
Abstract
Aims/hypothesis
This study investigated fibroblast growth factor 21 (FGF21)-mediated cardiac protection against apoptosis caused by diabetic lipotoxicity and explored the protective mechanisms involved.
Methods
Cardiac Fgf21 mRNA expression was examined in a diabetic mouse model using real-time PCR. After pre-incubation of palmitate-treated cardiac H9c2 cells and primary cardiomyocytes with FGF21 for 15 h, apoptosis and Fgf21-induced cell-survival signalling were investigated using small interfering (si)RNA and/or pharmacological inhibitors. We also examined the cardiac apoptotic signalling and structural and functional indices in wild-type and Fgf21-knockout (Fgf21-KO) diabetic mice.
Results
In a mouse model of type 1 diabetes, cardiac Fgf21 expression was upregulated about 40-fold at 2 months and 3–1.5-fold at 4 and 6 months after diabetes. FGF21 significantly reduced palmitate-induced cardiac apoptosis. Mechanistically, palmitate downregulated, but FGF21 upregulated, phosphorylation levels of extracellular signal-regulated kinase (ERK)1/2, mitogen-activated protein kinase 14 (p38 MAPK) and AMP-activated protein kinase (AMPK). Inhibition of each kinase with its inhibitor and/or siRNA revealed that FGF21 prevents palmitate-induced cardiac apoptosis via upregulating the ERK1/2-dependent p38 MAPK–AMPK signalling pathway. In vivo administration of FGF21, but not FGF21 plus ERK1/2 inhibitor, to diabetic or fatty-acid-infused mice significantly prevented cardiac apoptosis and reduced inactivation of ERK1/2, p38 MAPK and AMPK and prevented cardiac remodelling and dysfunction. The Fgf21-KO mice were more susceptible to diabetes-induced cardiac apoptosis, and this could be prevented by administration of FGF21. Deletion of Fgf21 did not further exacerbate cardiac dysfunction.
Conclusions/interpretation
These results demonstrate that FGF21 prevents lipid- or diabetes-induced cardiac apoptosis by activating the ERK1/2–p38 MAPK–AMPK pathway. FGF21 may be a therapeutic target for the treatment of diabetes-related cardiac damage.
Similar content being viewed by others
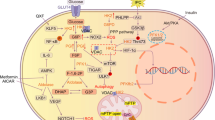

Introduction
Diabetic cardiomyopathy is attributed to multiple pathogenic factors, including hyperglycaemia, hyperlipidaemia, hypertension and inflammation [1–3]. Cardiomyopathy is the late consequence of diabetes-induced early cardiac responses. One of the key early cardiac responses is apoptosis [2, 4, 5]. Inhibition of the early cardiac apoptosis can prevent subsequent diabetic cardiomyopathy [4, 6]. Thus, reducing cardiac apoptosis may be beneficial to prevent diabetic cardiomyopathy.
Fibroblast growth factor 21 (FGF21) has been identified as a potent metabolic regulator with specific effects on glucose and lipid metabolism [7–9]. It has been predominantly investigated in the liver and adipose tissue [10]. However, FGF21 is also expressed in other tissues such as the myocardium [11]. FGF21 was found to be an acute response protein to protect tissues from acute toxicity [12, 13]. Additionally, the anti-apoptotic effects of FGF21 on islet beta cells and endothelial cells were also reported [14, 15]. However, the effect of FGF21 on the heart remains largely unknown. FGF21 functions through binding to fibroblast growth factor receptor (FGFR)1 and the co-factor β-Klotho [16]. The existence of FGFR1, β-Klotho [17] and FGF21 [11] in the myocardium implies that FGF21 may play certain physiological roles in the heart. Recently, several studies have demonstrated FGF21-mediated protection against myocardial ischaemia/reperfusion injury [18, 19] and isoprenaline-induced cardiac hypertrophy [20]. However, the effect of FGF21 on diabetic heart remains elusive.
In the present study, we examined whether FGF21 protects the heart from NEFA- or diabetes-induced cardiac apoptosis and dysfunction in vitro and in vivo.
Methods
Animal experiments
Five sets of animal studies were performed: (1) measuring cardiac Fgf21 mRNA expression; (2) assessing cardiac FGF21-mediated protection from lipotoxicity induced by NEFA infusion; (3) evaluating acute FGF21-mediated protection from diabetes-induced cardiac apoptosis; (4) testing whether Fgf21-knockout (Fgf21-KO) mice are susceptible to diabetic cardiac apoptosis; and (5) testing whether FGF21 prevents diabetic cardiomyopathy in a model of chronic diabetes. All animal experiments were conducted in accordance with the guideline of the Institutional Animal Care and Use committees of Wenzhou Medical University and University of Louisville. See electronic supplementary material (ESM) Methods for details.
Cardiac function and BP assay
Cardiac function and BP were measured by echocardiography and tail-cuff manometry, respectively [21, 22]. See ESM Methods for details.
Biochemical and histochemical assay
Plasma triacylglycerol was measured using a triacylglycerol assay kit. Cardiac apoptosis was detected by TUNEL or DNA fragmentation [23]. Cardiac fibrosis was examined using Sirius Red staining. The expression and/or phosphorylation of target genes was detected by western blot or real-time quantitative (q)PCR. See ESM Methods for details.
Primary cardiomyocyte isolation, cell culture, palmitate and FGF21 treatments and small interfering RNA transfection
Adult mouse cardiomyocytes were isolated as described previously [24]. H9c2 cells and/or cardiomyocytes were pre-treated with pharmaceutical inhibitors or specific small interfering (si)RNAs against Erk1 (also known as Mapk3)/Erk2 (also known as Mapk1), p38Mapk (also known as Mapk14) and Ampk with or without FGF21 (50 ng/ml), followed by palmitate treatment for 15 h. A pilot time-course study has been performed to optimise the transfection efficiency with p38Mapk siRNA in adult mouse cardiomyocytes (ESM Fig. 1). See ESM Methods for details.
Statistical analysis
Data were collected from three replicates of cell-culture experiments, or from n ≥ 5 mice per group for in vivo studies and presented as mean ± SD. One-way ANOVA was used to determine general differences, followed by a post hoc Tukey’s test for the difference between groups, using Origin 7.5 software (Northampton, MA, USA). Statistical significance was considered as p < 0.05.
Results
Fgf21 mRNA expression is increased in the heart of mice with diabetes
We found that fasting for 24 h increased Fgf21 mRNA expression about nine fold in the heart (ESM Fig. 2a) and 26-fold in the liver (ESM Fig. 2b). The endoplasmic reticulum stress inducer thapsigargin [25, 26] also significantly increased Fgf21 mRNA in the heart (ten fold) (ESM Fig. 2c) and liver (14-fold) (ESM Fig. 2d). In streptozotocin (STZ)-induced diabetic mice, the hepatic Fgf21 mRNA expression was significantly decreased at 2, 4 and 6 months after diabetes onset (ESM Fig. 2f), while cardiac Fgf21 mRNA was unexpectedly increased 40-fold at 2 months and 1.5–2.5-fold at 4–6 months (ESM Fig. 2e).
FGF21 prevents palmitate-induced cardiac apoptosis via ERK1/2-mediated pathways
To define the remarkable induction of cardiac Fgf21 expression at the early stage of diabetes as a protective response to diabetic lipotoxicity, H9c2 cells were treated with palmitate to mimic diabetic hyperlipidaemia [27]. Exposure of H9c2 cells to palmitate at 62.5 μmol/l for 15 h induced a significant increase of apoptosis, detected by caspase-3 cleavage and DNA fragmentation (Fig. 1a, b), which was prevented by FGF21 in a dose range of 25–100 ng/ml. The mechanistic study revealed that palmitate inhibited the phosphorylation of extracellular signal-regulated kinase (ERK)1/2 (Fig. 1c), mitogen-activated protein kinase 14 (p38 MAPK) (Fig. 1d) and AMP-activated protein kinase (AMPK) (Fig. 1e), and this effect was reduced by FGF21 treatment (Fig. 1c–e).

FGF21 prevents palmitate-induced cardiac apoptosis and inactivation of ERKg1/2, p38 MAPK and AMPK. Pre-treatment of H9c2 cells with FGF21 (50 ng/ml) for 1 h was followed by co-treatment with palmitate (62.5 μmol/l) for 15 h. Caspase-3 cleavage (a) and DNA fragmentation (b) were detected as markers of apoptosis, and phosphorylation levels of ERK1/2 (c), p38 MAPK (d) and AMPK (e) were examined as markers of FGF21-mediated protection. Some H9c2 cells were treated with both FGF21 (50 ng/ml) and PD98059 (20 μmol/l), SB203580 (20 μmol/l) or compound C (10 μmol/l) 1 h before and during 15 h palmitate treatment, and then caspase-3 cleavage (f–h) and PTEN phosphorylation (l) were examined by western blot. Primary cardiomyocytes were pre-treated with FGF21 (50 ng/ml, light grey bar) or PBS (white bar) for 1 h followed by co-treatment with palmitate (62.5 μmol/l; black bar, PBS + palmitate; dark grey bar, FGF21 + palmitate) in the presence of either siRNA against Erk1/2, p38Mapk or Ampk or control siRNA. Caspase-3 cleavage (i–k) was examined as above. Data were collected from at least three independent experiments and presented as mean ± SD. * p < 0.05 vs control; † p < 0.05 vs palmitate; ‡ p < 0.05 vs palmitate/FGF21; § p < 0.05 vs control in Erk1/2, p38Mapk or Ampk siRNA treatment. C-cas3, caspase-3 cleavage; Com C, compound C; Con, control; p38, p38 MAPK; Pal, palmitate; PD, PD98059; SB, SB203580
To establish whether the upregulation of these kinases is required for FGF21-mediated protection, H9c2 cells were treated with both FGF21 (50 ng/ml) and an inhibitor of ERK1/2 (PD98059, 20 μmol/l), p38 MAPK (SB203580, 20 μmol/l) or AMPK (compound C, 10 μmol/l) for 1 h before and during palmitate treatment for 15 h (Fig. 1f–h). The FGF21-mediated protection from palmitate-induced apoptosis was attenuated by each kinase inhibitor (Fig. 1f–h). To exclude a non-specific effect of an inhibitor, we further evaluated the role of these kinases in FGF21-mediated protection by using specific siRNAs against Erk1/2 (ESM Fig. 3a–c), p38Mapk (ESM Fig. 3d–f) and Ampk (ESM Fig. 3g–i). We found that the specific siRNA, but not the control, efficiently silenced the expression and phosphorylation of each kinase (ESM Fig. 3a–i), and also abolished FGF21-mediated protection from palmitate-induced apoptosis in adult cardiomyocytes (Fig. 1i–k). Inhibition of ERK1/2 or AMPK also increased phosphatase and tensin homologue (PTEN) phosphorylation (Fig. 1l).
Time-course studies showed that FGF21 treatment for 1–15 h increased ERK1/2 phosphorylation at 1–3 h and 9–12 h, peaking at 1 h and 9 h (ESM Fig. 4a), p38 MAPK phosphorylation at 3–9 h, peaking at 6 h (ESM Fig. 4b), and AMPK phosphorylation at 6–15 h, peaking at 12 h (ESM Fig. 4c). To dissect whether these kinases are in the same signalling pathway, H9c2 cells were treated with FGF21 and each inhibitor for 15 h (ESM Fig. 4d–l). PD98059 inhibited not only ERK1/2 (ESM Fig. 4d), but also p38 MAPK (ESM Fig. 4e) and AMPK (ESM Fig. 4f). SB203580 did not affect ERK1/2 (ESM Fig. 4g), but significantly inhibited p38 MAPK (ESM Fig. 4h) and AMPK phosphorylation (ESM Fig. 4i). Compound C inhibited only AMPK (ESM Fig. 4j–l). These results suggest that FGF21 activates an ERK1/2-mediated p38 MAPK–AMPK signalling pathway.
To rule out inhibitor non-specificity, the direct role of ERK1/2 in the top position of the signalling pathway was confirmed with Erk1/2 siRNA that almost completely abolished ERK1/2 phosphorylation (Fig. 2a, b) and expression (Fig. 2a, c) and the subsequent Fgf21-increased phosphorylation of p38 MAPK (Fig. 2a, d) and AMPK (Fig. 2a, e), which also abolished FGF21-mediated protection against palmitate-induced apoptosis (Fig. 2a, f). To further validate the role of ERK1/2–p38 MAPK–AMPK signalling in FGF21-mediated cardiac protection in a more physiological context, the specific siRNAs against Erk1/2 (ESM Fig. 3a–c) and p38Mapk (ESM Fig. 3d–f) were also used to knockdown these kinases in adult cardiomyocytes. We found that Erk1/2 siRNA, but not control, almost completely abolished the FGF21-mediated increase in p38 MAPK and AMPK phosphorylation (Fig. 2g, h), and p38Mapk siRNA had no significant effects on FGF21-mediated increase in ERK1/2 phosphorylation (Fig. 2i), but significantly abolished the FGF21-mediated increase in AMPK phosphorylation (Fig. 2j).

Inhibition of ERK1/2 or PTEN with siRNA attenuates FGF21-mediated anti-apoptotic function against palmitate. H9c2 cells were treated with FGF21 (50 ng/ml, light grey bar) or PBS (white bar) for 1 h followed by co-treatment with palmitate (62.5 μmol/l; black bar, PBS + palmitate; dark grey bar, FGF21 + palmitate) in the presence of either control or Erk1/2-specific siRNA and then western blotting was used to detect the phosphorylation levels of ERK1/2 (a–c), p38 MAPK (a, d) and AMPK (a, e), and caspase-3 cleavage (a, f). Primary cardiomyocytes were pre-treated with FGF21 (50 ng/ml, light grey bar) or PBS (white bar) for 1 h followed by co-treatment with palmitate (62.5 μmol/l; black bar, PBS + palmitate; dark grey bar, FGF21 + palmitate) in the presence of either siRNA against Erk1/2 (g, h) or p38Mapk (i, j) or control siRNA. p38 MAPK, AMPK and ERK1/2 phosphorylation was examined by western blot. Some H9c2 cells were treated with PBS (white bar) or palmitate (black bar) in the presence of control or Pten siRNA, and the phosphorylation (k, l) and levels of PTEN (k, m) and caspase-3 cleavage (k, n) were examined by western blot. Data collection and presentation are the same as in Fig. 1. * p < 0.05 vs control in the control siRNA group; † p < 0.05 vs palmitate in the control siRNA group; § p < 0.05 vs control in Erk1/2 siRNA group. Con, control; p38, p38 MAPK; Pal, palmitate
In addition, Pten siRNA almost completely abolished the phosphorylation (Fig. 2k, l) and abundance of PTEN (Fig. 2k, m), which also prevented palmitate-induced apoptosis (Fig. 2k, n), confirming the pivotal role of PTEN in the palmitate-induced apoptotic signalling pathway.
FGF21 prevents acute NEFA-infusion-induced cardiac apoptosis in mice
To see whether FGF21 can also protect the heart from lipotoxicity in vivo, mice were intraperitoneally given NEFA (0.1 g/10 g) with and without FGF21 (100 μg kg−1 day−1) for 10 days.
NEFA significantly increased serum triacylglycerol levels, and this was not affected by FGF21 (Fig. 3a). Significant cardiac apoptosis was observed in NEFA-infused mice, but not in mice with BSA or mice treated with FGF21 (Fig. 3b–d). FGF21 also significantly prevented the NEFA-mediated decrease in ERK1/2 (Fig. 3e, f), p38 MAPK (Fig. 3e, g) and AMPK phosphorylation (Fig. 3e, h) and increase in PTEN phosphorylation (Fig. 3i).

FGF21 prevents NEFA-infusion-induced cardiac apoptosis. Mice were infused with NEFA (0.1 g/10 g body weight) with or without FGF21 (100 μg kg−1 day−1) for 10 days. Plasma triacylglycerol was measured using an assay kit (a). Cardiac apoptosis was detected by TUNEL (b, c) and caspase-3 cleavage (d); arrows in micrographs indicate apoptotic nuclei. Phosphorylation of ERK1/2 (e, f), p38 MAPK (e, g), AMPK (e, h) and PTEN (e, i) was examined by western blot. Data are presented as mean ± SD. n ≥ 5 for each group. * p < 0.05 vs control; † p < 0.05 vs NEFA treatment. Scale bar, 100 μm. Con, control; p38, p38 MAPK; TAG, triacylglycerol
FGF21 prevents diabetes-induced cardiac apoptosis through ERK1/2 activation
To confirm whether the anti-apoptotic effect of FGF21 observed in the above studies can be replicated in type 1 diabetes, FGF21 (100 μg kg−1 day−1) or vehicle was administered to diabetic and control mice for 10 days. FGF21 slightly decreased the blood glucose in diabetes, but had no effect on controls (Fig. 4a). Additionally, FGF21 did not affect the plasma triacylglycerol level in either control or diabetic mice (Fig. 4b).

FGF21 prevention of diabetes-induced cardiac apoptosis is mediated by ERK1/2 activation. Diabetic and control mice were given FGF21 (100 μg kg−1 day−1) or PBS daily for 10 days. Blood glucose (a) and plasma triacylglycerol (b) levels were examined using a blood glucose monitor or a triacylglycerol assay kit. Cardiac apoptosis was assessed by TUNEL (c, d) and caspase-3 cleavage (e); arrows in micrographs indicate the apoptotic nuclei. Phosphorylation of ERK1/2 (f), p38 MAPK (g), AMPK (h) and PTEN (i) was examined by western blot. Some mice were treated with both FGF21 and the ERK1/2 inhibitor PD98059 for 10 days. Caspase-3 cleavage (j, k) and ERK1/2 (j, l), p38 MAPK (j, m), AMPK (j, n) and PTEN (j, o) phosphorylation were detected by western blot. Data are presented as mean ± SD. n = 8 for each group. * p < 0.05 vs control; † p < 0.05 vs diabetes; ‡ p < 0.05 vs diabetes/FGF21. Scale bar, 100 μm. C-cas3, caspase-3 cleavage; Con, control; DM, diabetes; p38, p38 MAPK; PD, PD98059; TAG, triacylglycerol
TUNEL staining (Fig. 4c, d) and caspase-3 cleavage (Fig. 4e) showed that FGF21 prevented diabetes-induced cardiac apoptosis. Diabetes also significantly decreased cardiac ERK1/2 (Fig. 4f), p38 MAPK (Fig. 4g) and AMPK (Fig. 4h) phosphorylation and increased PTEN phosphorylation (Fig. 4i), all of which were completely prevented by FGF21. These results suggest that FGF21 prevents diabetes-induced cardiac apoptosis via increasing ERK1/2, p38 MAPK and AMPK phosphorylation and inhibiting PTEN phosphorylation, as found in the in vitro study.
Inhibition of ERK1/2 by PD98059 (10 mg/kg daily, Fig. 4j, l) completely abolished FGF21-mediated prevention of diabetes-induced cardiac apoptosis (Fig. 4j, k), and FGF21-maintained cardiac p38 MAPK (Fig. 4j, m) and AMPK phosphorylation (Fig. 4j, n). Furthermore, inactivation of ERK1/2 also abolished FGF21-mediated prevention of diabetes-induced PTEN phosphorylation (Fig. 4j, o). These results demonstrated that FGF21 prevented diabetes-related inhibition of ERK1/2, resulting in a reduction of diabetes-induced PTEN-activation-mediated apoptosis.
Fgf21-KO mice are more susceptible to diabetes-induced cardiac apoptosis
To further define the protective role of endogenous FGF21, diabetes was induced in both Fgf21-KO and wild-type (WT) control mice. Compared with WT mice with diabetes, Fgf21-KO mice with diabetes showed a further elevation of blood glucose (Fig. 5a) and plasma triacylglycerol (Fig. 5b). Treatment of Fgf21-KO diabetic mice with FGF21 (100 μg kg−1 day−1 for 10 days) significantly decreased blood glucose compared with non-treated Fgf21-KO diabetic mice (Fig. 5a).

Fgf21-KO mice are more sensitive to diabetes-induced cardiac apoptosis. Fgf21-KO and WT mice were induced to develop diabetes (black bar) with STZ and treated with FGF21 (100 μg kg−1 day−1, light grey bar) or PBS (white bar) for 10 days. Blood glucose (a) and plasma triacylglycerol (b) levels were examined as above. Cardiac apoptosis was assessed by TUNEL (c, d) and caspase-3 cleavage (e, f); arrows in micrographs indicate the apoptotic nuclei. Phosphorylation of PTEN (e, g), ERK1/2 (e, h), p38 MAPK (e, i) and AMPK (e, j) was examined by western blot. n = 8 for each group.* p < 0.05 vs WT control; † p < 0.05 vs WT diabetes; ‡ p < 0.05 vs Fgf21-KO diabetes; § p < 0.05 vs Fgf21-KO control. Scale bar, 100 μm. C-cas3, caspase-3 cleavage; Con, control; DM, diabetes; TAG, triacylglycerol
Apoptosis was significantly more abundant in the heart of Fgf21-KO mice with diabetes than in WT mice with diabetes (Fig. 5c–f), which was same as the PTEN phosphorylation profile (Fig. 5e, g). The inhibition of ERK1/2 (Fig. 5e, h), p38 MAPK (Fig. 5e, i) and AMPK (Fig. 5e, j) was also more evident in heart from Fgf21-KO mice with diabetes than in heart from WT mice with diabetes. FGF21 treatment of Fgf21-KO mice with diabetes completely reversed diabetes-increased apoptosis and PTEN phosphorylation and diabetes-decreased ERK1/2, p38 MAPK and AMPK phosphorylation (Fig. 5c–j).
FGF21 prevents diabetes-induced cardiac remodelling and dysfunction through ERK1/2 activation
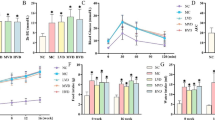
The next study examined whether FGF21 prevention of cardiac apoptosis can result in prevention of cardiac remodelling and dysfunction. WT and Fgf21-KO mice with diabetes chronically treated with FGF21 (100 μg kg−1 day−1) for 2 months showed a significant reduction in blood glucose, which was abolished by ERK1/2 inhibition in WT diabetes (Fig. 6a). However, FGF21 treatment did not affect plasma triacylglycerol, which was significantly higher in Fgf21-KO diabetic mice compared with WT diabetic mice; the presence or absence of ERK1/2 inhibition did not affect plasma triacylglycerol in either the WT or the Fgf21-KO diabetic mice (Fig. 6b).

FGF21 prevents diabetes-induced cardiac remodelling and dysfunction through ERK1/2 activation. Fgf21-KO and WT mice were induced as diabetic (black bar) with STZ, and treated with PBS (white bar) or FGF21 (100 μg kg−1 day−1, light grey bar) for 2 months. One set of WT diabetic mice were treated with FGF21 + ERK1/2 inhibitor (PD98059, dark grey bar). Blood glucose (a) and plasma triacylglycerol (b) were examined as above. Cardiac apoptosis was assessed by TUNEL (c) and caspase-3 cleavage (d, e). Phosphorylation of PTEN (d, f), ERK1/2 (d, g), p38 MAPK (d, h) and AMPK (d, i) was examined by western blot. Cardiac remodelling was examined by CTGF expression (d, j) and Sirius Red staining (k, l). Data are presented as mean ± SD. n = 8 for each group. * p < 0.05 vs control; † p < 0.05 vs WT diabetic mice; ‡ p < 0.05 vs diabetic mice receiving FGF21; § p < 0.05 vs Fgf21-KO control; ¶ p < 0.05 vs Fgf21-KO diabetic mice. Scale bar, 100 μm. Con, control; DM, diabetes; p38, p38 MAPK; TAG, triacylglycerol
Cardiac apoptosis was detected by TUNEL staining (Fig. 6c, ESM Fig. 5) and caspase-3 cleavage (Fig. 6d, e). There was significant cardiac apoptosis in diabetes, particularly in Fgf21-KO mice. Consistent with an apoptotic effect, PTEN phosphorylation was increased in diabetes (Fig. 6d, f), and ERK1/2 (Fig. 6d, g), p38 MAPK (Fig. 6d, h) and AMPK inhibition (Fig. 6d, i) were also more evident in Fgf21-KO diabetic mice than in WT diabetic mice. These diabetes-induced apoptosis and associated signalling changes were prevented by FGF21 treatment (Fig. 6a–i), but FGF21-mediated protection was significantly abolished by PD98059 treatment (Fig. 6a–i). Diabetes also induced cardiac remodelling, shown by an increase in connective tissue growth factor (CTGF) expression (Fig. 6d, j) and collagen accumulation (Fig. 6k, l), which was significantly different in Fgf21-KO diabetic mice than in WT diabetic mice.
Diabetes-induced hypertension was significantly prevented by FGF21 treatment in both WT and Fgf21-KO diabetic mice (Table 1). The preventive effects of FGF21 were abolished by ERK1/2 inhibition (Table 1). Diabetes significantly reduced cardiac function, indicated by decreased ejection fraction (EF) and fractional shortening (FS) in both WT and Fgf21-KO mouse hearts (Table 1). At the same time, diabetes also induced alterations in several structural indices (Table 1), including decreased left ventricular (LV) postal wall thickness, increased end systolic LV inner diameter and volume, together with a small decrease in LV mass, but a significant increase in LV mass index, which reflects a greater body weight loss, in both WT and Fgf21-KO diabetes (Table 1). All these cardiac functional and structural changes were mildly but significantly improved by FGF21 treatment in both WT and Fgf21-KO diabetic mice, an effect that was abolished by ERK1/2 inhibition (Table 1). Fgf21 deletion had no significant effects on cardiac structural and functional variables under basal conditions.
Discussion
The early upregulation of cardiac Fgf21 mRNA expression was found to gradually decrease with the progression of diabetes (ESM Fig. 2e). We hypothesised that, besides induction of lipolysis to meet cardiac energy requirements, the early upregulation of cardiac FGF21 expression may be a compensatory effect to protect from the acute toxic effect of the accumulated intermediates of lipid metabolism in the heart. Here, we provide direct evidence for the anti-apoptotic effect of FGF21 in cardiac tissue, with protection against palmitate in the cultured cardiac cell line and primary cardiomyocytes and STZ-induced diabetes or NEFA infusion in mice. We have also demonstrated that FGF21 supplementation prevents the progression of diabetes-induced cardiomyopathy in both WT and Fgf21-KO mice, a finding that is supportive of previous studies showing FGF21 protects against myocardial ischaemia/reperfusion injury [18, 19] and isoprenaline-induced cardiac hypertrophy [20].
Mice with Fgf21 deletion were more susceptible to diabetes-induced cardiac apoptosis, but cardiac structural and functional derangements were not exacerbated in these mice (Fig. 6a–1 and Table 1). This could be attributed to the relative time span of the diabetic model, as we have established that diabetes-induced cardiac apoptosis at an early stage (7–21 days) leads to the development of diabetic cardiomyopathy in later life (4–6 months) [28]. Therefore, we could see increased apoptosis in Fgf21-KO diabetic mice at the early stage (2 months), but could not yet see any exacerbation of diabetes-induced cardiac structural and functional changes.
The insulin-sensitising and lipid-manipulating effects of FGF21 [29, 30] may not be the predominant contributor to cardiac protection by FGF21 in mice with STZ-induced type 1 diabetes, as these mice have severe insulin deficiency. We also only observed a slight glucose-lowing effect (Figs 4a, 5a and 6a), but without global lipid-lowering potency (Figs 4b, 5b and 6b) at a lower FGF21 dose level (100 μg kg−1 day−1), which is consistent with previous reports [30, 31]. Based on our in vitro studies showing that FGF21 protects against palmitate-induced cardiac apoptosis in H9c2 cells and primary cardiomyocytes, and also previous reports indicating that liver- and adipose-tissue-secreted FGF21 protects the heart from ischaemia/reperfusion injury [18] and exogenous FGF21 protects the heart from isoprenaline-induced cardiac hypertrophy [20], we presume that cardiac protection from diabetes by FGF21 may predominantly be attributed to its direct actions on the heart.
The present study elucidated the intracellular mechanisms by which FGF21 prevents palmitate-mediated apoptosis in vitro and NEFA-infusion- or diabetes-mediated apoptosis in vivo. The activation of ERK1/2 by FGF21 in cardiac cells and heart plays a pivotal role in FGF21-mediated anti-apoptotic effects by activation of downstream p38 MAPK and AMPK (Figs 1f–k, 2, 4 and 6). These findings corroborate previous studies that provided indirect evidence for this mechanism. Chau et al demonstrated that FGF21 regulates mitochondrial activity and enhances oxidative capacity through an AMPK-dependent mechanism in adipocytes [32]. Ge et al reported that FGF21 increases glucose uptake through sequential activation of ERK1/2 and serum response factor (SRF)/ELK1, member of ETS oncogene family (ELK-1) in adipocytes [33]. Wente et al also documented the important role of ERK1/2 in the anti-apoptosis effects of FGF21 [14]. Here, we found that inhibition of ERK1/2, p38 MAPK or AMPK abolishes FGF21-mediated cardiac protection from palmitate-induced apoptosis in vitro (Figs 1f–k and 2a, f, k, m), and that inhibition of ERK1/2 abolished the protection by FGF21 against diabetes-induced cardiac apoptosis and cardiomyopathy (Figs 4j–o, 6c–l and Table 1). Although FGF21 activation of ERK1/2 has not been documented in cardiac cells and tissues, it has been widely studied in adipocyte, hepatocyte and beta cells, in which FGF21 induces heparin-independent tyrosine phosphorylation of FGFR substrate-2, a docking protein linking FGFRs to the Ras–MAPK pathway, and transient activation of MAPK, including ERK1/2 [14, 31, 34]. Whether FGF21 activation of ERK1/2 occurs in a similar manner in cardiac cells needs to be verified.
Clinical studies have demonstrated that elevated serum FGF21 is closely associated with hypertension [35–37]. Zhu et al revealed that FGF21 ameliorates BP in a fructose-induced hypertension model [38]. In the present study hypertension observed in a mouse model of type 1 diabetes was also prevented by FGF21 supplementation (Table 1). Therefore, FGF21-mediated anti-hypertension effects might provide an additional beneficial effect on the diabetic heart in addition to FGF21-mediated direct protection from diabetes. This important issue will be further defined in future studies.
We found that the FGF21-activated AMPK following inhibition of PTEN function provided the anti-apoptotic effect, as either inhibition or knockdown of ERK1/2 or AMPK could completely abolish the inactivation of PTEN by FGF21 (Figs 1l, 4j, o and 6d, f). PTEN is a lipid phosphatase that inhibits the phosphatidylinositol 3-kinase (PI3K) pathway, in which PI3K phosphorylates Akt to promote cell survival. The present study implies that the prevention of palmitate-induced cardiac apoptosis by FGF21 may partially depend on Akt. Our findings not only support previous studies showing that FGF21-mediated cardiac protection against ischaemia/reperfusion injury is mediated by PI3K/Akt cell survival signalling under both in vitro and in vivo conditions [18, 19], but also further establish FGF21-mediated cardiac protection against diabetic pathogenic changes via upregulation of ERK1/2–p38 MAPK–AMPK-mediated cell survival pathways.
It should be mentioned that the preventive effects of FGF21 on diabetic cardiac dysfunction were incomplete (84.5% for EF and 89.3% for LV mass index). This partial prevention may be related to the multiple pathogenic factors of diabetic cardiomyopathy [1–3]. Also, the prevention of diabetic cardiac apoptosis using FGF21 at 100 μg kg body weight−1 day−1 for 2 months may not be enough to completely prevent diabetes-induced cardiac dysfunction, and the dose will be optimised in future studies.
Conclusions
In summary, our study reveals the following new findings: (1) FGF21 is expressed in the heart, and increases in cardiac FGF21 expression in type 1 diabetes is a protective mechanism for lipotoxicity-induced cardiac apoptosis in vitro and in vivo; (2) the cardioprotective effect of FGF21 against diabetic damage is mediated by ERK1/2–p38 MAPK–AMPK cell survival pathways. This study lays the groundwork for the potential clinical use of FGF21 to treat heart disease, particularly in diabetes.
Abbreviations
- AMPK:
-
AMP-activated protein kinase
- CTGF:
-
Connective tissue growth factor
- EF:
-
Ejection fraction
- ERK:
-
Extracellular signal-regulated kinase (ERK)
- FGF21:
-
Fibroblast growth factor 21
- FGFR:
-
Fibroblast growth factor receptor
- Fgf21-KO:
-
Fgf21-knockout
- FS:
-
Fractional shortening
- LV:
-
Left ventricular
- p38 MAPK:
-
Mitogen-activated protein kinase 14
- PI3K:
-
Phosphatidylinositol 3-kinase
- PTEN:
-
Phosphatase and tensin homologue
- siRNA:
-
Small interfering RNA
- STZ:
-
Streptozotocin
- WT:
-
Wild-type
References
Bugger H, Abel ED (2014) Molecular mechanisms of diabetic cardiomyopathy. Diabetologia 57:660–671
Boudina S, Abel ED (2010) Diabetic cardiomyopathy, causes and effects. Rev Endocr Metab Disord 11:31–39
Cai L, Kang YJ (2001) Oxidative stress and diabetic cardiomyopathy: a brief review. Cardiovasc Toxicol 1:181–193
Cai L, Kang YJ (2003) Cell death and diabetic cardiomyopathy. Cardiovasc Toxicol 3:219–228
Boudina S, Abel ED (2007) Diabetic cardiomyopathy revisited. Circulation 115:3213–3223
Cai L, Wang J, Li Y et al (2005) Inhibition of superoxide generation and associated nitrosative damage is involved in metallothionein prevention of diabetic cardiomyopathy. Diabetes 54:1829–1837
Adams AC, Kharitonenkov A (2012) FGF21: the center of a transcriptional nexus in metabolic regulation. Curr Diabetes Rev 8:285–293
Cuevas-Ramos D, Almeda-Valdes P, Aguilar-Salinas CA, Cuevas-Ramos G, Cuevas-Sosa AA, Gomez-Perez FJ (2009) The role of fibroblast growth factor 21 (FGF21) on energy balance, glucose and lipid metabolism. Curr Diabetes Rev 5:216–220
Laeger T, Henagan TM, Albarado DC et al (2014) FGF21 is an endocrine signal of protein restriction. J Clin Invest 124:3913–3922
Mraz M, Bartlova M, Lacinova Z et al (2009) Serum concentrations and tissue expression of a novel endocrine regulator fibroblast growth factor-21 in patients with type 2 diabetes and obesity. Clin Endocrinol (Oxf) 71:369–375
Fon Tacer K, Bookout AL, Ding X et al (2010) Research resource: comprehensive expression atlas of the fibroblast growth factor system in adult mouse. Mol Endocrinol 24:2050–2064
Feingold KR, Grunfeld C, Heuer JG et al (2012) FGF21 is increased by inflammatory stimuli and protects leptin-deficient ob/ob mice from the toxicity of sepsis. Endocrinology 153:2689–2700
Ye D, Wang Y, Li H et al (2014) Fibroblast growth factor 21 protects against acetaminophen-induced hepatotoxicity by potentiating peroxisome proliferator-activated receptor coactivator protein-1alpha-mediated antioxidant capacity in mice. Hepatology 60:977–989
Wente W, Efanov AM, Brenner M et al (2006) Fibroblast growth factor-21 improves pancreatic beta-cell function and survival by activation of extracellular signal-regulated kinase 1/2 and Akt signaling pathways. Diabetes 55:2470–2478
Lu Y, Liu JH, Zhang LK et al (2010) Fibroblast growth factor 21 as a possible endogenous factor inhibits apoptosis in cardiac endothelial cells. Chin Med J (Engl) 123:3417–3421
Suzuki M, Uehara Y, Motomura-Matsuzaka K et al (2008) beta Klotho is required for fibroblast growth factor (FGF) 21 signaling through FGF receptor (FGFR) 1c and FGFR3c. Mol Endocrinol 22:1006–1014
Kurosu H, Choi M, Ogawa Y et al (2007) Tissue-specific expression of beta Klotho and fibroblast growth factor (FGF) receptor isoforms determines metabolic activity of FGF19 and FGF21. J Biol Chem 282:26687–26695
Liu SQ, Roberts D, Kharitonenkov A et al (2013) Endocrine protection of ischemic myocardium by FGF21 from the liver and adipose tissue. Sci Rep 3:2767
Cong WT, Ling J, Tian HS et al (2013) Proteomic study on the protective mechanism of fibroblast growth factor 21 to ischemia-reperfusion injury. Can J Physiol Pharmacol 91:973–984
Planavila A, Redondo I, Hondares E et al (2013) Fibroblast growth factor 21 protects against cardiac hypertrophy in mice. Nat Commun 4:2019
Zhou G, Li X, Hein DW et al (2008) Metallothionein suppresses angiotensin II-induced nicotinamide adenine dinucleotide phosphate oxidase activation, nitrosative stress, apoptosis, and pathological remodeling in the diabetic heart. J Am Coll Cardiol 52:655–666
Tan Y, Li X, Prabhu SD et al (2012) Angiotensin II plays a critical role in alcohol-induced cardiac nitrative damage, cell death, remodeling, and cardiomyopathy in a protein kinase C/nicotinamide adenine dinucleotide phosphate oxidase-dependent manner. J Am Coll Cardiol 59:1477–1486
Zhao Y, Tan Y, Xi S et al (2013) A novel mechanism by which SDF-1beta protects cardiac cells from palmitate-induced endoplasmic reticulum stress and apoptosis via CXCR7 and AMPK/p38 MAPK-mediated interleukin-6 generation. Diabetes 62:2545–2558
Luo J, Hill BG, Gu Y et al (2007) Mechanisms of acrolein-induced myocardial dysfunction: implications for environmental and endogenous aldehyde exposure. Am J Physiol Heart Circ Physiol 293:H3673–H3684
Cameron TL, Bell KM, Tatarczuch L et al (2011) Transcriptional profiling of chondrodysplasia growth plate cartilage reveals adaptive ER-stress networks that allow survival but disrupt hypertrophy. PLoS One 6:e24600
Schaap FG, Kremer AE, Lamers WH, Jansen PL, Gaemers IC (2013) Fibroblast growth factor 21 is induced by endoplasmic reticulum stress. Biochimie 95:692–699
Wang J, Song Y, Elsherif L et al (2006) Cardiac metallothionein induction plays the major role in the prevention of diabetic cardiomyopathy by zinc supplementation. Circulation 113:544–554
Cai L, Wang Y, Zhou G et al (2006) Attenuation by metallothionein of early cardiac cell death via suppression of mitochondrial oxidative stress results in a prevention of diabetic cardiomyopathy. J Am Coll Cardiol 48:1688–1697
Potthoff MJ, Inagaki T, Satapati S et al (2009) FGF21 induces PGC-1alpha and regulates carbohydrate and fatty acid metabolism during the adaptive starvation response. Proc Natl Acad Sci U S A 106:10853–10858
Xu J, Lloyd DJ, Hale C et al (2009) Fibroblast growth factor 21 reverses hepatic steatosis, increases energy expenditure, and improves insulin sensitivity in diet-induced obese mice. Diabetes 58:250–259
Kharitonenkov A, Shiyanova TL, Koester A et al (2005) FGF-21 as a novel metabolic regulator. J Clin Invest 115:1627–1635
Chau MD, Gao J, Yang Q, Wu Z, Gromada J (2010) Fibroblast growth factor 21 regulates energy metabolism by activating the AMPK-SIRT1-PGC-1alpha pathway. Proc Natl Acad Sci U S A 107:12553–12558
Ge X, Chen C, Hui X, Wang Y, Lam KS, Xu A (2011) Fibroblast growth factor 21 induces glucose transporter-1 expression through activation of the serum response factor/Ets-like protein-1 in adipocytes. J Biol Chem 286:34533–34541
Kouhara H, Hadari YR, Spivak-Kroizman T et al (1997) A lipid-anchored Grb2-binding protein that links FGF-receptor activation to the Ras/MAPK signaling pathway. Cell 89:693–702
Semba RD, Crasto C, Strait J, Sun K, Schaumberg DA, Ferrucci L (2013) Elevated serum fibroblast growth factor 21 is associated with hypertension in community-dwelling adults. J Hum Hypertens 27:397–399
Lin Z, Wu Z, Yin X et al (2010) Serum levels of FGF-21 are increased in coronary heart disease patients and are independently associated with adverse lipid profile. PLoS One 5:e15534
Chow WS, Xu A, Woo YC et al (2013) Serum fibroblast growth factor-21 levels are associated with carotid atherosclerosis independent of established cardiovascular risk factors. Arterioscler Thromb Vasc Biol 33:2454–2459
Zhu SL, Ren GP, Zhang ZY et al (2013) Therapeutic effect of fibroblast growth factor 21 on hypertension induced by insulin resistance. Yao Xue Xue Bao 48:1409–1414 (article in Chinese)
Acknowledgements
Some of the data were presented as an abstract at the 18th Annual Meeting of the Chinese Diabetes Society in 2014, Guangzhou, People’s Republic of China.
Funding
This study was supported in part by: the Junior Faculty Award (1-13-JF-53) from the American Diabetes Association; the Zhejiang Province Extremely Key Subject Building Project (Pharmacology and Biochemical Pharmaceutics 2009); the Starting-Up Fund for Chinese-American Research Institute for Diabetic Complications from Wenzhou Medical University (QTJ13007); grants from the National Science Foundation of China (81102486, 81273509, 81000294, 81370917, 81473261, 32500301), the Science Foundation of Zhejiang Province of China (Y14H070033), the Research Development Fund of Wenzhou Medical University (QTJ13005) and the Changjiang Innovation Team Program (2010R50042-17); a Key New Drug Development Grant (2012ZX09103-301-016); and Public Project of Science and Technology of Wenzhou City (Y2014073).
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Contribution statement
CZ, XiL and YT conceived and designed the experiments. CZ, ZH, JG, XY, XuL, SZ, MS, FZ, PC, WF, XiL and YT contributed to the acquisition of data. CZ, XiL, SW and YT analysed and interpreted the data. CZ, XiL and YT contributed to drafting the article. All authors have revised the manuscript critically for important intellectual content and approved the final version to be published.
Author information
Authors and Affiliations
Corresponding authors
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM Methods
(PDF 212 kb)
ESM Fig. 1
(PDF 212 kb)
ESM Fig. 2
(PDF 26 kb)
ESM Fig. 3
(PDF 153 kb)
ESM Fig. 4
(PDF 111 kb)
ESM Fig. 5
(PDF 130 kb)
Rights and permissions
About this article

Cite this article
Zhang, C., Huang, Z., Gu, J. et al. Fibroblast growth factor 21 protects the heart from apoptosis in a diabetic mouse model via extracellular signal-regulated kinase 1/2-dependent signalling pathway. Diabetologia 58, 1937–1948 (2015). https://doi.org/10.1007/s00125-015-3630-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-015-3630-8