicated comparisons
A candidate mediator of this TNKS-modulated adipocyte–hepatocyte crosstalk is adiponectin since TNKS inactivation increased plasma adiponectin and concomitantly decreased hepatic glucose output (Figs 3, 4 and 5). We therefore investigated whether adiponectin knockout could abrogate the impact of G007-LK on adipocyte–hepatocyte crosstalk. In line with the known anti-gluconeogenic effect of adiponectin [25], we found increased gluconeogenesis in hepatocytes exposed to CM from adiponectin-knockout vs wild-type female mouse adipocytes (Fig. 6b, bar 3 vs bar 1). However, G007-LK treatment of adipocytes, regardless of adiponectin genotype, conferred a comparable anti-gluconeogenic effect to the CM (bars 2 and 4). This ex vivo assay therefore suggested that upon short-term G007-LK treatment, adipocytes alter the secretion of factors apart from adiponectin to reduce gluconeogenesis in hepatocytes.
Discussion
The novel genetic model established here shows that in female mice, adipocyte-selective inactivation of the TNKS catalytic activity improves systemic glucose tolerance primarily by augmenting liver insulin sensitivity. Our ex vivo data implicate adipocyte-secreted factors in the indirect hepatic effect of adipose TNKS inactivation. A similar glucose phenotype, along with increased plasma adiponectin levels, is replicated by treating wild-type mice with the TNKS inhibitor G007-LK, suggesting that the expanding arsenal of TNKS inhibitors [16] may have potential as anti-hyperglycaemic agents. The clinical relevance of this study is supported by the association of polymorphisms near the human TNKS locus (8p23.1) with traits like diabetes [26–28], obesity [29, 30] and plasma adiponectin levels [31].
The increases in insulin sensitivity and adiponectin levels found in the CITA model are shared by our previous model of global TNKS protein ablation [17]. The similarity between the two models suggests that the impact of TNKS on systemic metabolism is largely mediated by its PARsylation activity in adipocytes. The two models do differ in phenotypic details. In contrast to the global model, CITA mice do not exhibit increased insulin sensitivity in muscle (Figs 4d and 5k). This difference could be because the CITA model does not target TNKS in muscle and was phenotyped in C57BL/6 female mice, whereas the global model was in male mice of a mixed genetic background [17]. It could also be that adipocyte functions are differentially affected in the two models since only the CITA model preserves the scaffolding function of TNKS (Fig. 2b).
Consistent with findings in cell-based studies [3], TNKS inactivation in the CITA adipose tissue results in axin stabilisation (Figs 2e and 5a). Consequently, this tissue exhibits certain transcriptional changes (Fig. 3e and ESM 5b) that mirror the reported effects of blocking Wnt signalling in adipocytes such as upregulation of GLUT4 [32, 33] and fatty acid synthase [34]. On the other hand, other changes in the CITA adipose tissue, including decreases in both overall adiposity (Fig. 3b) and inflammatory markers (Fig. 3e) go against the predicted effects of blocking Wnt signalling [34, 35]. Since TNKS can PARsylate many substrates besides axin [36], it is expected to have an impact on cellular processes unrelated to Wnt. For instance, TNKS can PARsylate the proteasome to enhance its proteolytic activity [37]. Because proteasomal inhibition can stabilise nascent adiponectin in the secretory pathway [38], the post-transcriptional increase in adiponectin observed in the plasma of CITA mice (Figs 3h and 5e) could reflect attenuation of proteasomal activity in the CITA adipocytes. Alternatively, the change in adiponectin secretion may be secondary to a direct TNKS effect on the inflammatory state or other processes in the adipocytes.
Unlike adipose tissue, the liver of the CITA mouse does not exhibit TNKS truncation (Fig. 2d) or its effects on axin and β-catenin (Fig. 2f). Thus, the metabolic phenotype of the CITA liver (Figs 4 and 5) is likely to be a remote effect of adipose TNKS inactivation mediated by adipokines. Indeed, gluconeogenesis in hepatocytes is attenuated by secreted factors from adipocytes where TNKS has been inactivated genetically or pharmacologically (Fig. 6). Based on the consequences of interventions that inhibit TNKS in adipocytes, we infer that a physiological function of TNKS catalysis in adipocytes is the enhancement of hepatic gluconeogenesis.
The specific adipokine(s) mediating the proposed TNKS-modulated communication with liver remains to be identified. Although our short-term ex vivo studies do not implicate adiponectin in this TNKS-modulated crosstalk (Fig. 6), it remains possible that sustained increase in circulating adiponectin in CITA mice and in G007-LK-treated mice might help improve glucose tolerance. Both our ex vivo and in vivo studies indicate that regulation of fat-to-liver communication by TNKS is more robust in female mice. Given the prevailing view that sexual dimorphism in metabolism results from the interplay between gonadal hormones and sex chromosome complements [39], we speculate that ovarian hormones accentuate the CITA phenotype through a synergism between oestrogen action and TNKS inhibition. A candidate molecular mechanism would be synergistic blockade of Wnt signalling, since TNKS inhibition [3] and oestrogen receptor action [40, 41] have separately been shown to block Wnt signalling. Future effort will attempt to explore mechanism(s) underlying the sex specificity of the CITA phenotype and to identify the adipokine(s) mediating TNKS-regulated communication between adipose tissue and liver.
Abbreviations
- CITA:
-
Catalytically inactive TNKS in adipose tissue
- HFD:
-
High-fat diet
- IRAP:
-
Insulin-responsive aminopeptidase
- PAR:
-
Poly-ADP-ribose
- PARP:
-
PAR polymerase
- RBP4:
-
Retinol-binding protein 4
- TNKS:
-
Tankyrase (TRF1-interacting ankyrin-related ADP-ribose polymerase)
References
Hsiao SJ, Smith S (2008) Tankyrase function at telomeres, spindle poles, and beyond. Biochimie 90:83–92
Sbodio JI, Chi NW (2002) Identification of a tankyrase-binding motif shared by IRAP, TAB182, and human TRF1 but not mouse TRF1. NuMA contains this RxxPDG motif and is a novel tankyrase partner. J Biol Chem 277:31887–31892
Huang SM, Mishina YM, Liu S et al (2009) Tankyrase inhibition stabilizes axin and antagonizes Wnt signalling. Nature 461:614–620
Chi NW, Lodish HF (2000) Tankyrase is a golgi-associated mitogen-activated protein kinase substrate that interacts with IRAP in GLUT4 vesicles. J Biol Chem 275:38437–38444
Zhong L, Yeh TY, Hao J et al (2015) Nutritional energy stimulates NAD+ production to promote tankyrase-mediated PARsylation in insulinoma cells. PLoS One 10, e0122948
Zhang Y, Liu S, Mickanin C et al (2011) RNF146 is a poly(ADP-ribose)-directed E3 ligase that regulates axin degradation and Wnt signalling. Nat Cell Biol 13:623–629
Bisht KK, Dudognon C, Chang WG, Sokol ES, Ramirez A, Smith S (2012) GDP-mannose-4,6-dehydratase is a cytosolic partner of tankyrase 1 that inhibits its poly(ADP-ribose) polymerase activity. Mol Cell Biol 32:3044–3053
Kim MK, Smith S (2014) Persistent telomere cohesion triggers a prolonged anaphase. Mol Biol Cell 25:30–40
Bai P (2015) Biology of poly(ADP-ribose) polymerases: the factotums of cell maintenance. Mol Cell 58:947–958
Yeh TY, Sbodio JI, Tsun ZY, Luo B, Chi NW (2007) Insulin-stimulated exocytosis of GLUT4 is enhanced by IRAP and its partner tankyrase. Biochem J 402:279–290
De Rycker M, Price CM (2004) Tankyrase polymerization is controlled by its sterile alpha motif and poly(ADP-ribose) polymerase domains. Mol Cell Biol 24:9802–9812
Guo HL, Zhang C, Liu Q et al (2012) The Axin/TNKS complex interacts with KIF3A and is required for insulin-stimulated GLUT4 translocation. Cell Res 22:1246–1257
MacDonald BT, Tamai K, He X (2009) Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev Cell 17:9–26
Chen B, Dodge ME, Tang W et al (2009) Small molecule-mediated disruption of Wnt-dependent signaling in tissue regeneration and cancer. Nat Chem Biol 5:100–107
Waaler J, Machon O, Tumova L et al (2012) A novel tankyrase inhibitor decreases canonical Wnt signaling in colon carcinoma cells and reduces tumor growth in conditional APC mutant mice. Cancer Res 72:2822–2832
Lehtio L, Chi NW, Krauss S (2013) Tankyrases as drug targets. FEBS J 280:3576–3593
Yeh TY, Beiswenger KK, Li P et al (2009) Hypermetabolism, hyperphagia, and reduced adiposity in tankyrase-deficient mice. Diabetes 58:2476–2485
Bluher M (2014) Adipokines—removing road blocks to obesity and diabetes therapy. Mol Metab 3:230–240
Li P, Fan W, Xu J et al (2011) Adipocyte NCoR knockout decreases PPARγ phosphorylation and enhances PPARγ activity and insulin sensitivity. Cell 147:815–826
Lau T, Chan E, Callow M et al (2013) A novel tankyrase small-molecule inhibitor suppresses APC mutation-driven colorectal tumor growth. Cancer Res 73:3132–3144
Voronkov A, Holsworth DD, Waaler J et al (2013) Structural basis and SAR for G007-LK, a lead stage 1,2,4-triazole based specific tankyrase 1/2 inhibitor. J Med Chem 56:3012–3023
Zemany L, Bhanot S, Peroni OD et al (2014) Transthyretin antisense oligonucleotides lower circulating RBP4 levels and improve insulin sensitivity in obese mice. Diabetes 64:1603–14
Jeffery E, Berry R, Church CD et al (2014) Characterization of Cre recombinase models for the study of adipose tissue. Adipocyte 3:206–211
He Z, Zhu HH, Bauler TJ et al (2013) Nonreceptor tyrosine phosphatase Shp2 promotes adipogenesis through inhibition of p38 MAP kinase. Proc Natl Acad Sci U S A 110:E79–E88
Miller RA, Chu Q, Le Lay J et al (2011) Adiponectin suppresses gluconeogenic gene expression in mouse hepatocytes independent of LKB1-AMPK signaling. J Clin Invest 121:2518–2528
Dunn JS, Mlynarski WM, Pezzolesi MG et al (2006) Examination of PPP1R3B as a candidate gene for the type 2 diabetes and MODY loci on chromosome 8p23. Ann Hum Genet 70:587–593
Pezzolesi MG, Nam M, Nagase T et al (2004) Examination of candidate chromosomal regions for type 2 diabetes reveals a susceptibility locus on human chromosome 8p23.1. Diabetes 53:486–491
Kim SH, Ma X, Weremowicz S et al (2004) Identification of a locus for maturity-onset diabetes of the young on chromosome 8p23. Diabetes 53:1375–1384
Scherag A, Dina C, Hinney A et al (2010) Two new loci for body-weight regulation identified in a joint analysis of genome-wide association studies for early-onset extreme obesity in French and German study groups. PLoS Genet 6, e1000916
Lindgren CM, Heid IM, Randall JC et al (2009) Genome-wide association scan meta-analysis identifies three loci influencing adiposity and fat distribution. PLoS Genet 5, e1000508
Ling H, Waterworth DM, Stirnadel HA et al (2009) Genome-wide linkage and association analyses to identify genes influencing adiponectin levels: the GEMS Study. Obesity 17:737–744
Gustafson B, Smith U (2010) Activation of canonical wingless-type MMTV integration site family (Wnt) signaling in mature adipocytes increases beta-catenin levels and leads to cell dedifferentiation and insulin resistance. J Biol Chem 285:14031–14041
Gustafson B, Smith U (2012) The WNT inhibitor Dickkopf 1 and bone morphogenetic protein 4 rescue adipogenesis in hypertrophic obesity in humans. Diabetes 61:1217–1224
Longo KA, Wright WS, Kang S et al (2004) Wnt10b inhibits development of white and brown adipose tissues. J Biol Chem 279:35503–35509
Wright WS, Longo KA, Dolinsky VW et al (2007) Wnt10b inhibits obesity in ob/ob and agouti mice. Diabetes 56:295–303
Guettler S, LaRose J, Petsalaki E et al (2011) Structural basis and sequence rules for substrate recognition by Tankyrase explain the basis for cherubism disease. Cell 147:1340–1354
Cho-Park PF, Steller H (2013) Proteasome regulation by ADP-ribosylation. Cell 153:614–627
Gu D, Wang Z, Dou X et al (2013) Inhibition of ERK1/2 pathway suppresses adiponectin secretion via accelerating protein degradation by ubiquitin-proteasome system: relevance to obesity-related adiponectin decline. Metabolism 62:1137–1148
Link JC, Chen X, Arnold AP, Reue K (2013) Metabolic impact of sex chromosomes. Adipocyte 2:74–79
Cleveland AG, Oikarinen SI, Bynote KK et al (2009) Disruption of estrogen receptor signaling enhances intestinal neoplasia in Apc Min/+ mice. Carcinogenesis 30:1581–1590
Chaudhary SC, Singh T, Talwelkar SS et al (2014) Erb-041, an estrogen receptor-beta agonist, inhibits skin photocarcinogenesis in SKH-1 hairless mice by downregulating the WNT signaling pathway. Cancer Prev Res 7:186–198
Acknowledgements
We thank J. Olefsky (UC San Diego) for providing aP2-Cre Salk mice and T.-Y. J. Yeh, K. Beiswenger (formerly at UC San Diego) and L. Slater (UC San Diego) for technical assistance.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This work was supported in part by grants from the Department of Veterans Affairs (5I01BX000702) and the American Diabetes Association (7-05-CD-05) to NWC.
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Contribution statement
LZ, NWC, JW, SK and SS designed the study. LZ, YD, JS, SAP, GB, SM, MZ, EB and SKM collected and analysed the data. The manuscript was written by LZ and revised by NWC, JW and SK. All authors edited the manuscript and approved the final version. NWC is responsible for the integrity of the work as a whole.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM Methods
(PDF 94 kb)
ESM Fig. 1
(PDF 529 kb)
ESM Fig. 2
(PDF 525 kb)
ESM Fig. 3
(PDF 631 kb)
ESM Fig. 4
(PDF 390 kb)
ESM Fig. 5
(PDF 372 kb)
ESM Table 1
(PDF 69 kb)
Rights and permissions
About this article

Cite this article
Zhong, L., Ding, Y., Bandyopadhyay, G. et al. The PARsylation activity of tankyrase in adipose tissue modulates systemic glucose metabolism in mice. Diabetologia 59, 582–591 (2016). https://doi.org/10.1007/s00125-015-3815-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-015-3815-1